

Abstract
Phosphatidylinositol (3–5) trisphosphate (PIP3) is a central regulator of diverse neuronal functions that are critical for seizure progression, however its role in seizures is unclear. We have recently hypothesised that valproic acid (VPA), one of the most commonly used drugs for the treatment of epilepsy, may target PIP3 signalling as a therapeutic mode of action. Here, we show that seizure induction using kainic acid in a rat in vivo epilepsy model resulted in a decrease in hippocampal PIP3 levels and reduced protein kinase B (PKB/AKT) phosphorylation, measured using ELISA mass assays and Western blot analysis, and both changes were restored following VPA treatment. These finding were reproduced in cultured rat hippocampal primary neurons and entorhinal cortex–hippocampal slices during exposure to the GABA(A) receptor antagonist pentylenetetrazol (PTZ), which is widely used to generate seizures and seizure-like (paroxysmal) activity. Moreover, VPA's effect on paroxysmal activity in the PTZ slice model is blocked by phosphatidylinositol 3-kinase (PI3K) inhibition or PIP2 sequestration by neomycin, indicating that VPA's efficacy is dependent upon PIP3 signalling. PIP3 depletion following PTZ treatment may also provide a positive feedback loop, since enhancing PIP3 depletion increases, and conversely, reducing PIP3 dephosphorylation reduces paroxysmal activity and this effect is dependent upon AMPA receptor activation. Our results therefore indicate that PIP3 depletion occurs with seizure activity, and that VPA functions to reverse these effects, providing a novel mechanism for VPA in epilepsy treatment.
Keywords: Kainic acid, Pentylenetetrazol, PKB/AKT, mTOR2, PIP3, Seizure control, Valproic acid (VPA)
Graphical abstract

Highlights
-
•
In vivo seizure induction (using kainic acid) reduces hippocampal PIP3 levels.
-
•
In vivo seizure induction (using kainic acid) reduces hippocampal phospho-PKB levels.
-
•
Valproic acid protects against these reductions under seizure conditions only.
-
•
Similar regulation is seen with PTZ-induced in vitro seizure activity.
-
•
Seizure-induced PIP3 reduction causes a feedback activation of seizure activity.
Introduction
Despite the emergence of many new antiepileptic drugs in the last couple of decades (Bialer and White, 2010), approximately one-third of all epilepsy patients continue to have poorly controlled seizures (Kwan and Brodie, 2006). As a result, there has been considerable interest in defining the cellular and molecular changes in the brain that contribute to the occurrence of spontaneous seizures and epilepsy (Bialer et al., 2010). Understanding these will lead to therapeutic strategies not only for controlling the development of epilepsy (epileptogenesis) after a brain injury, but also for identifying new targets for treating seizures.
The phosphoinositide, phosphatidylinositol (3–5)-trisphosphate (PIP3), contributes to cell signalling and has an important regulatory role not only in acute cellular physiology (e.g. synaptic transmission) but also in maintaining basal cellular activity (Vanhaesebroeck et al., 2012). Phosphoinositide levels are controlled by a complex range of lipid kinases and phosphatases (Andrews et al., 2007, Cantley, 2002, Di and De, 2006, Wu and Hu, 2010). In the case of PIP3, the final step in its production is catalysed by phosphatidylinositol 3 kinase (PI3K) and the cellular effects of PIP3 are inhibited by dephosphorylation through phosphatases, including the SH2 domain-containing inositol 5′-phosphatase 2 (SHIP2) (Andrews et al., 2007, Suwa et al., 2009) and the phosphatase and tensin homolog (PTEN). The cellular function of PIP3 is mediated through direct binding to a wide range of proteins and the downstream regulation of the protein kinase B (PKB or AKT) signalling pathway (Andrews et al., 2007, Wu and Hu, 2010).
Several recent studies have implicated the PI3K/PIP3/AKT pathway in seizure generation and epilepsy. The pro-apoptotic activity of the Bcl-2 interacting mediator of cell death (Bim) pathway is regulated by PIP3-dependent phosphorylation of AKT, which is upregulated after seizures in animal models, and is altered in the hippocampus of patients with intractable epilepsy (Shinoda et al., 2004). The mammalian target of rapamycin (mTOR) pathway, which is a target of AKT and thus is also activated by PIP3, plays an important role in epileptogenesis in models of chronic epilepsy and acute seizure activity (Buckmaster and Lew, 2011, Zeng et al., 2009, Zhang and Wong, 2012). Finally, deletion of PTEN in dentate granule cells results in spontaneous seizures and abnormal electroencephalogram (EEG) activity (Backman et al., 2001, Ljungberg et al., 2009). Therefore, PIP3 and the phosphoinositide pathway in general, provide excellent candidate targets for regulating ictogenesis and epileptogenesis.
VPA (valproic acid, 2-propylpentanoic acid) is a commonly used broad-spectrum antiepileptic drug (Loscher, 1999) with multiple mechanisms (Balding and Geller, 1981, Boeckeler et al., 2006, Chang et al., 2010, Elphick et al., 2011, Lagace et al., 2005, Terbach and Williams, 2009). A number of these mechanisms could be explained by an action of VPA on phosphoinositides. We have recently shown that VPA rapidly attenuates the turnover of phosphoinositides in a simple biomedical model, Dictyostelium (Chang et al., 2012, Xu et al., 2007), and this predicts seizure control activity in mammalian in vitro seizure models (Chang et al., 2012, Chang et al., 2013). However, an established role for VPA in regulating seizure-dependent phosphoinositide turnover has yet to be established.
Here, we investigated the regulation of PIP3 in an in vivo kainic acid induced seizure model, in an in vitro neuronal culture model for PTZ-induced burst activity and in an in vitro model of induced paroxysmal activity in ex-vivo slices using PTZ, using radio-labelled inositol, PIP3 ELISA mass assays, and Western blot analysis. Using these readouts, we show that PIP3 levels decrease in all these models and that VPA restores PIP3 level, providing a novel mechanistic insight into VPA function. We further show that modulating phosphoinositide signalling regulates both paroxysmal activity and the efficacy of VPA in regulating these seizure-associated activities. Together our results indicate that PIP3 depletion is a critical step in PTZ/kainic acid-induced seizure progression and that VPA acts on this pathway, providing a novel mechanistic target for seizure control.
Methods
Chemicals
All chemicals were provided by Sigma Pty Ltd (unless otherwise stated).
Animals
Male Sprague–Dawley rats (SD) were kept under controlled environmental conditions (24–25 °C; 50–60% humidity; 12 h light/dark cycle) with free access to food and water. All the experiments were approved by a local ethics committee, the UK home office and performed in accordance with the guidelines of the Animals (scientific procedure) Act 1986.
Kainic acid treatment to induced status epilepticus
Male Sprague–Dawley rats (300–350 mg) were given kainic acid (Tocris Biosciences) at a dose of 10 mg/kg (Gupta et al., 2002), or saline by intraperitoneal injection. Experimental animals were than monitored to determine the severity of seizures. The rating of the severity of seizures was based on the Racine scale (stage 1, mouth and facial movements; stage 2, head nodding and more severe facial and mouth movements; stage 3, forelimb clonus; stage 4, rearing and bilateral forelimb clonuses; stage 5, rearing and falling, with loss of postural control, full motor seizure) (Racine et al., 1972). Onset of seizures occurred 30–100 min after kainic acid injection. One hour after the animals reached stage 5 behavioural seizures, single doses of either saline or VPA (400 mg/kg) were separately administered intraperitoneally and 1 h after drug application, the animals were sacrificed by being placed in a CO2 chamber (10 L volume chamber with a flow rate of 4 L/min). The hippocampi were then collected for further analysis, including PIP3 assay (using total protein as a loading control) and Western blot analysis (as described below).
Western blot analysis
Brain tissue was homogenized by 10 up-and-down strokes of a homogenizer in 10 times the brain tissue volume of aCSF (in mM: NaCl 119, KCl 2.5, MgSO4 1.3, CaCl2 2.5, NaH2PO4 1, NaHCO3 26.2 and glucose 16.6). After centrifugation, cells were washed with ice-cold phosphate buffered saline (PBS), followed by lysis for 5 min in ice-cold RIPA Buffer supplemented with protease (Complete mini EDTA free, Roche) and phosphatase inhibitors (PhosStop, Roche). Cell lysates were centrifuged at 12,000 g for 5 min, and equal amounts of protein supernatant (20 μg) were separated by 10% SDS-PAGE and transferred onto PVDF membrane (Immobilon ®-FL transfer membrane, Millipore). Membranes were blocked with Tris-buffered saline–tween-20 (TBST) containing 5% BSA and incubated with primary antibodies (AKT and phospho-AKT(Ser473), Cell Signalling 587 F11) overnight at 4 °C. After washing with TBST, membranes were incubated with secondary antibodies (IRDye 800CW Goat anti-Rabbit and IRDye 800CW Goat anti-Mouse, Odyssey) for 1 h at room temperature. After a second round of washing with TBST, the immuno-reactive bands were visualized using an Odyssey Infrared Imaging System.
PI(3–5)P3 ELISA mass assay
A PIP3 ELISA mass assay (Echelon Biosciences, Inc.) was used as an independent means to determine the relative amount of PIP3 present in treated or untreated neuronal cell cultures. For primary neuronal cultures, tissue culture media were removed from cells and the experiments were stopped by the addition of 0.5 mL of ice-cold 0.5 M TCA. For hippocampal slice experiments, brain tissue was homogenized in 10 times the brain tissue volume of aCSF by 10 up-and-down strokes of a tissue homogenizer (Dounce tissue grinder). Lipid extraction was carried out as described below, and for each condition, the PIP3 analysis was carried out as described in the manufacturer's instructions, with the colorimetric signal measured by a plate reader at 450 nm.
Hippocampal neuronal cultures
Neuronal cultures were prepared from the hippocampi of P0–P3 postnatal rats. Brain dissection was performed in ice-cold dissection solution (in mM: NaCl 137, 5.4 KCl, Na2HPO4 0.25, KH2PO4 0.44, CaCl2 1.3, MgSO4 1.0, NaHCO3 4.2, HEPES 5, and 20%FBS). The dissected tissue was then incubated for 10 min in 0.25% (w/v) trypsin in 37 °C and washed twice with washout solution (in mM: NaCl 137, KCl 5.4, Na2HPO4 0.25, KH2PO4 0.44, CaCl2 1.3, MgSO4 1.0, NaHCO3 4.2, HEPES 5) and dissociated by trituration in the presence of DNase (150 mg/mL). Cells were then plated on poly-l-lysine-coated 12-well plates with neurobasal A medium, containing supplement B27 (and 25 μM glutamine), and AraC (1 μM) was added to the cultures within 48 h of plating. The medium was changed once per week. Experiments were carried out 14 days after neuron cell preparation.
Cell radiolabelling and detection
Primary hippocampal cells in 12-well plates were labelled by addition of 0.1 μM myo-2-[3H]-inositol (20 Ci/mmol) (Hartmann Analytic GmbH) to the medium overnight. Cells were then washed with unlabelled medium prior experimentation. To analyse PTZ-induced activity, cells were treated with 5 mM PTZ for 20 min (Sugaya et al., 1989), followed by addition of drugs for 30 min. Following PTZ-induction and drug treatment, the reactions were terminated by replacing media with 500 μL of ice-cold 0.5 M trichloroacetic acid (TCA), cells were then scraped, collected, and cell pellets were washed twice with 5 mL of 300 μM TCA containing 1 mM EDTA.
Analysis of 3H-inositol distribution involved the extraction of labelled cell pellets with 0.75 mL of chloroform/methanol/12 N HCl (2/4/0.1, v/v). After thorough mixing, chloroform (0.25 mL) and water (0.25 mL) were then added and vortexed. The samples were centrifuged at a low speed for 20 min, with 100 μL of the resulting aqueous phase and organic phase used for [3H]-inositol phosphate and [3H]-inositol lipid, analysis respectively by scintillation counting. The remaining lower organic phase containing the [3H]-inositol lipids was also collected, dried under vacuum, and analysed by thin-layer chromatography (TLC). Lipids were separated in: chloroform/acetone/methanol/acetic acid/water (40/13/15/12/7 v/v), using TLC plates (silica gel 60 (VWR Pty Ltd)), pretreated by coating with a mixture of methanol/water (2/3 v/v) containing 1% potassium oxalate (w/v) followed by desiccated at 100 °C for 20–30 min before use. Phosphoimaging plates (Fuji BAS-TR2040) were used to monitor [3H]-inositol lipid distribution by at least 14 day exposure, prior to visualization on a Typhoon phosphoimager. Individual lipid spots were then quantified by liquid-scintillation (Beckman Pty Ltd). Authentic standards were used for PIP2, PIP, and Pl, and the lipids were visualized using 0.2% (w/v in methanol) 8-Anilino-1-naphthalenesulfonic acid ammonium salt, or 10% cupric sulphate in 8% aqueous phosphoric acid, allowed to dry 10 min at room temperature, and then placed into a 145 °C oven for 10 min (Supplementary Fig. 1).
Supplementary Fig. 1.

Quantification of 3H-inositol labelled phosphoinositide derived from primary hippocampal neurons using known standards. (A) Separation of known tritiated phospholipids (left and right) and 3H-inositol labelled phosphoinositide derived from hippocampal neurons (centre). Individual lipid spots were quantified by liquid-scintillation (Beckman Pty Ltd). The right hand panel (A) shows authentic standards used for PIP2, PIP, and Pl, with lipids visualized using 10% cupric sulphate in 8% aqueous phosphoric acid, allowed to dry for 10 min at room temperature, and then placed into a 145 °C oven for 10 min. The left hand panel (A) shows specific labelled lipids prepared using defined phosphoinositides in lipid vesicles incubated with 32P-ATP in the presence of phospholipid kinase (Xu et al., 2007). The central panel (A) shows example lipid extracts from primary hippocampal neurons following TLC separation and phosphoimaging. Phosphoimager plates (Fuji BAS-TR2040) were used to monitor [3H]-inositol lipids distribution imaged on a Typhoon phosphoimager. (B) Calculated RF values and assigned phospholipid species.
In vitro electrophysiology
The preparation of entorhinal cortex-hippocampus slices and electrophysiological recording in CA1 has been described previously (Armand et al., 1998, Chang et al., 2010). In brief, SD rats (P20–P30, 50–150 g) were decapitated after killing by intraperitoneal injection with an overdose of pentobarbitone (500 mg/kg). The brain was removed and placed in oxygenated ice-cold sucrose solution (in mM: NaCl 87, KCl 2.5, MgCl2 7, CaCl2 0.5, NaH2PO4 1.25, NaHCO3 26.2, sucrose 75 and glucose 3). Slices (350 μm) were prepared with a Vibratome (Vibratome® 1500 sectioning system, Intracell) and were then stored in an interface chamber containing aCSF for at least 1 h. During the experiment, slices were transferred from the interface chamber into a submerged recording chamber and continuously perfused using gravity feed at 3-6 mL/min with prewarmed (36 °C) oxygenated aCSF (95% O2, 5% CO2). Field potentials were recorded with a glass microelectrode (1–2 MΩ) filled with aCSF solution placed in stratum radiatum of CA1 and were filtered at 1 kHz and digitized at 2 kHz (using an npi EXT-02 F extracellular amplifier recorded with WinEDR software). To induce epileptiform (paroxysmal) activity, PTZ (2 mM) was added to the perfusate and [K+] was increased (to 6 mM). Once the frequency of the paroxysmal activity was stable for at least 10 min, compounds were applied to the perfusate for the following 40 min, and washed out for a remaining 20 min. The anticonvulsant effects were evaluated by measuring the change in the frequency of the discharges at minute intervals. The discharge frequency was then averaged every 5 min during the experiment and normalised to baseline. The compounds PI103 (Echelon Biosciences, Inc.), AS 1949490 (Tocris Bioscience Pty Ltd), LY 294002 (Echelon Biosciences, Inc.), CNQX and kynurenic acid were prepared as 1000 times stocks in dimethylsulfoxide (DMSO). VPA was dissolved in distilled water as a 1000 times stock. Stocks were dissolved in aCSF to achieve their final concentrations during experiments.
Data analysis
Results are presented as mean ± SEM. Statistical comparisons were performed by using the paired Student's t-test or one way ANOVA followed by Tukey for post-hoc analysis for significant main effects by using GraphPad Prism. Statistical significance was taken as follows: ∗ p < 0.05; ∗∗p < 0.01, and ∗∗∗p < 0.005.
Results
An in vivo decrease in hippocampal pSer473 AKT and PIP3 levels in a kainic acid-induced status epilepticus model is restored by VPA
To examine a role for PIP3 during seizure activity and following VPA treatment, we first employed an in vivo seizure model where status epilepticus was induced using kainic acid. In these experiments, repeated intraperitoneal injection of kainic acid (10 mg/kg) (Gupta et al., 2002) was used until animals developed stage 5 behaviour seizures, and animals were then administered with intraperitoneal VPA (400 mg/kg) or saline control, and hippocampi were removed after 1 h. As reported previously (Chang et al., 2013), VPA caused a significant reduction in behavioural seizure score, as described by the Racine scale, in these experiments (Supplementary Fig. 2). We then determined the effect of VPA on PIP3 levels in vivo, both using Western analysis to monitor the phosphorylation state of AKT and by direct monitoring of PIP3 levels using an ELISA mass assay technique.
Supplementary Fig. 2.

The effect of VPA on seizure severity during self-sustaining status epilepticus induced by kainic acid from data presented in Fig. 1. For each animal, the Racine score was recorded before and after application of compounds. There are two animals (two out of six) that received saline and in which the behavioural seizure ceased/decreased over 1 h ; these were excluded from the results. Summary data of the effects of application of VPA (400 mg/kg) on seizure behaviour over 1 h post-administration. Two way ANOVA test with Tukey's multiple comparisons test was used for statistical analysis. ***p < 0.001.
Since PIP3 is critical for the recruitment of cytosolic AKT to the plasma membrane, enabling Thr308 phosphorylation by phosphoinositide-dependent kinase 1, and subsequent Ser473 phosphorylation by the rapamycin insensitive mTORC2 complex (Alessi et al., 1997, Apsel et al., 2008, Sarbassov et al., 2005, Stokoe et al., 1997), the phosphorylation state of AKT can be used as a readout for PIP3 levels (Huang et al., 2011). To assess the phosphorylation state of AKT during seizures and following VPA treatment, hippocampi were extracted from animals and analysed by Western blot using total AKT and pSer473 AKT antibodies (Figs. 1A, B). These experiments showed a significant decrease in AKT phosphorylation after seizure induction (49.9 ± 9.3% of control, N = 4, p = 0.045)(Figs. 1A, B). Treatment of animals during seizure activity with VPA restored AKT phosphorylation to control levels (101.0 ± 13.6% of control, N = 4, p = 0.005 compared to kainic acid treatment only). However, VPA treatment in the absence of convulsant did not significantly alter AKT phosphorylation levels (102.0 ± 5.5% of control, N = 3). These data suggest that seizure activity causes a reduction in PIP3 levels and VPA acts to block this reduction.
Fig. 1.

In vivo status epilepticus activity induced by kainic acid decreases Ser473 AKT phosphorylation and PIP3 levels but these are restored by VPA. Animals were induced to stage 5 (Racine) behavioural seizures using kainic acid (KA; 10 mg/kg), single doses of either saline or VPA (400 mg/kg) were then administered intraperitoneally, and hippocampi were extracted and analysed. (A) Western immunoblot assays were used to determine levels of phosopho-Ser473 (pAKT) and total (tAKT) AKT protein during status activity or following VPA treatment, and (B) summarised for control (non-induced; N = 3), control with VPA (N = 3), kainic acid (N = 4), and kainic acid with VPA (N = 4) treated animals, with pAKT normalised to tAKT levels. (C) Derived samples were also analysed by direct PIP3 ELISA mass assay. Graphs show means ± SEM, with individual data point illustrated. Statistical analysis was performed by post-hoc Tukey test, following ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001).
We then confirmed the accuracy of our Western analysis by directly measuring hippocampal PIP3 levels using a PIP3 ELISA mass assay technique (Fig. 1C). In these parallel experiments, PIP3 was extracted from in vivo hippocampi samples of treated animals and analysed, where we observed a significant decrease in PIP3 after seizure induction (69.1 ± 6.5% of control, N = 4, p = 0.003) (Fig. 1C), consistent with the decrease shown for phosphorylated AKT levels. Treatment of animals during seizure activity with VPA also restored PIP3 to control levels (111.0 ± 7.4% of control, N = 4, p = 0.005 compared to kainic acid treatment only). VPA treatment in the absence of convulsant did not significantly alter PIP3 levels (98.5 ± 4.6% of control, N = 3). These experiments indicate a decrease in vivo hippocampal PIP3 levels during kainic acid-induced seizure activity that is restored by treatment with VPA, but also indicate that VPA in the absence of such activity does not change PIP3 levels. Interestingly, separation of data derived from kainate animals treated with saline only injections (control) between animals that continued to seize 1 h post-injection and those that showed reduced seizure-like behaviour suggests that the reduction in PIP3 levels is dependent on the presence of seizure activity (Supplementary Fig. 3).
Supplementary Fig. 3.

State dependence of PIP3 regulation in in vivo kainic acid-induced status epilepticus. Six animals reached stage 5 behavioural seizure following kainic acid induction (KA; 10 mg/kg), and a single dose of saline was then administered intraperitoneally, and animals were observed for another hour. From these animals, 2 out of 6 showed a decrease or cessation of seizure behaviour over this period, and these were compared. (A) Western immunoblot assays were performed to determine levels of phosopho-Ser473 (pAKT) and total (tAKT) AKT protein, and summarised for control (non-induced; N = 3), kainic acid (N = 4), and kainic acid with a decrease or cessation of seizure behaviour (N = 2), with pAKT normalised to tAKT levels. (B) Derived samples were also analysed by direct PIP3 ELISA mass assay. Graphs show means ± SEM with individual data point illustrated.
PIP3 levels decrease in hippocampal neurons during PTZ-induced activity and are restored by VPA
In order to investigate the role of phosphoinositide turnover generally in seizure progression and as a target for VPA, we then employed an in vitro model of bursting activity, using PTZ applied to cultured rat primary hippocampal neurons. In these experiments we labelled the cultured rat primary hippocampal neurons with myo-[3H]-inositol which is incorporated into phosphoinositides (PI, PIP, PIP2, and PIP3) or cleaved from PIP3 by PLC to release the inositol phosphate, InsP3 (Fig. 2A). Phosphoinositides were quantified by lipid extraction and separation by thin layer chromatography (TLC) (Fig. 2B), using purified phosphoinositides as controls (Supplementary Fig. 1). Phosphoinositide turnover in these experiments was determined by measuring the change of [3H]-labelled phosphoinositide distribution following application of the convulsant PTZ (5 mM) (Sugaya et al., 1989). Previous studies have shown that application of PTZ induces bursting activity in primary cultured neurons, in a similar manner to seizure-like activity in hippocampal slices or in vivo (Sugaya et al., 1989). Repeating this approach, we observed a significant decrease in PIP3 after application of PTZ (62.8 ± 3.8% of control, N = 18, p = 0.001 compared to control) (Figs. 2B, C), whereas there was no change in PI, PIP, and PIP2 levels, nor in cytosolic inositol phosphates (Supplementary Fig. 4). Similar to the in vivo results, application of VPA (1 mM; a physiologically relevant level commonly used in these studies (Chang et al., 2012, Chang et al., 2013)), under burst-inducing conditions, restored PIP3 levels to those of control (92.2 ± 7.7% of control, N = 15, p = 0.012 compared to PTZ) (Figs. 2B, C). However (as observed in the in vivo experiments), application of VPA under control conditions (in the absence of PTZ) did not change PIP3 levels (106.0 ± 11.7% of control, N = 9) suggesting an effect of VPA on PIP3 levels only under burst-inducing conditions.
Fig. 2.

Seizure-inducing conditions in primary rat hippocampal neurons decrease PIP3 levels but this is restored by VPA. (A) Phosphoinositide synthesis and interconversion can be monitored by the use of radio-labelled myo-inositol (MI) that is incorporated into phosphatidylinositol (PI), then phosphatidylinositol 4 phosphate (PIP) and phosphatidylinositol 4,5 bisphosphate (PIP2) by sequential phosphorylation of the inositol ring by 4′ kinases (PI4K) and 5′ kinases (PI5K), respectively. Red circles represent phosphates. PIP2 is cleaved by phospholipase C (PLC) to produce aqueous inositol trisphosphate (IP3) or is phosphorylated by 3′ kinases (PI3K) to produce PIP3. (B) Example of phosphoinositide labelling in primary hippocampal neurons under seizure-inducing conditions (PTZ at 5 mM), imaged following separation by TLC. (C) Comparison of level of PIP3 in primary hippocampal neurons under control condition (N = 18) and with VPA alone (1 mM, N = 9), or in seizure-inducing conditions in the absence (N = 18) or presence of VPA (1 mM, N = 14) measured by quantification of TLC separated phosphoinositides in comparison with standards (Supplementary Fig. 1). (D) Comparison of PIP3 level in primary hippocampal neurons under control conditions (control, N = 7) or with VPA alone (N = 5), or in seizure-inducing conditions in the absence (N = 4) or presence of VPA (1 mM; N = 5), or in control condition with PI3K inhibitor (10 μM, LY-294002, LY, N = 3) measured by PIP3 ELISA mass assay. Graphs show means ± SEM, with individual data point illustrated. Statistical analysis was performed by post-hoc Tukey test, following ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001). Data for other inositol phosphate species are provided in Supplementary Fig. 4.
Fig. 4.

VPA control of epileptiform activity is PIP3-dependent. (A) Example trace recording of epileptiform activity (burst discharges) in hippocampal slices induced by application of PTZ (PTZ 2 mM, K+ 6 mM) under control conditions (DMSO) and with VPA (1 mM); or (B) with VPA (1 mM) after treatment with PI3K inhibitors (PI-103 (10 μM); LY-294002 (LY, 20 μM)) and following sequestration of PIP2 with Neomycin (Neo, 100 μM) for 30 min. (C) Summary of PTZ-induced epileptiform discharge data for hippocampal slices with VPA treatment only (control) or with prior application of PI-103 (N = 4), LY-294002 (N = 4), or Neomycin (N = 5), where VPA was applied between 0 and 40 min. (D) Comparison of the mean frequency of epileptiform activity under different conditions, averaged from 20 to 40 min post-treatment. Graphs show means ± SEM, with individual data point illustrated. Statistical analysis was performed by post-hoc Tukey test, following ANOVA (**p < 0.01, ***p < 0.001).
We also repeated these experiments using the PIP3 ELISA mass assay to confirm the accuracy of our [3H]-labelling experiments. Here, we reproduced the cultured rat primary hippocampal neuron experiments, and exposed cells to PTZ (5 mM) in the presence or absence of VPA (Fig. 2D). Consistent with the previous experiments, administration of PTZ significantly decreased PIP3 level (44.9 ± 8.7% of control, N = 4 independent experiments in triplicate, p = 0.000, compared to control). Application of VPA (1 mM) restored PIP3 levels (81.9 ± 10.23% of control, N = 5 independent experiments in triplicate, p = 0.049), whereas application of VPA (1 mM) in control conditions (in the absence of PTZ) had no significant effect on PIP3 level (93.0 ± 12.4% of control, N = 5 independent experiments in triplicate). As a control we confirmed that inhibition of PI3K activity (using 10 μM LY294002) (Lee et al., 2005, Workman et al., 2010) resulted in a large reduction in PIP3 level (45.0 ± 6.9% of control, N = 3 independent experiments in triplicate).
pSer473 AKT and PIP3 levels decrease in vitro in hippocampal slices during PTZ-induced paroxysmal activity and are restored by VPA
We then correlated paroxysmal activity with PIP3 levels in an in vitro seizure model using ex vivo rat hippocampal slices treated with PTZ. Paroxysmal activity in these slices was recorded by field potential monitoring and pSer473 AKT phosphorylation and PIP3 levels were measured, pre- and post-VPA treatment. In these experiments, epileptiform discharges (paroxysmal activity) appeared 10–30 min after application of PTZ and increased extracellular [K+] (to 6 mM) (Sugaya et al., 1989) (Fig. 3A). Epileptiform discharges consisted of a positive field potential on which population spikes were superimposed. In this model, VPA caused a small but significant decrease in the frequency of PTZ-induced epileptiform discharges, 20–40 min after application (to 76.5 ± 2.2% of baseline N = 5, control 99.8 ± 3.5% of baseline N = 5) (Figs. 3B, C), as has been previously shown (Chang et al., 2012, Heinemann et al., 1994). Changes in PIP3 levels in hippocampal slices during seizure induction were then measured by Western blot analysis using total AKT and pSer473 AKT antibodies, and by PIP3 ELISA mass (Figs. 3D, E). These experiments demonstrated that application of PTZ significantly decreased pAKT levels (49.3 ± 4.7% of control, N = 5, p = 0.00, compared to control) with no significant change in total AKT levels (104.7 ± 5.7% of control, N = 5) (Fig. 3D). Application of VPA (1 mM) to the slices during paroxysmal activity induction partially restored pAKT levels (77.1 ± 8.6% of control, N = 5, p = 0.012, compared to PTZ). Application of VPA (1 mM) in control condition had no effect on pAKT and total AKT levels (90.7 ± 4.7% of control, N = 5, 101.7 ± 6.3% of control, N = 5, respectively), consistent with the PIP3 data that the effect of VPA was only evident during paroxysmal activity. Using the ELISA mass assay approach, these hippocampal slices showed a significant decrease in PIP3 (41.3 ± 7.4% of control, N = 13) under bursting conditions (Fig. 3E). Application of VPA restored PIP3 levels to that of control (88.9 ± 13.9% of control, N = 11). Furthermore, as shown in the dissociated primary neurons in culture experiments, VPA did not change the level of PIP3 under control conditions (91.91 ± 12.66% of control, N = 9), confirming that the effect of VPA on PIP3 was only evident during paroxysmal activity.
Fig. 3.

Epileptiform activity in rat hippocampal slices decreases Ser473 AKT phosphorylation and PIP3 levels but these are restored by VPA. (A) Example trace recording of epileptiform activity (burst discharges) in hippocampal slices induced by application of PTZ (PTZ 2 mM, K+ 6 mM) and (B) following addition of VPA (1 mM). (C) Comparison of the mean frequency of burst discharges in the presence or absence of VPA (1 mM), expressed as a percentage of the baseline value (N = 5). (D) Western immunoblot assays show levels of phosopho-Ser473 (pAKT) and total (tAKT) AKT protein during epileptiform activity in hippocampal slices with summary data (N = 5) showing pAKT normalised to tAKT levels. Graphs show means ± SEM with individual data point illustrated. (E) Comparison of PIP3 level in the same hippocampal slices (control (N = 15), VPA (1 mM, N = 9)), and in seizure condition in the absence (N = 13), or presence (N = 11) of VPA measured by PIP3 ELISA mass assay. Graphs show means ± SEM, with individual data point illustrated. Statistical analysis was performed by post-hoc Tukey test, following ANOVA (**p < 0.01, ***p < 0.001). Quantification of washout epileptiform activity is shown in Supplementary Fig. 5.
VPA's effect on paroxysmal activity control depends on PIP3 regulation
Having reproduced these in vivo PIP3 regulatory effects in vitro, we then asked if VPA's efficacy at decreasing paroxysmal activity is dependent on PIP3 regulation. To do this, we tested the efficacy of VPA on controlling paroxysmal activity following a reduction in PI3K activity (using two PI3K inhibitors; PI-103 (Bechard et al., 2012) and LY294002 (Lee et al., 2005, Workman et al., 2010)), and following sequestration of PIP2 by neomycin (Gabev et al., 1989, Haughey et al., 1999, Lee et al., 2005). In these experiments, paroxysmal activity was induced in hippocampal slices (as previously with PTZ and elevated K+) for 30 min prior to exposure to PI-103 (10 μM) for 50 min with a subsequent stable baseline recording for over 20 min, prior to the addition of VPA for an additional 40 min (Fig. 4). In these experiments, VPA alone significantly decreased the frequency of epileptiform discharges (VPA only: 76.5 ± 2.2% of baseline, N = 5, p = 0.002 compared to control). However, in the presence of PI-103, VPA efficacy against PTZ-induced paroxysmal activity control was abolished (PI-103 + VPA: 109.8 ± 4.1% of baseline (PI-103 only), N = 4, p = 0.000 compared to VPA only). Similar results were observed in the presence of LY294002 (20 μM) (LY294002 + VPA: 97.8 ± 7.0% of baseline (LY294002 only), N = 4, p = 0.008 compared to VPA only). We then repeated these experiments with neomycin, a compound that sequesters PIP2 with a high affinity (Gabev et al., 1989) and thus reduces PIP3 production. In these experiments, neomycin (100 μM) application also blocked the paroxysmal activity control effect of VPA on the frequency of epileptiform activity (98.3 ± 1.8% of baseline (neomycin only), N = 5, p = 0.004 compared to VPA only). These results suggest that the anticonvulsant effect of VPA is dependent on modulation of PIP3 signalling.
Inhibiting PIP3 production or dephosphorylation in hippocampal slices alters burst discharges during PTZ treatment
We next asked whether changes in PIP3 levels altered seizure progression. We tested the effect of the pharmacological reduction of PIP3 levels using a specific class I PI3K inhibitor (PI-103) (Fig. 5A), in influencing the burst discharges in the in vitro PTZ-induced paroxysmal activity model. PI-103 is a pyridofuropyrimidine lead compound (Hayakawa et al., 2006), showing specificity for class IAα, β, and δ isoforms of p110 PI3K and class IB isoform p110γ, with significantly improved specificity compared to other PI3K inhibitors such as Wortmannin and LY294002 (Workman et al., 2010). Application of PI-103 (10 μM) to hippocampal slices during PTZ treatment significantly increased the frequency of burst activity to 138.9 ± 5.1% of baseline, N = 5 (DMSO control: 99.1 ± 3.0% of baseline, N = 4, p = 0.005, unpaired t-test) (Figs. 5A, B, C). Since PI-103 does not induce increased burst discharges in the absence of PTZ, this suggests that a reduction in PIP3 production during paroxysmal activity results in enhanced paroxysmal activity.
Fig. 5.

Epileptiform activity in rat hippocampal slices is regulated by PIP3. (A) Pharmacological regulation of PIP3 can be used to reduce the conversion of PIP2 into PIP3 via PI3K inhibition (using PI-103) and to reduce the conversion of PIP3 to PIP2 by SHIP2 inhibition (using As1949490). Red circles represent phosphates on phosphoinositides. (B) Example trace recording of epileptiform activity (burst discharges) in hippocampal slices induced by application of PTZ (PTZ 2 mM, K+ 6 mM) under control conditions (DMSO) and after inhibition of PI3K activity (PI-103, 10 μM) or SHIP2 activity (As, As1949490 10 μM). (C) Summary of the effect of PI-103 (N = 5) and (D) AS1949490 (N = 5) in PTZ-induced burst discharges in comparison with control (DMSO), where averaged data is presented from 20 to 40 min post-treatment. Graphs show means ± SEM, with individual data point illustrated. Statistical analysis was performed by unpaired Student's t-tests (*p < 0.05, ***p < 0.001). Quantification of washout epileptiform activity is shown in Supplementary Fig. 6.
Since reducing PIP3 levels enhanced paroxysmal activity, we then examined the reverse relationship, by reducing PIP3 dephosphorylation in this model using AS1949490, a selective SHIP2 (SH2 domain-containing inositol 5′-phosphatase 2) inhibitor (Suwa et al., 2009) (Fig. 5A). SHIP2 catalyzes the dephosphorylation of PIP3 to PIP2 and is strongly expressed in the mammalian brain (Dyson et al., 2005). Administration of AS1949490 (10 μM) to hippocampal slices following treatment with PTZ significantly suppressed the frequency of burst discharges to 82.7 ± 2.8% of baseline (N = 5, p = 0.01 compared to DMSO, unpaired t-test) (Figs. 5A, B, D). This result, in combination with that shown for PI-103, supports a role for PIP3 in providing an important protective role in paroxysmal activity associated with seizure progression.
AMPA receptor activity is necessary for PIP3 depletion
Together our data indicate a causal relationship between PIP3 depletion and paroxysmal activity (related to seizure progression), and moreover, imply that VPA's action is dependent upon the PIP3 pathway. However, our data does not preclude a role for seizure activity itself leading to depletion of PIP3, which then further enhances the seizure activity as a positive feedback loop. To examine this, we employed two glutamate receptor antagonists, kynurenic acid, which is a broad-spectrum glutamate receptor antagonist (Perkins and Stone, 1982, Stone and Burton, 1988), and CNQX, which is a competitive AMPA/kainate receptor antagonist (Honore et al., 1988, Watkins et al., 1990); both have been shown to attenuate seizures (Baraban et al., 2005, Galvan et al., 2000, Godukhin et al., 2002) and are not expected to regulate PIP3 levels. Again using the hippocampal slice/PTZ-induced paroxysmal activity model, application of kynurenic acid (1 mM) significantly decreased the frequency of epileptiform discharges (Kyn: 67.4 ± 2.2% of baseline, N = 4, p = 0.01, compared to control) (Figs. 6A, B), and CNQX (20 μM) almost abolished the frequency of burst activity (2.0 ± 2.3% of baseline, N = 4, p = 0.00, compared to control), 20–40 min after PTZ application. Analysis of PIP3 levels in these hippocampal slice samples using the PIP3 ELISA mass assay showed that kynurenic acid (1 mM) and CNQX (20 μM) treatment during seizure induction blocked PIP3 reduction caused by PTZ treatment (Fig. 6C). These results indicate that AMPA receptor activity is necessary for paroxysmal activity progression and the consequent reduction in PIP3 levels during this process.
Fig. 6.

Glutamate receptor inhibitors reduce epileptiform activity and restore PIP3 levels in rat hippocampal slices. (A) Example trace recording of epileptiform activity (burst discharges) in hippocampal slices induced by application of PTZ (PTZ 2 mM, K+ 6 mM) followed by treatment with kynurenic acid (Kyn, 1 mM), a board spectrum glutamate receptor inhibitor, or CNQX (20 μM), an AMPA receptor inhibitor. (B) Comparison of the mean frequency of epileptiform activity induced by PTZ application (N = 5), or following kynurenic acid (N = 4) or CNQX (N = 4) treatment expressed as a percentage of the baseline value. (C) Comparison of PIP3 level in the same hippocampal slices following seizure induction with PTZ (N = 5) or following kynurenic acid (N = 5) or CNQX (N = 5) treatment measured by PIP3 ELISA mass assay. Graphs show means ± SEM with individual data point illustrated. Statistical analysis was performed by post-hoc Tukey test, following ANOVA (**p < 0.01, ***p < 0.001). Quantification of washout epileptiform activity is shown in Supplementary Fig. 7.
Discussion
In the present study, we show that PIP3 levels decrease following in vivo seizures induced with kainic acid and during application of the convulsant PTZ in two in vitro models, and this reduction is restored by the widely used anti-epileptic drug, VPA (Fig. 7). We show that the mechanism of VPA's action is likely to be dependent upon the regulation of PIP3, since blocking PIP3 production (with pharmacological inhibitors or by sequestration of PIP2) blocks VPA-dependent epileptiform activity control. Alterations in PIP3 levels are likely to have an important effect on cell behaviour during seizures since we show PIP3 regulates downstream cell signalling in the PKB/AKT pathway. Seizure-dependent PIP3 reduction is also likely to be involved in seizure progression, since attenuating PIP3 production during PTZ-induced paroxysmal activity causes increased epileptiform activity and reduced dephosphorylation reduces this activity. Inhibiting paroxysmal activity through blocking AMPA receptors prevents PIP3 depletion, indicating that the reduction in PIP3 is not caused by PTZ, but rather by the generation of paroxysmal (burst) activity. This leads to a paradigm in which seizure-like activity is associated with a reduction in PIP3, giving rise to a positive feedback amplification of this activity. These data therefore implicate, for the first time, a reduction in PIP3 levels in neurons during paroxysmal activity as a therapeutic target for seizure control.
Fig. 7.
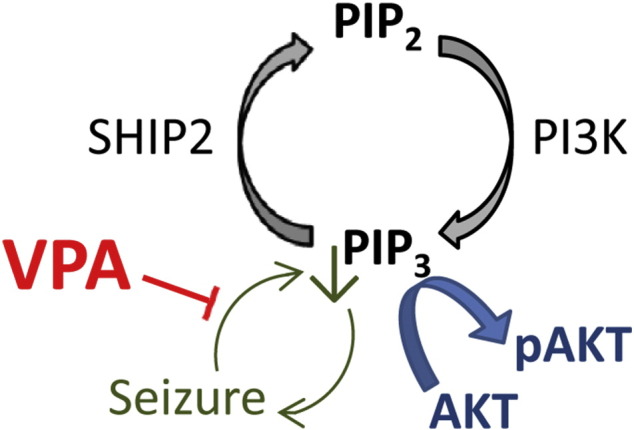
Action of valproic acid (VPA) in regulating PIP3 during seizure activity. Phosphoinositide signalling (black) involves the phosphorylation of PIP2 to produce PIP3 by PI3K activity, and where the reverse reaction is catalysed by SHIP2 activity. PIP3 functions to regulate a range of cellular effects including the activation of downstream AKT activity (blue) by phosphorylation. Seizure activity (green) triggers a decrease in PIP3 giving rise to a feed-back amplification effect. VPA functions to attenuate the seizure-dependent decrease in PIP3 causing a resultant reduction in seizure activity.
One important corollary of the data described here is that VPA may have specific effects only visible during seizure activity, as VPA did not affect PIP3 levels under control conditions. This observation is consistent with previous studies that show that VPA only affects frequency facilitation of synaptic transmission in epileptic but not control animals (Chang and Walker, 2011). The impact of this is that it may be essential to employ seizure-inducing conditions in subsequent experiments to identify the primary target(s) of current treatments and to develop new therapies for seizure control.
As PIP3 is crucial for the regulation of neuron excitability via multiple mechanisms (Vanhaesebroeck et al., 2012), there are a range of means by which a reduction in PIP3 could be involved in seizure activity. During receptor-stimulated activation, PIP3 is asymmetrically distributed on the plasma membrane and interacts with proteins involved in cytoskeleton assembly and membrane fusion during regulated endocytosis and exocytosis (Schmid, 1997, Spiliotis and Nelson, 2003), suggesting that PIP3 plays an important role in neurotransmission, receptor trafficking and membrane repair. PIP3 is also crucial in the modulation of voltage-gated calcium channels (Viard et al., 2004) and directly regulates the inwardly rectifying ATP-sensitive K+ channels (MacGregor et al., 2002). Increasing PIP3 production promotes translocation of ion channels to the plasma membrane, including nonselective cation and calcium-dependent potassium channels (Kanzaki et al., 1999, Lhuillier and Dryer, 2002). Furthermore, PIP3 regulates AMPA receptor localization within the synapse by modulation of scaffold proteins, such that PIP3 depletion enhances mobility and dispersion of AMPA receptors in spines and reduces presynaptic activity (Arendt et al., 2010). Seizure-induced depletion of PIP3 would also interfere in homeostatic synaptic plasticity that precisely maintains basal synaptic activity (Davis, 2006, Wang et al., 2012). These wide and varied functions of PIP3 in neuronal and synaptic function, together with our demonstrated seizure-dependent reduction in PIP3, suggest that seizure activity fundamentally changes neuronal function.
One downstream target of PIP3 is the AKT/mTOR pathway and as such, PIP3 regulation is likely to play a major role in the function of this pathway. Post-seizure, AKT activation has been widely shown to be increased in both animal seizure models and in the hippocampi of epileptic patients (Shinoda et al., 2004, Zhang and Wong, 2012). Blocking pathway activation post-seizure also prevents further seizure development, thus implicating a transient increase in the pathway activation after seizures in promoting epileptogenesis (MacGregor et al., 2002, Meikle et al., 2008, Zeng et al., 2009). Suppression of post-seizure mTOR activation in other seizure models (kainate and pilocarpine) also blocks pathologic changes such as mossy fibre sprouting and synapse formation (Buckmaster and Lew, 2011, Buckmaster et al., 2009, Zeng et al., 2010) and spontaneous seizures (Zeng et al., 2010) implicating this effect as a mechanism leading to chronic epilepsy. The effect of VPA observed by us on the AKT/mTOR pathway is also supported by a range of other studies, including in vitro (Lamarre and Desrosiers, 2008, Wu and Shih, 2011) and in vivo studies (Bates et al., 2012). Thus our data suggest that seizure activity may reduce AKT/mTOR activation, which then increases post-seizure to cause pathogenic changes and epileptogenesis, and that this initial decrease is blocked by VPA thereby possibly reducing post-seizure pathway elevation. It remains to be determined if this PIP3-dependent action contributes to the neuroprotective action of VPA (Bolanos et al., 1998, Brandt et al., 2006, Li and El Mallahk, 2000, Mora et al., 1999, Wilot et al., 2007).
A range of different models are used in epilepsy research, where models often show different sensitivities to epilepsy treatments, presumably due to different molecular mechanisms. In the in vitro studies described here, we have employed PTZ, which is thought to function through regulating GABA(A) signalling, to induce paroxysmal activity associated with seizure progression (Huang et al., 2001), and have shown that this induction mechanism causes a reduction in PIP3, which is rescued by VPA. We have also induced status epilepticus in vivo using kainic acid, where kainic acid functions as a specific agonist for a class of glutamate receptors (kainate receptors) (Nadler, 1981), and have again shown a decrease in PIP3 during seizure activity and that this decrease is also rescued by VPA. These results thus suggest a common reduction in PIP3 levels during seizure-like activity following exposure to multiple independent convulsants. Subsequent studies will be necessary to investigate this mechanism in other seizure models, and to determine if this PIP3 rescue is related to the anti-epileptogenic effect of VPA (Bolanos et al., 1998, Brandt et al., 2006).
VPA provides a range of therapeutic roles in addition to seizure control, including bipolar disorder and migraine prophylaxis (Terbach and Williams, 2009). It is currently unclear if the mechanism of action for VPA identified here is common to these conditions. Recent studies in a simple biomedical model system (Chang et al., 2012, Xu et al., 2007) suggest that a VPA-dependent attenuation of phosphoinositides is not associated with a decrease in cytosolic inositol levels. This site of action contrasts with the function of VPA in bipolar disorder treatment (Eickholt et al., 2005) where VPA is thought to act through a cytosolic mechanism in the depletion of inositol phosphates (Berridge et al., 1989, Terbach and Williams, 2009), possibly through the indirect inhibition of de novo inositol synthesis (Eickholt et al., 2005, Shaltiel et al., 2007, Vaden et al., 2001).
We have therefore shown for the first time an effect of seizure activity on PIP3 levels and that restoring these levels may have an anti-seizure effect. Moreover, targeting this pathway with VPA reduces paroxysmal activity. These findings suggest a novel approach to the treatment of seizures and potentially other conditions for which VPA is effective such as migraine and bipolar disorder.
The following are the supplementary data related to this article.
Supplementary Fig. 4.

3H-inositol labelled phosphoinositide and inositol phosphate distribution from primary hippocampal neurons under seizure-inducing conditions (PTZ at 5 mM) following separation by TLC and quantification by scintillation counting, control condition (N = 18) and with VPA (1 mM, N = 9), or in seizure-inducing conditions in the absence (N = 18) or presence of VPA (1 mM, N = 14). Graphs show means ± SEM, with individual data point illustrated. No statistical difference was shown by post-hoc Tukey test, following ANOVA analysis.
Supplementary Fig. 5.

Quantification of washout epileptiform activity in rat hippocampal slices in control or VPA-treated slices from data presented in Fig. 3. Graphs show means ± SEM, with individual data point illustrated.
Supplementary Fig. 6.

Quantification of washout epileptiform activity in rat hippocampal slices in control experiments or following treatment with the PI3K specific antagonist PI-103 or the SHIP2 antagonist AS1949490 as detailed in Fig. 6. Graphs show means ± SEM, with individual data point illustrated.
Supplementary Fig. 7.

Quantification of washout epileptiform activity in rat hippocampal slices in control experiments or following treatment with the broad spectrum glutamate receptor inhibitor kynurenic acid or the AMPA receptor specific inhibitor CNXQ, as detailed in Fig. 6. Graphs show means ± SEM, with individual data point illustrated.
Acknowledgments
We gratefully acknowledge an NC3Rs grant G0ss900775 to RSBW and MW.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Matthew C. Walker, Email: m.walker@ucl.ac.uk.
Robin S.B. Williams, Email: robin.williams@rhul.ac.uk.
References
- Alessi D.R., James S.R., Downes C.P., Holmes A.B., Gaffney P.R., Reese C.B., Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Andrews S., Stephens L.R., Hawkins P.T. PI3K class IB pathway. Sci. STKE. 2007;2007:cm2. doi: 10.1126/stke.4072007cm2. [DOI] [PubMed] [Google Scholar]
- Apsel B., Blair J.A., Gonzalez B., Nazif T.M., Feldman M.E., Aizenstein B., Hoffman R., Williams R.L., Shokat K.M., Knight Z.A. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt K.L., Royo M., Fernandez-Monreal M., Knafo S., Petrok C.N., Martens J.R., Esteban J.A. PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat. Neurosci. 2010;13:36–44. doi: 10.1038/nn.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armand V., Louvel J., Pumain R., Heinemann U. Effects of new valproate derivatives on epileptiform discharges induced by pentylenetetrazole or low Mg2 + in rat entorhinal cortex–hippocampus slices. Epilepsy Res. 1998;32:345–355. doi: 10.1016/s0920-1211(98)00030-8. [DOI] [PubMed] [Google Scholar]
- Backman S.A., Stambolic V., Suzuki A., Haight J., Elia A., Pretorius J., Tsao M.S., Shannon P., Bolon B., Ivy G.O., Mak T.W. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat. Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- Balding F., Jr., Geller H.M. Sodium valproate enhancement of gamma-aminobutyric acid (GABA) inhibition: electrophysiological evidence for anticonvulsant activity. J. Pharmacol. Exp. Ther. 1981;217:445–450. [PubMed] [Google Scholar]
- Baraban S.C., Taylor M.R., Castro P.A., Baier H. Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience. 2005;131:759–768. doi: 10.1016/j.neuroscience.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Bates R.C., Stith B.J., Stevens K.E. Chronic central administration of valproic acid: increased pro-survival phospho-proteins and growth cone associated proteins with no behavioral pathology. Pharmacol. Biochem. Behav. 2012;103:237–244. doi: 10.1016/j.pbb.2012.08.023. [DOI] [PubMed] [Google Scholar]
- Bechard M., Trost R., Singh A.M., Dalton S. Frat is a phosphatidylinositol 3-kinase/Akt-regulated determinant of glycogen synthase kinase 3beta subcellular localization in pluripotent cells. Mol. Cell. Biol. 2012;32:288–296. doi: 10.1128/MCB.05372-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M.J., Downes C.P., Hanley M.R. Neural and developmental actions of lithium: a unifying hypothesis. Cell. 1989;59:411–419. doi: 10.1016/0092-8674(89)90026-3. [DOI] [PubMed] [Google Scholar]
- Bialer M., White H.S. Key factors in the discovery and development of new antiepileptic drugs. Nat. Rev. Drug Discov. 2010;9:68–82. doi: 10.1038/nrd2997. [DOI] [PubMed] [Google Scholar]
- Bialer M., Johannessen S.I., Levy R.H., Perucca E., Tomson T., White H.S. Progress report on new antiepileptic drugs: a summary of the Tenth Eilat Conference (EILAT X) Epilepsy Res. 2010;92:89–124. doi: 10.1016/j.eplepsyres.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Boeckeler K., Adley K., Xu X., Jenkins A., Jin T., Williams R.S. The neuroprotective agent, valproic acid, regulates the mitogen-activated protein kinase pathway through modulation of protein kinase A signalling in Dictyostelium discoideum. Eur. J. Cell Biol. 2006;85:1047–1057. doi: 10.1016/j.ejcb.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Bolanos A.R., Sarkisian M., Yang Y., Hori A., Helmers S.L., Mikati M., Tandon P., Stafstrom C.E., Holmes G.L. Comparison of valproate and phenobarbital treatment after status epilepticus in rats. Neurology. 1998;51:41–48. doi: 10.1212/wnl.51.1.41. [DOI] [PubMed] [Google Scholar]
- Brandt C., Gastens A.M., Sun M., Hausknecht M., Loscher W. Treatment with valproate after status epilepticus: effect on neuronal damage, epileptogenesis, and behavioral alterations in rats. Neuropharmacology. 2006;51:789–804. doi: 10.1016/j.neuropharm.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Buckmaster P.S., Lew F.H. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J. Neurosci. 2011;31:2337–2347. doi: 10.1523/JNEUROSCI.4852-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster P.S., Ingram E.A., Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J. Neurosci. 2009;29:8259–8269. doi: 10.1523/JNEUROSCI.4179-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley L.C. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Chang P., Walker M.C. Valproate decreases frequency facilitation at mossy fiber—CA3 synapses after status epilepticus. Epilepsy Res. 2011;93:192–196. doi: 10.1016/j.eplepsyres.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Chang P., Chandler K.E., Williams R.S., Walker M.C. Inhibition of long-term potentiation by valproic acid through modulation of cyclic AMP. Epilepsia. 2010;51:1533–1542. doi: 10.1111/j.1528-1167.2009.02412.x. [DOI] [PubMed] [Google Scholar]
- Chang P., Orabi B., Deranieh R.M., Dham M., Hoeller O., Shimshoni J.A., Yagen B., Bialer M., Greenberg M.L., Walker M.C., Williams R.S. The antiepileptic drug valproic acid and other medium-chain fatty acids acutely reduce phosphoinositide levels independently of inositol in Dictyostelium. Dis. Model Mech. 2012;5:115–124. doi: 10.1242/dmm.008029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P., Terbach N., Plant N., Chen P.E., Walker M.C., Williams R.S. Seizure control by ketogenic diet-associated medium chain fatty acids. Neuropharmacology. 2013;69:105–114. doi: 10.1016/j.neuropharm.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis G.W. Homeostatic control of neural activity: from phenomenology to molecular design. Annu. Rev. Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Di P.G., De C.P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Dyson J.M., Kong A.M., Wiradjaja F., Astle M.V., Gurung R., Mitchell C.A. The SH2 domain containing inositol polyphosphate 5-phosphatase-2: SHIP2. Int. J. Biochem. Cell Biol. 2005;37:2260–2265. doi: 10.1016/j.biocel.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Eickholt B.J., Towers G., Ryves W.J., Eikel D., Adley K., Ylinen L., Chadborn N., Harwood A., Nau H., Williams R.S. Effects of valproic acid derivatives on inositol trisphosphate depletion, teratogenicity, GSK-3β inhibition and viral replication — a screening approach for new bipolar disorder drugs based on the valproic acid core structure. Mol. Pharmacol. 2005;67:1–8. doi: 10.1124/mol.104.009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elphick L.M., Pawolleck N., Guschina I.A., Chaieb L., Eikel D., Nau H., Harwood J.L., Plant N.J., Williams R.S. Conserved valproic acid-induced lipid droplet formation in Dictyostelium and human hepatocytes (huh7) identifies structurally active compounds. Dis. Model Mech. 2011;5:231–240. doi: 10.1242/dmm.008391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabev E., Kasianowicz J., Abbott T., McLaughlin S. Binding of neomycin to phosphatidylinositol 4,5-bisphosphate (PIP2) Biochim. Biophys. Acta. 1989;979:105–112. doi: 10.1016/0005-2736(89)90529-4. [DOI] [PubMed] [Google Scholar]
- Galvan C.D., Hrachovy R.A., Smith K.L., Swann J.W. Blockade of neuronal activity during hippocampal development produces a chronic focal epilepsy in the rat. J. Neurosci. 2000;20:2904–2916. doi: 10.1523/JNEUROSCI.20-08-02904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godukhin O., Savin A., Kalemenev S., Levin S. Neuronal hyperexcitability induced by repeated brief episodes of hypoxia in rat hippocampal slices: involvement of ionotropic glutamate receptors and L-type Ca(2 +) channels. Neuropharmacology. 2002;42:459–466. doi: 10.1016/s0028-3908(02)00005-9. [DOI] [PubMed] [Google Scholar]
- Gupta Y.K., Briyal S., Chaudhary G. Protective effect of trans-resveratrol against kainic acid-induced seizures and oxidative stress in rats. Pharmacol. Biochem. Behav. 2002;71:245–249. doi: 10.1016/s0091-3057(01)00663-3. [DOI] [PubMed] [Google Scholar]
- Haughey N.J., Holden C.P., Nath A., Geiger J.D. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J. Neurochem. 1999;73:1363–1374. doi: 10.1046/j.1471-4159.1999.0731363.x. [DOI] [PubMed] [Google Scholar]
- Hayakawa M., Kaizawa H., Moritomo H., Koizumi T., Ohishi T., Okada M., Ohta M., Tsukamoto S., Parker P., Workman P., Waterfield M. Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg. Med. Chem. 2006;14:6847–6858. doi: 10.1016/j.bmc.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Heinemann U., Draguhn A., Ficker E., Stabel J., Zhang C.L. Strategies for the development of drugs for pharmacoresistant epilepsies. Epilepsia. 1994;35(Suppl. 5):S10–S21. doi: 10.1111/j.1528-1157.1994.tb05959.x. [DOI] [PubMed] [Google Scholar]
- Honore T., Davies S.N., Drejer J., Fletcher E.J., Jacobsen P., Lodge D., Nielsen F.E. Quinoxalinediones: potent competitive non-NMDA glutamate receptor antagonists. Science. 1988;241:701–703. doi: 10.1126/science.2899909. [DOI] [PubMed] [Google Scholar]
- Huang R.Q., Bell-Horner C.L., Dibas M.I., Covey D.F., Drewe J.A., Dillon G.H. Pentylenetetrazole-induced inhibition of recombinant gamma-aminobutyric acid type A (GABA(A)) receptors: mechanism and site of action. J. Pharmacol. Exp. Ther. 2001;298:986–995. [PubMed] [Google Scholar]
- Huang B.X., Akbar M., Kevala K., Kim H.Y. Phosphatidylserine is a critical modulator for Akt activation. J. Cell Biol. 2011;192:979–992. doi: 10.1083/jcb.201005100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki M., Zhang Y.Q., Mashima H., Li L., Shibata H., Kojima I. Translocation of a calcium-permeable cation channel induced by insulin-like growth factor-I. Nat. Cell Biol. 1999;1:165–170. doi: 10.1038/11086. [DOI] [PubMed] [Google Scholar]
- Kwan P., Brodie M.J. Refractory epilepsy: mechanisms and solutions. Expert. Rev. Neurother. 2006;6:397–406. doi: 10.1586/14737175.6.3.397. [DOI] [PubMed] [Google Scholar]
- Lagace D.C., O'Brien W.T., Gurvich N., Nachtigal M.W., Klein P.S. Valproic acid: how it works. Or not. Clin. Neurosci. Res. 2005;4:215–225. [Google Scholar]
- Lamarre M., Desrosiers R.R. Up-regulation of protein l-isoaspartyl methyltransferase expression by lithium is mediated by glycogen synthase kinase-3 inactivation and beta-catenin stabilization. Neuropharmacology. 2008;55:669–676. doi: 10.1016/j.neuropharm.2008.05.033. [DOI] [PubMed] [Google Scholar]
- Lee C.C., Huang C.C., Wu M.Y., Hsu K.S. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J. Biol. Chem. 2005;280:18543–18550. doi: 10.1074/jbc.M414112200. [DOI] [PubMed] [Google Scholar]
- Lhuillier L., Dryer S.E. Developmental regulation of neuronal K(Ca) channels by TGFbeta1: an essential role for PI3 kinase signaling and membrane insertion. J. Neurophysiol. 2002;88:954–964. doi: 10.1152/jn.2002.88.2.954. [DOI] [PubMed] [Google Scholar]
- Li R., El Mallahk R.S. A novel evidence of different mechanisms of lithium and valproate neuroprotective action on human SY5Y neuroblastoma cells: caspase-3 dependency. Neurosci. Lett. 2000;294:147–150. doi: 10.1016/s0304-3940(00)01559-7. [DOI] [PubMed] [Google Scholar]
- Ljungberg M.C., Sunnen C.N., Lugo J.N., Anderson A.E., D'Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis. Model Mech. 2009;2:389–398. doi: 10.1242/dmm.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W. Valproate: a reappraisal of its pharmacodynamic properties and mechanisms of action. Prog. Neurobiol. 1999;58:31–59. doi: 10.1016/s0301-0082(98)00075-6. [DOI] [PubMed] [Google Scholar]
- MacGregor G.G., Dong K., Vanoye C.G., Tang L., Giebisch G., Hebert S.C. Nucleotides and phospholipids compete for binding to the C terminus of KATP channels. Proc. Natl. Acad. Sci. U. S. A. 2002;99:2726–2731. doi: 10.1073/pnas.042688899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L., Pollizzi K., Egnor A., Kramvis I., Lane H., Sahin M., Kwiatkowski D.J. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J. Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora A., Gonzalez-Polo R.A., Fuentes J.M., Soler G., Centeno F. Different mechanisms of protection against apoptosis by valproate and Li + Eur. J. Biochem. 1999;266:886–891. doi: 10.1046/j.1432-1327.1999.00919.x. [DOI] [PubMed] [Google Scholar]
- Nadler J.V. Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci. 1981;29:2031–2042. doi: 10.1016/0024-3205(81)90659-7. [DOI] [PubMed] [Google Scholar]
- Perkins M.N., Stone T.W. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982;247:184–187. doi: 10.1016/0006-8993(82)91048-4. [DOI] [PubMed] [Google Scholar]
- Racine R.J., Gartner J.G., Burnham W.M. Epileptiform activity and neural plasticity in limbic structures. Brain Res. 1972;47:262–268. doi: 10.1016/0006-8993(72)90268-5. [DOI] [PubMed] [Google Scholar]
- Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schmid S.L. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annu. Rev. Biochem. 1997;66:511–548. doi: 10.1146/annurev.biochem.66.1.511. [DOI] [PubMed] [Google Scholar]
- Shaltiel G., Mark S., Kofman O., Belmaker R.H., Agam G. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol. Rep. 2007;59:402–407. [PubMed] [Google Scholar]
- Shinoda S., Schindler C.K., Meller R., So N.K., Araki T., Yamamoto A., Lan J.Q., Taki W., Simon R.P., Henshall D.C. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. J. Clin. Invest. 2004;113:1059–1068. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiliotis E.T., Nelson W.J. Spatial control of exocytosis. Curr. Opin. Cell Biol. 2003;15:430–437. doi: 10.1016/s0955-0674(03)00074-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokoe D., Stephens L.R., Copeland T., Gaffney P.R., Reese C.B., Painter G.F., Holmes A.B., McCormick F., Hawkins P.T. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- Stone T.W., Burton N.R. NMDA receptors and ligands in the vertebrate CNS. Prog. Neurobiol. 1988;30:333–368. doi: 10.1016/0301-0082(88)90027-5. [DOI] [PubMed] [Google Scholar]
- Sugaya E., Sugaya A., Takagi T., Tsuda T., Kajiwara K., Yasuda K., Komatsubara J. Pentylenetetrazole-induced changes of the single potassium channel in primary cultured cerebral cortical neurons. Brain Res. 1989;497:239–244. doi: 10.1016/0006-8993(89)90268-0. [DOI] [PubMed] [Google Scholar]
- Suwa A., Yamamoto T., Sawada A., Minoura K., Hosogai N., Tahara A., Kurama T., Shimokawa T., Aramori I. Discovery and functional characterization of a novel small molecule inhibitor of the intracellular phosphatase, SHIP2. Br. J. Pharmacol. 2009;158:879–887. doi: 10.1111/j.1476-5381.2009.00358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terbach N., Williams R.S. Structure–function studies for the panacea, valproic acid. Biochem. Soc. Trans. 2009;37:1126–1132. doi: 10.1042/BST0371126. [DOI] [PubMed] [Google Scholar]
- Vaden D.L., Ding D., Peterson B., Greenberg M.L. Lithium and valproate decrease inositol mass and increase expression of the yeast INO1 and INO2 genes for inositol biosynthesis. J. Biol. Chem. 2001;276:15466–15471. doi: 10.1074/jbc.M004179200. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B., Stephens L., Hawkins P. PI3K signalling: the path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- Viard P., Butcher A.J., Halet G., Davies A., Nurnberg B., Heblich F., Dolphin A.C. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat. Neurosci. 2004;7:939–946. doi: 10.1038/nn1300. [DOI] [PubMed] [Google Scholar]
- Wang G., Gilbert J., Man H.Y. AMPA receptor trafficking in homeostatic synaptic plasticity: functional molecules and signaling cascades. Neural Plast. 2012;2012:825364. doi: 10.1155/2012/825364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins J.C., Krogsgaard-Larsen P., Honore T. Structure–activity relationships in the development of excitatory amino acid receptor agonists and competitive antagonists. Trends Pharmacol. Sci. 1990;11:25–33. doi: 10.1016/0165-6147(90)90038-a. [DOI] [PubMed] [Google Scholar]
- Wilot L.C., Bernardi A., Frozza R.L., Marques A.L., Cimarosti H., Salbego C., Rocha E., Battastini A.M. Lithium and valproate protect hippocampal slices against ATP-induced cell death. Neurochem. Res. 2007;32:1539–1546. doi: 10.1007/s11064-007-9348-3. [DOI] [PubMed] [Google Scholar]
- Workman P., Clarke P.A., Raynaud F.I., van Montfort R.L. Drugging the PI3 kinome: from chemical tools to drugs in the clinic. Cancer Res. 2010;70:2146–2157. doi: 10.1158/0008-5472.CAN-09-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P., Hu Y.Z. PI3K/Akt/mTOR pathway inhibitors in cancer: a perspective on clinical progress. Curr. Med. Chem. 2010;17:4326–4341. doi: 10.2174/092986710793361234. [DOI] [PubMed] [Google Scholar]
- Wu J.B., Shih J.C. Valproic acid induces monoamine oxidase A via Akt/forkhead box O1 activation. Mol. Pharmacol. 2011;80:714–723. doi: 10.1124/mol.111.072744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Muller-Taubenberger A., Adley K.E., Pawolleck N., Lee V.W., Wiedemann C., Sihra T.S., Maniak M., Jin T., Williams R.S. Attenuation of phospholipid signaling provides a novel mechanism for the action of valproic acid. Eukaryot. Cell. 2007;6:899–906. doi: 10.1128/EC.00104-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.H., Rensing N.R., Wong M. Developing antiepileptogenic drugs for acquired epilepsy: targeting the mammalian target of rapamycin (mTOR) pathway. Mol. Cell. Pharmacol. 2009;1:124–129. doi: 10.4255/mcpharmacol.09.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.H., McDaniel S., Rensing N.R., Wong M. Regulation of cell death and epileptogenesis by the mammalian target of rapamycin (mTOR): a double-edged sword? Cell Cycle. 2010;9:2281–2285. doi: 10.4161/cc.9.12.11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Wong M. Pentylenetetrazole-induced seizures cause acute, but not chronic, mTOR pathway activation in rat. Epilepsia. 2012;53:506–511. doi: 10.1111/j.1528-1167.2011.03384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]