

Abstract
Cell cycle exit is an obligatory step for the differentiation of oligodendrocyte progenitor cells (OPCs) into myelinating cells. A key regulator of the transition from proliferation to quiescence is the E2F/Rb pathway, whose activity is highly regulated in physiological conditions and deregulated in tumors. In this paper we report a lineage-specific decline of nuclear E2F1 during differentiation of rodent OPC into oligodendrocytes (OLs) in developing white matter tracts and in cultured cells. Using chromatin immunoprecipitation (ChIP) and deep-sequencing in mouse and rat OPCs, we identified cell cycle genes (i.e., Cdc2) and chromatin components (i.e., Hmgn1, Hmgn2), including those modulating DNA methylation (i.e., Uhrf1), as E2F1 targets. Binding of E2F1 to chromatin on the gene targets was validated and their expression assessed in developing white matter tracts and cultured OPCs. Increased expression of E2F1 gene targets was also detected in mouse gliomas (that were induced by retroviral transformation of OPCs) compared with normal brain. Together, these data identify E2F1 as a key transcription factor modulating the expression of chromatin components in OPC during the transition from proliferation to differentiation.
Keywords: brain, cell cycle, chromatin, development, tumor
Introduction
A molecular understanding of the relationship between cell cycle and differentiation in oligodendrocyte progenitor cells (OPCs) is of great significance for a wide variety of human neurological and psychiatric disorders, ranging from schizophrenia to premature brain pathologies, and from demyelinating disorders to glioma. Proliferation is essential for maintenance of the OPC pool, whereas cell cycle exit is required for differentiation (Casaccia-Bonnefil and Liu, 2003; Frederick and Wood, 2004; Sohn et al., 2006). These highly regulated events have important implications for brain development, when the number of myelinating cells has to adequately match the number of axons (Raff et al., 1998), and for repair from demyelinating conditions, when progenitors divide to generate an appropriate number of oligodendrocytes (OLs) for remyelination. Loss of regulatory control occurs in cancer, which is defined by the acquisition of proliferative advantage (typically achieved by escaping the cell cycle regulatory circuitry), changes in chromatin structure, and aberrant transcriptional regulation (Brock et al., 2007; Watanabe and Maekawa, 2010; Nair and Kumar, 2012). Recent studies identified distinct subtypes of glioma based on expression profiling, with the “proneural” subtype sharing a substantial overlap with the OPC transcriptome (Verhaak et al., 2010). Furthermore, injecting retroviruses that express mitogens (i.e., PDGF) into the subcortical white matter stimulated the massive proliferation of OPCs and induced the formation of tumors that closely resembled proneural glioblastoma (Lei et al., 2011). Therefore, understanding how cell cycle exit is coupled with differentiation is of great importance and direct relevance to dysmyelinating disorders and to mechanisms of gliomagenesis.
In broad terms, cell cycle entry is regulated by a series of “molecular switches,” regulating the expression of genes affecting DNA synthesis and replication. A critical regulator of the cell cycle genes is the transcription factor E2F1 which, in its unbound state, forms complexes with the Dp1 partner and binds avidly to DNA (Martinez-Balbas et al., 2000). The bioavailability of unbound nuclear E2F1 is dependent on the phosphorylation state of the retinoblastoma protein Rb. When unphosphorylated, Rb sequesters E2F1 (Dyson, 1998; Nevins, 1998). Mitogens increase Rb phosphorylation by upregulating the activity of cyclin-dependent kinase (CDK) complexes (Sherr, 1994; Weinberg, 1995), thereby releasing E2F1. Inhibitors of CDK activity (i.e., CDKI), which include members of the INK (i.e., p15Ink4b, p16Ink4a, p18Ink4c and p19Ink4d) and Kip (i.e., p21Cip1, p27Kip1, p57Kip2) family, negatively regulate Rb phosphorylation (Ruas and Peters, 1998; Sherr and Roberts, 1999). The importance of Rb pathway regulation in OPCs was suggested by the role of CDKIs in modulating proliferation (Tikoo et al., 1998; Ghiani et al., 1999; Tang et al., 1999) and differentiation, since genetic ablation of the genes encoding for p21Cip1 (Zezula et al., 2001), p18 Ink (Tokumoto et al., 2002), or p27Kip1 (Casaccia-Bonnefil et al., 1997, 1999; Durand et al., 1997) impaired the progression of OPC into myelin-forming cells. Because increased CDKI activity results in loss of E2F1 activation, we focused our analysis on the identification of its target genes and performed an unbiased genome-wide analysis of chromatin occupancy in proliferating and differentiating OPCs.
Materials and Methods
Animals.
Use of animals of either sex in this research was strictly compliant with the guidelines set forth by the US Public Health Service in their policy on Human Care and Use of Laboratory Animals, and in the Guide for the Care and Use of Laboratory Animals. Mice were maintained under a pathogen-free environment at Mount Sinai School of Medicine animal facility. All procedures received prior approval from the Institutional Animal Care and Use Committee (IACUC). Timed pregnancy Sprague Dawley rats and mice were purchased from Charles River Laboratory. Animal handling and experiments on rats and mice of either sex were performed according to the German animal protection laws (LANUV Nordrhein-Westfalen AZ 8.87-51.05.20.10.262).
Cell culture and treatment.
Rat oligodendrocyte progenitors were isolated from the cortices of postnatal day 1 (P1) rats and cultured as previously described (Shen et al., 2008). Differential shaking followed by magnetic beads immunoselection was used to isolate progenitors. Briefly, cells were incubated with the A2B5 antibody and purified using magnetic beads (Miltenyi Biotec), and then plated at a density of 2 × 105 cells/ml on poly-d-lysine-coated 10 cm plates. Progenitors were maintained as proliferating by the addition of bFGF and PDGFAA (10 ng/ml) to the culture medium (DMEM, 100 μg/ml albumin, 100 μg/ml apotransferrin, 16 μg/ml putrescine, 0.06 ng/ml progesterone, 40 ng/ml selenium, 5 μg/ml insulin, 1 mm sodium pyruvate, 2 mm l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin). Differentiation was initiated by placing the cells in mitogen-free cultured medium and by adding l-3,3′,5-triiodothyronine sodium salt (T3 hormone, 45 nm) to induce oligodendrocyte differentiation or BMP4 (50 ng/ml) to generate astrocytes (Wu et al., 2012). Mouse oligodendrocyte progenitors were isolated from P6–P8 C57BL6 mice and cultured as previously described (Cahoy et al., 2008). Briefly, dissociated mouse forebrains were resuspended in panning buffer. To deplete microglia, the single-cell suspension was sequentially panned on BSL1 panning plates and then incubated on PDGFRα plates. The adherent cells were trypsinized and plated onto poly-d-lysine-coated plates. The cultures were maintained under proliferating conditions by addition of PDGFAA (10 ng/ml) and bFGF (20 ng/ml) and then differentiated by adding T3 hormone (45 nm). The mouse oligodendrocyte precursor cell line Olineu (Jung et al., 1995) was grown on poly-ornithine-coated culture dishes. The immature Olineu cells were maintained in growth medium consisting of DMEM supplemented with 2 mm l-glutamine, 1 mm sodium pyruvate, 10 ng/ml biotin, 100 mg/ml apotransferrin, 100 mm putrescine, 20 nm progesterone, 30 nm sodium selenite, 5 mg/ml insulin, 1% horse serum, 100 U/ml penicillin, and 100 mg/ml streptomycin. Differentiation was induced by switching the cells to a serum-free medium containing 45 nm T3. The Pten;p53 knock-out glioma cell line, obtained as described by Lei et al., 2011, was maintained in culture in growth medium consisting of DMEM supplemented with 2 mm l-glutamine, 1 mm sodium pyruvate, 10 ng/ml biotin, 100 mg/ml apotransferrin, 100 mm putrescine, 20 nm progesterone, 30 nm sodium selenite, 5 mg/ml insulin, 1% horse serum, 100 U/ml penicillin, and 100 mg/ml streptomycin.
Tissue collection and sectioning.
Mice were perfused intracardially with 4% paraformaldehyde in 0.1 m phosphate buffer. Brains were removed from the skulls, postfixed overnight, and cryopreserved by sequential immersion of 10%, 20%, and 30% sucrose solution in 0.1 m phosphate buffer, pH 7.4. Brains were then embedded in OCT (Fisher Scientific) and sectioned (12 μm).
Immunohistochemistry and immunocytochemistry.
Floating brain sections from mice at P2, P7, and P18 were immunostained with antibodies against E2F1 (1:500, Sc-193, Santa Cruz Biotechnology), PDGFRα (1:100, SC-338, Santa Cruz Biotechnology) and CC1 (1:250, OP80, Calbiochem). Sections were incubated with antibodies overnight at 4°C primarily diluted in 0.1 m PBS, pH 7.4, containing 0.01% Triton X-100 (v/v) and 5% normal goat serum (v/v). For secondary antibodies we used TRICI-conjugated AffiniPure goat antibody to mouse IgG and CY5-conjugated AffiniPure goat antibody to rabbit. Sections were incubated with secondary antibodies for 1 h at 22–25°C, then washed and mounted on the slides. For cell counting, z-stacks of 1 μm-thick single plane images were captured through the entire thickness of the slice using a confocal microscope (5100 Zeiss) and collapsed before using NIH ImageJ. Measurements were taken from 15–20 μm tissue sections obtained from three to four mice per group. Cells were quantified using unbiased stereological morphometric analysis for white matter to obtain the estimate of the total number of positive cells. The percentage of cells expressing the above markers was estimated by scoring the number of cells double-labeled with the markers. Statistical analysis was determined using Student's t test.
For immunocytochemistry, cells were fixed with 4% paraformaldehyde and washed three times before incubation with primary antibodies, including anti-Ki67 (ab15580, Abcam) and anti-E2F1 (Sc-193, Santa Cruz Biotechnology) at 4°C overnight. For staining of O4 and O1, cells were incubated with appropriate antibodies for 30 min, followed by wash and fixation.
BrdU incorporation and labeling.
Proliferating cells were labeled by intraperitoneal BrdU (Sigma-Aldrich) injections. Mice at P2, P7, and P18 were injected 2 h before being killed with 100 μg/g BrdU. After injection, animals were anesthetized with isoflurane and transcardially perfused with 0.1 m PBS, pH 7.4, followed by 4% paraformaldehyde. Brains were postfixed in 4% paraformaldehyde overnight. Serial coronal and sagittal sections (50 μm) were cut using a microtome (American Optical), collected in PBS, pH 7.4, and stored at 4°C until use. For BrdU labeling, the tissue was pretreated with 2 N HCl and neutralized in 0.1 m boric acid, pH 8.5. After washing, sections were incubated with primary antibody (1:50 anti-BrdU, BD Biosciences) overnight and then with the secondary antibody (1:200 TRITC-conjugated AffiniPure goat anti-mouse, Jackson ImmunoResearch Laboratories) for 1 h. After washing in PBS, pH 7.4, sections were mounted and analyzed by confocal microscopy (Zeiss).
RNA isolation and quantitative reverse transcription-PCR analysis.
Primary cells or tissue derived from corpus callosum were homogenized in TRIzol Reagent, and RNA was isolated following the manufacturer's instruction and cleaned using the RNeasy Mini kit (Qiagen). Total RNA (500 ng) was used in 20 μl of reverse transcription reaction, using qScript cDNA SuperMix (Quanta BioSciences). Quantitative reverse transcription (qRT)-PCR was performed using PerfeCTa SYBR Green FastMix (Quanta BioSciences) in an Applied Biosystems 7900HT Sequence Detection PCR System. The melting curve of each sample was measured to ensure the specificity of the products. Data were normalized to the internal control Gapdh or 18S and analyzed using a Pfaffl ΔΔCt method. Primers used in quantitative PCR are given in Table 1 for rat transcripts and Table 2 for murine transcript.
Table 1.
Primers used in quantitative PCR for rat transcripts
| Rat RT primers | Forward | Reverse |
|---|---|---|
| Cnpase | TGCTGCACTGTACAACCAAATTCT | CATCCTGTTGGGCATATTCTTCT |
| Mag | TCAACAGTCCCTACCCCAAG | GAGAAGCAGGGTGCAGTTTC |
| Mbp | AAATCGGCTCACAAGGGATTC | CTCCCAGCTTAAAGATTTTGGAAA |
| Plp | GGCGACTACAAGACCACCAT | CCTAGCCATTTTCCCAAACA |
| Mog | GAGGGACAGAAGAACCCACA | CAGTTCTCGACCCTTGCTTC |
| Pdgrfα | ACCATTTCTGTTCACGAGAAAGG | GCAGGTTCACAGTTTCCAGATG |
| E2f1 | GACCCTGCAGAACAAATGGT | AATCCCCTCTGCACTTTCCT |
| E2f3 | GGCACGTACATCCAGATCCT | CTCTGCTGGAGGGTCCTTGT |
| E2f4 | GTCCCATTGAGGTCCTGCTA | CTGCTTGGACGAGAGGTTTC |
| E2f5 | ACACAACCTTCCTCCCAGT | GCCGTAAGAGAGGAAACACG |
Table 2.
Primers used in quantitative PCR for murine transcripts
| Mouse RT primers | Forward | Reverse |
|---|---|---|
| Cdc2 | GCCAGAGCGTTTGGAATACC | CAGATGTCAACCGGAGTGGAGTA |
| Uhrf1 | TGAAGCGGATGACAAGACTG | CAGGGCTCGTCCTCAGATAG |
Clustering and ontology analysis.
The expression data for the time course of oligodendrocyte differentiation used for the clustering analysis was previously published (Swiss et al., 2011). Ontology analysis of each coexpressed group of genes was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) ontology software as previously described (Dennis et al., 2003; Huang et al., 2009; Swiss et al., 2011) using the whole genome as the background dataset and an EASE score < 10−3 considered highly enriched ontology groups. ChIP Enrichment Analysis (ChEA) was performed by interrogating ChEA software (http://amp.pharm.mssm.edu/lib/chea.jsp) as described previously (Lachmann et al., 2010). The microarray data where described previously (Swiss et al., 2011). Data where obtained from cultures containing >95% oligodendrocyte (OL)-lineage cells. Upon differentiation, these cultures contained 90–95% GalC+/MBP+ OLs. Less than 1% of the cells in the culture expressed the neuronal marker Neurofilament-H and the remaining 5% were astrocytes.
Chromatin immunoprecipitation and ChIP-Seq.
Chromatin was isolated from rat or mouse OPCs (>10 × 106 cells) and from oligodendrocytes (>10 × 106 cells) maintained in differentiation medium for 1, 3, or 5 d as previously described (Liu et al., 2010). The size of the DNA in the sheared chromatin fragments was tested before precipitation by Bioanalyzer to ensure that the majority of the fragment size was 200–400 bp. Immunoprecipitation was performed with 2.4 μg of anti-E2F1 (Sc-193, Santa Cruz Biotechnology)/1 unit OD260 readings. A total of 2.5 units of chromatin were used per condition. The DNA recovered from chromatin that was not immunoprecipitated was used as input. Chromatin that was immunoprecipitated with protein A-Dynabeads (Invitrogen, CA) in the absence of primary antibody (no Ab) was used as a negative control. The input and ChIP samples were sequenced by Illumina HiSeq 2000. The read sequences were generated by analyzing images and base calling using the Illumina data analysis pipeline. After quality control filtering by FASTAX (http://hannonlab.cshl.edu/fastx_toolkit/), only the read sequences with a quality score Q20 in at least 90% of bases were included for analysis. The reads from both Input and ChIP samples were first aligned to Rat Reference genome (build rn4 from University of California, Santa Cruz) using Bowtie, an ultrafast alignment method (Langmead et al., 2009). The peaks in the ChIP sample in reference to the input sample at day 0 (D0) and D1 were called from read alignments by the MACS algorithm (Zhang et al., 2008) and then annotated with gene information including the distance to Transcription Start Site (TSS) and locations in genes from genome mapping information of RefSeq transcripts. The peaks within 100 kb of TSS of the gene region for D0 and D1 were compared and only the peaks with >2.5 peak enrichment in D0 versus D1 were considered. The unique gene list characterized by a >2-fold decrease upon OL differentiation was then annotated using the software package DAVID (Dennis et al., 2003; Huang et al., 2009). The genes were clustered using the functional annotation clustering tool, and we only analyzed the ontology terms found in “Biological process” libraries. Primers used for validation in quantitative PCR (qPCR) after ChIP are given in Table 3 for murine genes and Table 4 for rat genes.
Table 3.
Primers used for quantitative PCR after ChIP for murine transcripts
| Mouse ChIP primers | Forward | Reverse |
|---|---|---|
| Cdc2 | ACAGAGCTCAAGAGTCAGTTGGC | CGCCAATCCGATTGCACGTAGA |
| Uhrf1 (−1792) | GCCAATCAGGAATCAAGAGC | AGTGGCTGAAGACCCAAGTG |
| Uhrf1 (−1272) | ATCCGAGTTGTGTCCTCCAC | CCCAGGAGAGTGAAAAACCA |
| Uhrf1 (−948) | CGAGGTCATCTAGGCCAATC | CTAACTTCCCCTCGGGACAC |
Table 4.
Primers used for quantitative PCR after ChIP for rat transcripts
| Rat ChIP primers | For | Rev |
|---|---|---|
| Uhrf1 (−112) | TCCCCAACCACCATTCTAAC | CCATTGGTTTCTAGACCAGGA |
| Uhrf1 (+836) | TGAGTGACCCAGAAGGGAAC | CAGCCAATCAGGAATCAAGA |
| Uhrf1 (+1345) | AAAACCAAGTGCAGTGTGGAG | TTCCATGTTGAGCCTTCCTC |
Silencing of E2F1.
Pten;p53 knock-out glioma cells (2 × 105) were infected in vitro with GIPZ E2F1 shRNA viral particles (VGH5526-EG1869, Thermo Scientific) at multiplicity of infection = 5 in proliferation medium. Turbo GFP expression marked cells expressing the shRNA. After 48 h, infected cells were selected with puromycin (1 μg/ml) and cells were finally harvested for analysis after 72 h postinfection.
Silencing of E2F4.
After immunopanning, 2 × 104 OPCs were plated onto each well of an 8 well chamber slide. The following day 100 nmol/L siRNA was transfected into OPCs using Dharmacon TR#3 (Thermo Fisher Scientific, T-2003-01), siGenome Smartpool targeting E2f4 (Thermo Fisher Scientific, M-054294-000) and nontargeting siRNA pool (Thermo Fisher Scientific, D-001206-13). Transfection procedure was done according to the manufacturer's instructions. After 4 h of transfection, complexes were washed off cells and medium was changed to proliferation medium.
Mouse glioma model.
Proneural gliomas were generated as described by Lei et al., 2011. Briefly, PDGF-IRES-Cre retrovirus was injected into the subcortical white matter of adult mice that harbor floxed tumor suppressors (Pten and p53) and stop-floxed reporters. We used the coordinates of 2.1 mm lateral, 2.2 mm rostral, and 1.8 mm deep (2.1 mm + 2.2 mm + 1.8 mm) with bregma as the reference point. Using a stereotaxis platform, 0.4 μl of retrovirus was injected into the brain with a Hamilton syringe (flow rate 0.1 μl/min). All injections were done in mice between 6 and 8 weeks of age. All mice developed tumors with retrovirus injection. Following IACUC guidelines, animals were killed at the first sign of tumor-induced morbidity.
Microarray-based expression profiling of gliomas and validation by RT-PCR.
Normal adult mouse forebrain and end stage mouse tumors were dissected and snap frozen, and then shipped on dry ice to the Bionomics facility at Rutgers University for expression array analysis. The platform used was Affymetrix GeneChip Mouse Genome 430A 2.0 Array. The microarray labeling, hybridization, and quality controls were performed by following Affymetrix protocol. Raw data were then normalized and summarized by robust multichip average (RMA). A heat map of the expression data was generated using MeV version 4.8 (Saeed et al., 2003). Gene expression levels were normalized across samples for each gene. Red corresponds to high expression and green corresponds to low expression.
Statistics.
One-way ANOVAs were performed to determine significance for immunocytochemistry and immunohistochemistry at different time points. Assuming that significant main effects were observed (p < 0.05), Bonferroni post hoc tests were used to compare selected groups. Student's t tests were used for all other comparisons, including immunocytochemistry and qPCR analysis. ChIP-Seq-related statistics are described in the main text. All values included in the figure legends represent mean ± SEM. To identify the statistically significant differentially expressed genes between the two mouse models and normal mouse brain samples, we used the limma package (Smyth, 2004).
Results
Temporal relationship between OPC exit from the cell cycle and initiation of transcriptional program of differentiation
Differentiation of OPCs requires the coordinated exit from the cell cycle and initiation of a transcriptional program, modulated by chromatin reorganization. To better define the events regulating this critical transition between proliferation and differentiation, we first characterized the time course of cell cycle exit in a homogeneous population of OPCs. The proliferative state of the cells was monitored by assessing the expression of Ki67, a cellular marker of all the active phases of the cell cycle (G1, S, G2, and mitosis) and absent from resting cells (G0; Scholzen, 2000). Differentiation was initiated by the removal of the PDGFAA and the addition of thyroid hormone (T3). In agreement with previous reports (Ferret-Sena et al., 1990; Barres et al., 1994; Pasquini and Adamo, 1994; Gao et al., 1998; Tokumoto et al., 1999; Kaka et al., 2012), T3 treatment for more than 1 d favored the generation of O4+ late progenitors, decreased Ki67 nuclear expression, and decreased the percentage of double-positive Ki67+/O4+ during differentiation (Fig. 1A). These data suggested that in cultured OPCs, the time course of cell cycle exit coincided with the initiation of differentiation toward a myelinating phenotype. The time course of differentiation was defined by decreased levels of the progenitor marker Pdgfrα (Fig. 1B) and the corresponding increase of the myelin transcripts 2′,3′-cyclic nucleotide 3′-phosphodiesterase (Cnpase), myelin-associated glycoprotein (Mag), proteolipid protein (Plp), myelin-binding protein (Mbp), and myelin oligodendrocyte glycoprotein (Mog; Fig. 1C) in RNA samples collected from primary OPCs differentiated in the absence of mitogens and in the presence of T3 for 1 (D1), 3 (D3), and 5 (D5) d. Together, these results suggested that the time course of cell cycle exit coincided with the initiation of differentiation toward a myelinating phenotype.
Figure 1.
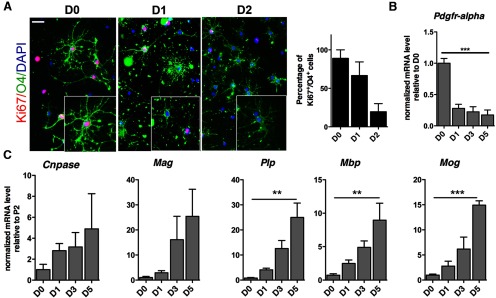
The time course of OPC differentiation correlates with cell cycle exit in vitro. A, Immunofluorescence image of the proliferative marker Ki67 (red) in rat primary OPCs, identified by immunoreactivity for the late oligodendrocyte progenitor marker O4+ (green). The bar graphs represent the percentage of double-positive Ki67+/O4+ cells relative to the total number of DAPI+ nuclei at the indicated data points. D0 denotes the presence of mitogens, while D1 and 2 indicate the days in culture in the absence of mitogens and in the presence of thyroid hormone. B, Transcript levels of the progenitor marker Pdgfrα detected by qRT-PCR of RNA samples isolated from OPC cultured in mitogens (D0) or in differentiation conditions for 1, 3, or 5 d. C, Transcript levels of myelin genes analyzed by quantitative RT-PCR in the same samples as in B. Values were normalized by the levels of Gapdh and shown as relative to the levels detected at D0. Error bars represent SD (n = 3, **p < 0.01, ***p < 0.005). Scale bar in A, 20 μm.
To determine whether a similar temporal relationship between proliferation and differentiation also occurred in vivo, we studied the behavior of OPCs in the developing corpus callosum. We used immunohistochemistry for Ki67 to detect proliferating cells and NG2 expression as a marker for oligodendrocyte progenitors (Nishiyama et al., 2009) at P2, P10, and P18. In agreement with the in vitro results, we detected a progressive reduction in the number of double-positive NG2+/Ki67+ cells occurring over time (Fig. 2A,B). These data were consistent with a similar time course of decreased expression of Pdgfrα transcripts (Fig. 2C) and concomitant increase of myelin gene transcripts (Cnpase, Mag, Plp, Mbp, Mog), starting during the first week and progressively increasing during the second and third postnatal week (Fig. 2D). Together, these results support the notion that OPCs stop expressing progenitor markers (i.e., PDGFRα and NG2) and lose their proliferative capacity as they differentiate into oligodendrocytes.
Figure 2.
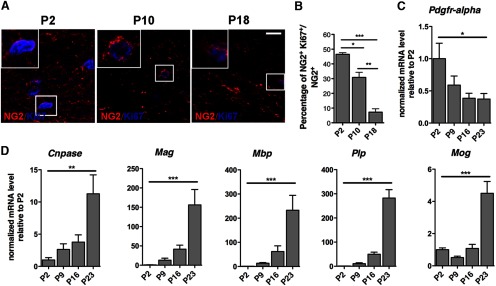
The time course of OPC differentiation correlates with cell cycle exit in the developing corpus callosum. A, Confocal images of OPCs colabeled by an antibody specific for the progenitor marker NG2 (red) and for the proliferative marker Ki67 (blue), in the developing corpus callosum at P2, P10, and P18. Note the progressive decrease of both NG2 and Ki67 with development. Insets show higher magnification of cells identified by the square. B, Quantification of proliferative NG2 cells over time is represented as bar graphs of NG2+/Ki67+ double-positive cells over total NG2+. Note the reduction of the proliferative capacity of NG2+ during differentiation and the fewer NG2 progenitors detected at P18 compared with P2. C, Transcript levels of the progenitor marker Pdgfrα detected by qRT-PCR of RNA samples isolated from the developing corpus callosum at the indicated time points. D, Transcript levels of myelin genes analyzed by quantitative RT-PCR in the same samples as in C. Values were normalized by the levels of Gapdh and shown as relative to the levels detected at P2. The bar graphs represent the average values and error bars represent SD (n = 3, *p < 0.05 **p < 0.01, ***p < 0.005). Scale bar in A, 50 μm.
OPC differentiation correlates with a lineage-specific decline of nuclear E2F1 and with cell cycle exit
The decision of progenitors to proliferate or growth arrest is modulated by the activity of CDKs, which regulate the phosphorylation status of the pocket protein Rb and ultimately determine the sequestration or release of the transcription factor E2F1 (Dimova and Dyson, 2005). Of the several members belonging to the E2F family, only E2F1 to E2F5 have the ability to bind to Rb (Frolov and Dyson, 2004) and have been reported to have cell context-specific roles (Swiss and Casaccia, 2010). To determine the expression of E2F family members during the progression of OPC to distinct lineages, we assessed the transcript levels in proliferating OPCs cultured either in the presence of T3 to induce oligodendrocyte differentiation or in the presence of BMP4 to induce astrocytic differentiation (Fig. 3A,B). Notably, the levels of Pdgfrα were reduced in both conditions, whereas the upregulation of myelin genes was detected only in T3-treated cells (Fig. 3A). Of the distinct E2F family members within the rat genome (E2f1, E2f3, E2f4, E2f5) most of the transcript levels did not differ between the two lineages, with the exception of E2f1, whose dramatic downregulation was detected only in cells differentiating toward oligodendrocytes (Fig. 3B). To better define the temporal regulation of E2F1 during OPC differentiation, we also tested the transcript levels in RNA samples collected at multiple time points during oligodendrocyte differentiation. A dramatic reduction of E2f1 expression was detected as OPC exited the cell cycle, and its levels remained low throughout differentiation (Fig. 3C). These changes in transcripts were associated with decreased protein levels. Immunocytochemistry of E2F1 revealed predominant nuclear immunoreactivity in primary proliferating OPCs (Fig. 3D) and cytoplasmic localization after cell cycle exit followed by a progressive decline in expression (Fig. 3D,E). Overall, these results identify E2F1 as an important regulator of the transition between OPC proliferation and differentiation.
Figure 3.
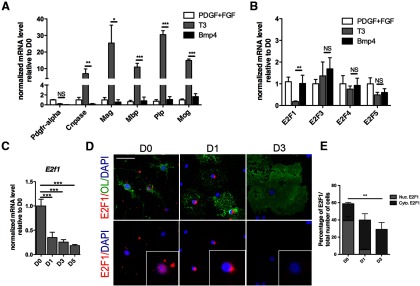
Lineage-specific decline of E2F1 nuclear levels during oligodendrocyte differentiation. A, Transcript levels of Pdgfrα, Cnpase, Mag, Mbp, Plp, Mog in rat OPCs cultured for 5 d in the presence of mitogens (PDGF + FGF2) or in conditions favoring differentiation into oligodendrocytes (T3) or astrocytes (BMP4) were analyzed by quantitative RT-PCR. B, Transcript levels of E2fs (E2f1, E2f3, E2f4, E2f5) were analyzed by quantitative RT-PCR in the same samples described in A. C, E2f1 transcript level in RNA samples isolated from OPC either kept in the presence of mitogens (D0), or differentiated for 1, 3, or 5 d. Transcript levels were normalized by the levels of Gapdh and shown as relative to the levels detected at D0. Bar graphs represent the average values and error bars represent SD (n = 3, *p < 0.05 **p < 0.01, ***p < 0.005). D, Protein expression and subcellular localization of E2F1 during rat OPC differentiation in cultured cells. OPCs were stained for E2F1 (red) and for the OL marker A2B5 (green in D0) or O4 (green in D1 and D3). Note the nuclear localization of E2F1 at D0 followed by its cytoplasmic localization as cells exited the cell cycle (D1). E, Bar graphs denote the percentage of cells with nuclear or cytosolic localization of E2F1+ during rat OPC differentiation at the indicated time points. **p < 0.005, two-way ANOVA. Scale bar in D, 20 μm.
A similar decrease of E2f1 transcript levels was detected in RNA samples extracted from the corpus callosum at P2, P9, P16, and P23 (Fig. 4A). This finding nicely correlated with the reduced number of E2F1+ cells colabeled with the proliferation marker BrdU (Fig. 4B,C). Notably, the percentage of E2F1+ BrdU+ cells decreased during differentiation, indicating that E2F1 was expressed only in proliferating cells and its levels declined as cells exited from the cell cycle and started to differentiate.
Figure 4.
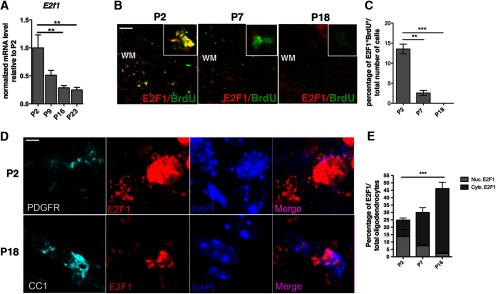
Decreased E2F1 expression and nucleo-cytoplasmic relocalization in oligodendrocyte progenitors in developing white matter tracts. A, E2f1 transcripts measured by quantitative RT-PCR in the developing corpus callosum at the indicated developmental time points. Values were normalized by the levels of Gapdh and shown as relative to the levels detected at P2. The bar graphs represent the average values and error bars represent SD (n = 3, **p < 0.01). B, Proliferating cells were labeled by an acute pulse of BrdU, which was administered 2 h before animals were killed at postnatal day 2, 7, or 18. E2F1 immunoreactivity (red) and BrdU staining (green) were imaged in the developing corpus callosum at the indicated postnatal time points (P2, P7, P18), using confocal microscopy. The insets provide a higher-magnification view of the BrdU-positive cells highlighted by a white box in the low-magnification panel. C, Bar graphs indicate the percentage of proliferating cells expressing E2F1 which is represented as number of double-positive BrdU/E2F1 cells relative to total cell number in the developing corpus callosum (n = 3, **p < 0.01, ***p < 0.05). D, E2F1 protein expression (red) and PDGFRα (light blue at P2) or CC1 (light blue at P18) reveals the nuclear localization (DAPI = blue nuclear staining) of E2F1 in progenitor cells at P2 and the progressive cytosolic shift in CC1+ cells at P18. E, Bar graphs denote the relative proportion of oligodendrocyte cells with nuclear or cytosolic E2F1 immunolabeling at the indicated time points. ***p < 0.0001, 2way ANOVA. Scale bars: A, 50 μm; D, 10 μm.
In addition, we detected a progressive nuclear-cytoplasmic localization of E2F1 in vivo (Fig. 4D,E). At postnatal day2, E2F1 was mainly found in the nucleus of PDGFRα+ OPC, whereas at postnatal day 18, E2F1 immunoreactivity appeared diffuse and cytoplasmic, in CC1+ oligodendrocytes. Therefore, decreased nuclear E2F1 staining in progenitors correlated with increased E2F1 cytoplasmic staining in differentiating cells and with onset of myelination. Together, these results support the notion that nuclear E2F1 is predominantly detected in proliferating OPCs and that it becomes cytoplasmic as the cells proceed toward a differentiated phenotype, suggesting that E2F1-nuclear signaling might prevent the changes in gene expression required for oligodendrocyte maturation.
An unbiased genome-wide approach identifies cell cycle genes and epigenetic modulators as key transcriptional targets for E2F1
The relationship between cell cycle regulators and dynamics of global gene expression during oligodendrocyte differentiation was further strengthened by the identification of E2F1 as a key transcription factor in a clustering analysis of coregulated genes. Putative target genes included those that were highly expressed in progenitors, downregulated during differentiation (Swiss et al., 2011), and functionally related to cell cycle control and nuclear structure as principal gene ontology categories (Fig. 5A). The identification of E2F1 as a key regulatory node was defined on the basis of the statistical enrichment of the ChIP-validated E2F binding sites within the gene list (Fig. 5B), using a web-based software system, ChIP Enrichment Analysis (ChEA) (Müller and Helin, 2000; Lachmann et al., 2010). Two additional cell cycle regulators (i.e., Myc and p53) and stem cell-related transcriptional activators (i.e., Sox2, Pou5f1) were also identified as transcription factors regulating similar clusters, albeit with lower ranking (Cole and McMahon, 1999; Müller and Helin, 2000; Vogelstein et al., 2000; Graham et al., 2003; Evans and Liu, 2008).
Figure 5.

Genome-wide analysis of E2F1 gene targets during oligodendrocyte differentiation. A, The graph on the left shows the kinetic profile of expression of genes coregulated with cell cycle genes during oligodendrocyte differentiation in vitro, while the bar graph on the right identifies the main gene ontology categories of the 309 downregulated genes. B, Bar graphs identify the most significant transcription factors with ChIP-validated binding sites in the coregulated genes (i.e., ChEA). C, Flow chart depicting the method of analysis of the E2F1 ChIP-Seq data: from the raw reads to genome alignment and from peak detection to filtering based on genome positioning, enrichment, and biological significance. The total number of reads, peaks, and genes is indicated below each text box. Ontology analysis was conducted by using the software package DAVID. D, Bar graphs representing the two most significant ontology categories, plotted on the basis of their p-value. The total number of genes in each category is shown at the end of each bar. E, Representative list of genes characterized by E2F1 binding in proliferating cells but not in differentiating cells (defined in terms of enrichment of binding at D0 compared with D1).
To clearly define the E2F1 target genes in an unbiased fashion, we adopted a genome-wide screen. Chromatin was cross-linked in proliferating and differentiating OPCs, immunoprecipitated with anti-E2F1 antibodies, and then processed for deep sequencing. This approach provided a snapshot of the biologically relevant E2F1 gene targets in proliferating and differentiating cells by addressing the chromatin occupancy in live cells. After alignment of the reads to the rat genome using Bowtie software, regions of binding were defined using a peak-calling software called MACS (Zhang et al., 2008). To identify critical targets, we focused our analysis on genes with E2F1 binding peaks located within 100 kb of TSS and with >2.5-fold enrichment in proliferating cells compared with differentiating cells (Fig. 5C). This analysis identified 236 genes that were further screened by overlap with genes characterized by a >2-fold decrease upon oligodendrocyte differentiation (Dugas et al., 2006; Swiss et al., 2011). This stringent analysis identified 87 genes with strong E2F1 binding and high transcript levels in proliferating OPCs and with low E2F1 binding and transcript downregulation during differentiation. Remarkably, the gene ontology categories revealed by this unbiased genome-wide analysis identified cell cycle and chromatin regulation as the top-ranking categories (Fig. 5D). Among relevant genes (Fig. 5E) in these categories we identified cell cycle components (Ccne, Ccne1, Ccna2, Ccnb1, Ccnd2, Ccnb2, Cdc20, Cdkn3, Cdkn2c), molecules regulating DNA methylation (Dnmt1, Uhrf1), chromatin components (Hmgn1, Hmgn2, Smarcb1), and RNA transcription (Pola1, Taf9, Hrnpd).
To validate the results of the E2F1-ChIP-Seq, we performed quantitative ChIP on chromatin samples isolated from murine Oli-Neu cells and immunoprecipitated with anti-E2F1 antibody (Fig. 6A). We selected two target genes, one for the cell cycle category (i.e., Cdc2a) and one for the chromatin regulation category (i.e., Uhrf1). In both cases, E2F1 binding was found on the chromatin at the identified target genes in proliferating cells, but not in differentiating cells. This pattern of chromatin occupancy was associated with transcriptional changes in gene transcripts, with decreased levels of Cdc2a and Uhrf1 in differentiating mouse OPCs both in vitro (Fig. 6B) and in vivo (Fig. 6C). Although we cannot formally exclude the contribution of other cell types, it is worth mentioning that our interpretation of the oligodendrocyte lineage-selective downregulation of transcripts in vivo was detected in previously published datasets (Cahoy et al., 2008). The transcript levels for Cdc2, for instance, did not change in neurons during the analyzed postnatal period, whereas a dramatic reduction of Cdc2 was detected as OPCs differentiated into oligodendrocytes and only a modest reduction could be ascribed to astrocytes. For Uhrf1 the oligodendrocyte-specific downregulation was entirely in agreement with previous data and further supported by the fact that this transcript is almost undetectable in neurons and relatively stable in astrocytes during the time period analyzed. Together, these data identify the transcription factor E2F1 as a key modulator of the transition between proliferation and differentiation by coregulating genes affecting cell cycle control and those affecting chromatin component and epigenetic modulators.
Figure 6.

Validation of E2F1 binding to target genes. A, Chromatin immunoprecipitation to validate binding of E2F1 on two mouse gene targets: the cell cycle gene Cdc2 (left) and the chromatin/DNA modulator Uhfr1 (right). Chromatin was isolated from murine Olineu cells either kept in proliferating conditions (D0) or cultured in differentiation conditions for 1 or 3 d (D1, D3) and immunoprecipitated with antibodies specific for E2F1. After reverse cross-linking, the bound DNA was amplified using primer sets specific for the mouse genomic regions (GRC m38/mm10 mouse genome) identified by the base pair distance from the TSS and drawn in gray on top of the genes. B, Validation of E2F1 target gene expression in primary cultures of mouse OPCs in proliferating (D0) and differentiating (D3) conditions. The transcript values of Cdc2 and Uhrf1 were measured by qRT-PCR, normalized by the levels of m18S and shown as relative to the levels detected at D0. C, Validation of E2F1 target gene expression in mouse corpus callosum at different developmental time points. The transcript values of Cdc2 and Uhrf1 were measured by qRT-PCR, normalized by the levels of m18S, and shown as relative to the levels detected at P2. The bar graphs represent the average values and error bars represent SD (n = 3, *p < 0.05 **p < 0.01, ***p < 0.005).
To further define the relationship between E2F1 and its target genes, we attempted to manipulate its expression using a silencing approach in primary OPC cultures. Unfortunately, all our attempts to directly target E2f1 in primary cells were unsuccessful and ended up in massive apoptosis (data not shown). To circumvent this issue, we decided to use a reverse approach and silence the repressive E2F4. The rationale behind this choice was based on the detection of an interesting switch between E2F1 and E2F4 binding to chromatin of the target genes of rat primary OPC (Fig. 7A–C). Whereas E2F1 was found at the promoter of target genes (i.e., Uhrf1) in proliferating cells (Fig. 7A), it was replaced by E2F4 and HDAC1 binding at the same sites as cells exited the cell cycle (Fig. 7B,C). These data suggested that E2F4/HDAC1 complexes might repress E2F1 targets as OPCs differentiate. For this reason, we considered a silencing approach of E2f4 in rat OPCs, as functionally equivalent to overexpressing E2f1. Indeed, siRNAs specific for E2f4 were effective in reducing its levels and resulted in modest upregulation of Ef1 and dramatic increase in Uhrf1 levels (Fig. 7D). Overall, these data support the model of E2F1-direct regulation of epigenetic modifiers.
Figure 7.

A developmental switch from E2F1 activating complexes to E2F4/HDAC1 repressive complexes during oligodendrocyte development. Immunoprecipitation of chromatin isolated from rat primary cultures of OPC with antibodies specific for E2F1 (A), E2F4 (B), and HDAC1 (C). Samples were collected in proliferating conditions (D0) and at multiple time points in differentiation conditions (in the absence of mitogens and in the presence of T3). After reverse cross-linking, the bound DNA was amplified using primer sets specific for the rat genomic regions (Baylor3.4/rn4 rat genome) identified by the base pair distance from the TSS and drawn in gray on top. The bar graphs indicate the relative binding enrichment at each of the three regions amplified by each primer set (white = −112; black = +836; gray = +1345). Note that binding of E2F1 is detected in proliferating conditions and is replaced by the recruitment of E2F4 and HDAC1 to the same regions at the transition between proliferation and differentiation. D, Effect of silencing E2f4 on the transcript levels of E2f1 and its target gene Uhrf1 was assessed by quantitative RT-PCR of RNA samples isolated from cultured silenced OPCs and referred to as fold changes over the transcript values measured in control cultures infected with scrambled control silencing constructs. The bar graphs represent the average values and error bars represent SD (n = 3, *p < 0.05 **p < 0.01, ***p < 0.005).
E2F1 target genes are deregulated in glioma tissues
Gliomas represent the most common and lethal adult brain tumors. They include a wide spectrum of malignancies ranging from slow-growing to highly aggressive tumors (Louis et al., 2007). A critical property of tumor cells is their loss of the ability to properly respond to the environmental cues, thus deregulating the mechanisms of proliferation and differentiation. Altered expression of cell cycle genes has been highly associated with gliomagenesis. For instance, the expression levels of genes encoding for positive regulators of cell cycle (cyclins, E2F1) have been show to be upregulated compared with controls (Tamiya et al., 2001; Alonso et al., 2005), whereas cell cycle inhibitors like p27Kip1 and p16Ink4a/p19Arf are downregulated (Kirla et al., 2003; Arifin et al., 2006; Hidaka et al., 2009). Given our results on the role of E2F1, not only in regulating cell cycle components, but also affecting genes with dramatic implications for chromatin structure and DNA methylation, we asked whether target genes identified in developing cells were deregulated in cancer. To this end we took advantage of brain tumors obtained by PDGF-IRES-Cre retrovirus injection into the subcortical white matter of adult mice that harbor floxed tumor suppressors (Pten in combination with Trp53) and stop-floxed reporters (Lei et al., 2011). Brain tumors with the histological features of glioblastomas formed with 100% penetrance in this mouse model, with a median survival of 27 dpi (Lei et al., 2011). As previously reported, microarray-based expression analysis showed that the Pten;Trp53 mouse tumors closely resembled the proneural subtype of human glioblastoma multiforme (GBM) (Verhaak et al., 2010) and were characterized by high levels of OPC genes including Pdgfra, Cspg4 (i.e., NG2), and Olig2 (Lei et al., 2011). A heat map from these microarray data revealed the relative upregulation of E2F family members (E2f1, E2Ff2, E2f3, E2f4), and their gene targets (H2afz, Hmgn1, Nup35, Pola1, Uhrf1, Ccnd2, Cdc2) in tumors compared with normal tissue (Fig. 8A). As expected, myelin genes (Plp1, Mbp, Mog) showed an opposite trend, being downregulated in the Pten;Trp53 tissue compared with control (Fig. 8B). To further validate the role of E2F1 in driving a specific transcriptional program in glial progenitors, we silenced E2f1 expression in glioma cells isolated from Pten;p53 mice (Sonabend et al., 2013). This allowed us to also overcome the apoptotic effect of E2f1 silencing in normal cells. Indeed, glioma cell lines well survived the infection with shRNA specific for E2f1 (Fig. 8C). A 50% reduction of E2f1 levels was detected in the E2f1-silenced cells compared with cells infected with scrambled controls (Fig. 8E) and nicely correlated with the percentage of infected cells (Fig. 8D). The effect of silencing was specific since, in addition to E2f1, the expression levels of the other E2f members were not significantly affected (data not shown; Fig. 8E). The expression of all the E2F1 gene targets (Uhrf1, H2Az, Hmgn1, Pola1) was also dramatically decreased, and this was accompanied by the detection of higher transcript levels for oligodendrocyte differentiation genes, such as Mog (Fig. 8E). Together, these results provide further support to the concept of gliomagenesis arising from loss of a critical element of regulation between cell cycle control and differentiation.
Figure 8.

Aberrant regulation of E2f1 target genes in glial tumors. A, Heat map representation of the RNA-Seq showing different expression profiling for E2f family members (E2f1, E2f2, E2f3, E2f4), E2F1 target genes (H2az, Hmgn1, Nup35, Pola1, Uhrf1, Ccnd2 and Cdk1), and myelin genes (Plp, Mbp, Mog) in control adult mouse brain tissues and in glioma tissue isolated from the Pten;p53 mouse. Color key with respect to the expression levels revealed low levels of E2F family members and target genes and high levels of myelin transcripts in normal brain tissue, and a reversal in tumor tissue. B, Bar graphs showing the logarithm of fold decrease (white bars for myelin genes) or fold increase (black bars for E2F1 targets) of the indicated genes in Pten;Trp53 tumors compared with normal brain tissue. Limma was used for the differential expression statistical analysis (Smyth, 2004). C, Phase-contrast micrographs and GFP+ epifluorescence of Pten;Trp53 glioma cells infected with a double-cistronic lentivirus expressing GFP and either scrambled shRNA or shRNA for E2f1. D, Bar graphs indicate the efficiency of lentivirus infection as measured by the percentage of GFP+ cells over total cells at 72 h postinfection. E, Bar graphs indicate the transcript levels measured by quantitative RT-PCR of E2F family members (E2f1, E2f4), E2F1 target genes (Uhrf1, H2az, Hmgn1, Pola1), and myelin genes (Mog) in E2f1-silenced cells compared with scrambled-infected controls. Values were normalized to the levels of the transcript for the 18S ribosomal subunit and shown as relative to the levels detected in shRNA scrambled-infected cells. The bar graphs represent the average values and error bars represent SD (*p < 0.05 **p < 0.01, ***p < 0.005). Note the downregulation of the identified target genes (black bars) upon E2f1 silencing as opposed to increased expression of the myelin gene Mog (white bars).
Discussion
Differentiation of progenitors into myelinating oligodendrocytes is a complex process requiring the tight coordination of proliferation, differentiation, and survival (Casaccia-Bonnefil and Liu, 2003; Raff, 2007; Emery, 2010). To achieve such a level of complexity, cell cycle exit, RNA processing, and synthesis of myelin protein and lipids need to be highly regulated, and this is achieved by a complex interplay between transcription factors, chromatin components, and microRNAs (Lau et al., 2008; Gobert et al., 2009). By focusing on the identification of the downstream targets of the transcriptional regulator E2F1, this study contributes to a better understanding of brain development and repair.
It is now well accepted that controlled proliferation is associated with physiological expansion of the progenitor pool followed by cell cycle exit and differentiation, as detected in the developing brain and in the adult brain in response to damage. Loss of regulatory control of glial proliferation, in contrast, is associated with lack of differentiation and gliomagenesis, which is often accompanied by dramatic changes in DNA methylation and global gene expression. This study identifies E2F1-regulated pathways of gene expression as relevant to normal OPC differentiation and deregulated in glial neoplasms that arise from malignant transformation of OPCs.
E2F1 was the first protein found to bind the tumor suppressor Rb and, to date, it remains the best characterized among the E2Fs (Bagchi et al., 1991). E2F family members can positively and negatively regulate the expression of genes necessary for G1-S phase transition as well as many other genes necessary for cell-cycle progression in a variety of different cell types (Dimova and Dyson, 2005). E2F1, as do other members of the E2F family (belonging to the “classical group”), forms heterodimers with DP proteins and binds to DNA with high affinity (Bandara et al., 1993). Activity of classical E2F complexes is mainly regulated by their ability to interact with pocket proteins (Rb, p107, p130). When pocket proteins are in their hypophosphorylated status, they sequester the activation domain (Helin et al., 1993) and prevent transcriptional changes (Blais and Dynlacht, 2007). On the contrary, hyperphosphorylated pocket proteins are unable to interact with E2Fs (E2F1–E2F5), thereby releasing their activity and leading to the recruitment of chromatin modifiers and activation of gene transcription (DeGregori et al., 1995). Despite the importance of E2F1 as a molecular switch to control cell-cycle entry, little is known about its role in OPCs or oligodendrocytes. Previous studies of rat OPCs suggested that the proliferative effect of NT-3 was secondary to stimulation of the nuclear binding activity of E2F1 (Kumar et al., 1998), whereas other studies reported direct repression of E2f1 level by thyroid hormone receptors, which are known to favor myelination in vivo (Nygård et al., 2003). In addition, Rb downregulation has been associated with rat OPC differentiation (Huang et al., 2002).
The rationale for focusing this study on the role of E2F1 as key regulator of the proliferation/differentiation transition was based on a series of converging evidence. First, E2f1 transcript levels are downregulated when OPCs differentiate into oligodendrocytes, but not when they are pushed toward the astrocytic fate by stimuli such as BMP4. Second, the expression pattern of E2f1 is inversely correlated with that of late differentiation genes (i.e., myelin transcripts). Third, E2F1 protein is exported into the cytoplasm upon differentiation of OPCs, both in cultured cells and in developing white matter tracts. Given the role of E2F1 as nuclear transcription factor, we concluded that its nuclear-cytoplasm shuttling might be required to dampen the E2F1-driven pattern of expression and allow the differentiation process to occur. Finally, E2F1 was identified as a critical regulatory node of gene clusters that are coregulated with cell cycle genes (Swiss et al., 2011).
In this study we adopted genome-wide ChIP-Seq to identify the transcriptional targets of E2F1 at the transition between proliferation and differentiation in OPC, and identified chromatin components (i.e., Hmgn1, Hmgn2, H2Az) and modulators of DNA methylation (i.e., Uhrf1, Dnmt1) among the most significant target genes coregulated with modulators of the cell cycle (i.e., Cdc2, Ccnd2, Ccne, etc.). The discovery that transcription factors involved in cell cycle control also play a pivotal role in regulating genes with important roles in chromatin structure and DNA methylation underscores the relevance of maintaining a tight control between cell cycle and differentiation in physiological conditions and its deregulation in cancer. It is interesting to note that the E2F1 target chromatin proteins, Hmgn1 and 2, belong to the high-mobility group of proteins and have been implicated in nucleosomal rearrangement (Körner et al., 2003; Reeves, 2010; Kato et al., 2011; Barkess et al., 2012; Kugler et al., 2012, 2013), especially during processes related to DNA damage and repair (Birger et al., 2005; Masaoka et al., 2012). Uhrf1 is a molecule previously reported to be expressed in OPCs (Dugas et al., 2006). In proliferating cells it binds to hemi-methylated DNA and has been proposed to couple methylation of DNA to histone methylation (Hashimoto et al., 2010; Liu et al., 2013), and has been implicated as a regulator of the fidelity to accurately transmit DNA methylation during the process of DNA replication (Bostick et al., 2007; Sharif et al., 2007; Arita et al., 2008; Avvakumov et al., 2008; Zhang et al., 2011).
The identification of chromatin modulators as E2F1 gene targets would predict that the consequences of altering E2F1 are greater than just altering the expression of cyclins or CDKIs. Indeed, increased expression of cyclins and downregulation of CDKIs, including p27Kip1 and p16Ink4a/p19Arf, have been described in gliomas (Tamiya et al., 2001; Alonso et al., 2005; Arifin et al., 2006), but altered expression of Rb (which is likely associated with increased bioavailability of nuclear E2F1) has been associated with worse prognosis (Hilton et al., 2004). Our data suggest an important role of E2F1 in the transition between proliferation and differentiation of OPC and reveal the existence of an HDAC1-dependent switch during the downregulation of critical E2F1 targets, such as Uhrf1. The switch of gene regulation due to distinct E2F family members has been reported on the promoter of cell cycle genes (Takahashi et al., 2000; Wells et al., 2000), and is supported by genome-wide analysis revealing a large overlap of E2F1 and E2F4 targets (Xu et al., 2007). It is likely that downregulation of gene targets may result from a series of events, including nuclear-cytoplasmic shuttling of E2F1 (Ivanova et al., 2007) and possibly deacetylation of E2F1. Indeed, we report that silencing of E2f1 in glioma cells—characterized by lack of Pten and p53 (and thereby more resistant to apoptosis)—resulted in dramatic downregulation of the identified gene targets. Studies in different cellular systems described acetylation of E2F1, mediated by p300, Cbp, and PCAF, as critical for its DNA binding activity and function as activator of gene expression (Martinez-Balbas et al., 2000; Marzio et al., 2000). Deacetylation of E2F1 is mediated by HDAC1 (Marzio et al., 2000), an enzyme that we and others (Marin-Husstege et al., 2002; Shen et al., 2008) previously reported as necessary for oligodendrocyte differentiation. We previously identified YY1 as an important transcription factor binding to HDAC1 and recruiting it to specific genomic loci (He et al., 2007). Here we show that HDAC1 is recruited to E2F binding sites on the genome, possibly exerting repression of its target genes by deacetylating E2F1 and modulating histone deacetylation at the transition between proliferation and differentiation.
Overall, these results identify E2F1 as crucial for oligodendrocyte development and provide insights into the important relation between cell cycle exit and chromatin modulation, with clear implications for potential mechanisms of tumorigenesis.
Footnotes
This work was supported by NIH-NINDS Grants R37NS42925, R01NS52738 (to P. Casaccia), R01NS066955 (to P. Canoll), and R01NS056427 and P30HD40677 (to V.G.).
The authors declare no competing financial interests.
References
- Alonso MM, Fueyo J, Shay JW, Aldape KD, Jiang H, Lee OH, Johnson DG, Xu J, Kondo Y, Kanzawa T, Kyo S, Bekele BN, Zhou X, Nigro J, McDonald JM, Yung WK, Gomez-Manzano C. Expression of transcription factor E2F1 and telomerase in glioblastomas: mechanistic linkage and prognostic significance. J Natl Cancer Inst. 2005;97:1589–1600. doi: 10.1093/jnci/dji340. [DOI] [PubMed] [Google Scholar]
- Arifin MT, Hama S, Kajiwara Y, Sugiyama K, Saito T, Matsuura S, Yamasaki F, Arita K, Kurisu K. Cytoplasmic, but not nuclear, p16 expression may signal poor prognosis in high-grade astrocytomas. J Neuro-oncol. 2006;77:273–277. doi: 10.1007/s11060-005-9037-5. [DOI] [PubMed] [Google Scholar]
- Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C, Arrowsmith CH, Dhe-Paganon S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822–825. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- Bagchi S, Weinmann R, Raychaudhuri P. The retinoblastoma protein copurifies with E2F-I, an E1A-regulated inhibitor of the transcription factor E2F. Cell. 1991;65:1063–1072. doi: 10.1016/0092-8674(91)90558-G. [DOI] [PubMed] [Google Scholar]
- Bandara LR, Buck VM, Zamanian M, Johnston LH, La Thangue NB. Functional synergy between DP-1 and E2F-1 in the cell cycle-regulating transcription factor DRTF1/E2F. EMBO J. 1993;12:4317–4324. doi: 10.1002/j.1460-2075.1993.tb06116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkess G, Postnikov Y, Campos CD, Mishra S, Mohan G, Verma S, Bustin M, West KL. The chromatin-binding protein HMGN3 stimulates histone acetylation and transcription across the Glyt1 gene. Biochem J. 2012;442:495–505. doi: 10.1042/BJ20111502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Lazar MA, Raff MC. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development. 1994;120:1097–1108. doi: 10.1242/dev.120.5.1097. [DOI] [PubMed] [Google Scholar]
- Birger Y, Catez F, Furusawa T, Lim JH, Prymakowska-Bosak M, West KL, Postnikov YV, Haines DC, Bustin M. Increased tumorigenicity and sensitivity to ionizing radiation upon loss of chromosomal protein HMGN1. Cancer Res. 2005;65:6711–6718. doi: 10.1158/0008-5472.CAN-05-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais A, Dynlacht BD. E2F-associated chromatin modifiers and cell cycle control. Curr Opin Cell Biol. 2007;19:658–662. doi: 10.1016/j.ceb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick M, Kim JK, Estève PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- Brock MV, Herman JG, Baylin SB. Cancer as a manifestation of aberrant chromatin structure. Cancer J. 2007;13:3–8. doi: 10.1097/PPO.0b013e31803c5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Liu A. Relationship between cell cycle molecules and onset of oligodendrocyte differentiation. J Neurosci Res. 2003;72:1–11. doi: 10.1002/jnr.10565. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Tikoo R, Kiyokawa H, Friedrich V, Jr, Chao MV, Koff A. Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27Kip1. Genes Dev. 1997;11:2335–2346. doi: 10.1101/gad.11.18.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Hardy RJ, Teng KK, Levine JM, Koff A, Chao MV. Loss of p27Kip1 function results in increased proliferative capacity of oligodendrocyte progenitors but unaltered timing of differentiation. Development. 1999;126:4027–4037. doi: 10.1242/dev.126.18.4027. [DOI] [PubMed] [Google Scholar]
- Cole MD, McMahon SB. The Myc oncoprotein: a critical evaluation of transactivation and target gene regulation. Oncogene. 1999;18:2916–2924. doi: 10.1038/sj.onc.1202748. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–2826. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Tai YC, Speed TP, Ngai J, Barres BA. Functional genomic analysis of oligodendrocyte differentiation. J Neurosci. 2006;26:10967–10983. doi: 10.1523/JNEUROSCI.2572-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand B, Gao FB, Raff M. Accumulation of the cyclin-dependent kinase inhibitor p27/Kip1 and the timing of oligodendrocyte differentiation. EMBO J. 1997;16:306–317. doi: 10.1093/emboj/16.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330:779–782. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- Evans PM, Liu C. Roles of Krupel-like factor 4 in normal homeostasis, cancer and stem cells. Acta Biochim Biophys Sin. 2008;40:554–564. doi: 10.1111/j.1745-7270.2008.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferret-Sena V, Sena A, Besnard F, Fressinaud C, Rebel G, Sarliève LL. Comparison of the mechanisms of action of insulin and triiodothyronine on the synthesis of cerebroside sulfotransferase in cultures of cells dissociated from brains of embryonic mice. Dev Neurosci. 1990;12:89–105. doi: 10.1159/000111838. [DOI] [PubMed] [Google Scholar]
- Frederick TJ, Wood TL. IGF-I and FGF-2 coordinately enhance cyclin D1 and cyclin E-cdk2 association and activity to promote G1 progression in oligodendrocyte progenitor cells. Mol Cell Neurosci. 2004;25:480–492. doi: 10.1016/j.mcn.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci. 2004;117:2173–2181. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- Gao FB, Apperly J, Raff M. Cell-intrinsic timers and thyroid hormone regulate the probability of cell-cycle withdrawal and differentiation of oligodendrocyte precursor cells. Dev Biol. 1998;197:54–66. doi: 10.1006/dbio.1998.8877. [DOI] [PubMed] [Google Scholar]
- Ghiani CA, Yuan X, Eisen AM, Knutson PL, DePinho RA, McBain CJ, Gallo V. Voltage-activated K+ channels and membrane depolarization regulate accumulation of the cyclin-dependent kinase inhibitors p27(Kip1) and p21(CIP1) in glial progenitor cells. J Neurosci. 1999;19:5380–5392. doi: 10.1523/JNEUROSCI.19-13-05380.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert RP, Joubert L, Curchod ML, Salvat C, Foucault I, Jorand-Lebrun C, Lamarine M, Peixoto H, Vignaud C, Frémaux C, Jomotte T, Françon B, Alliod C, Bernasconi L, Abderrahim H, Perrin D, Bombrun A, Zanoguera F, Rommel C, Hooft van Huijsduijnen R. Convergent functional genomics of oligodendrocyte differentiation identifies multiple autoinhibitory signaling circuits. Mol Cell Biol. 2009;29:1538–1553. doi: 10.1128/MCB.01375-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39:749–765. doi: 10.1016/S0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Vertino PM, Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics. 2010;2:657–669. doi: 10.2217/epi.10.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Sandoval J, Casaccia-Bonnefil P. Events at the transition between cell cycle exit and oligodendrocyte progenitor differentiation: the role of HDAC and YY1. Neuron Glia Biol. 2007;3:221–231. doi: 10.1017/S1740925X08000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Molecular and cellular biology. 1993;13:6501–6508. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka T, Hama S, Shrestha P, Saito T, Kajiwara Y, Yamasaki F, Sugiyama K, Kurisu K. The combination of low cytoplasmic and high nuclear expression of p27 predicts a better prognosis in high-grade astrocytoma. Anticancer Res. 2009;29:597–603. [PubMed] [Google Scholar]
- Hilton DA, Penney M, Pobereskin L, Sanders H, Love S. Histological indicators of prognosis in glioblastomas: retinoblastoma protein expression and oligodendroglial differentiation indicate improved survival. Histopathology. 2004;44:555–560. doi: 10.1111/j.1365-2559.2004.01887.x. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang Z, Tang XM, Cambi F. Down-regulation of the retinoblastoma protein (rb) is associated with rat oligodendrocyte differentiation. Mol Cell Neurosci. 2002;19:250–262. doi: 10.1006/mcne.2001.1077. [DOI] [PubMed] [Google Scholar]
- Ivanova IA, Vespa A, Dagnino L. A novel mechanism of E2F1 regulation via nucleocytoplasmic shuttling: determinants of nuclear import and export. Cell Cycle. 2007;6:2186–2195. doi: 10.4161/cc.6.17.4650. [DOI] [PubMed] [Google Scholar]
- Jung M, Krämer E, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, Khazaie K, Chlichlia K, von Blankenfeld G, Kettenmann H, Trotter J. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur J Neurosci. 1995;7:1245–1265. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- Kaka GR, Tiraihi T, Delshad A, Arabkheradmand J, Kazemi H. In vitro differentiation of bone marrow stromal cells into oligodendrocyte-like cells using triiodothyronine as inducer. Int J Neurosci. 2012;122:237–247. doi: 10.3109/00207454.2011.642037. [DOI] [PubMed] [Google Scholar]
- Kato H, van Ingen H, Zhou BR, Feng H, Bustin M, Kay LE, Bai Y. Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR. Proc Natl Acad Sci U S A. 2011;108:12283–12288. doi: 10.1073/pnas.1105848108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirla RM, Haapasalo HK, Kalimo H, Salminen EK. Low expression of p27 indicates a poor prognosis in patients with high-grade astrocytomas. Cancer. 2003;97:644–648. doi: 10.1002/cncr.11079. [DOI] [PubMed] [Google Scholar]
- Körner U, Bustin M, Scheer U, Hock R. Developmental role of HMGN proteins in Xenopus laevis. Mech Dev. 2003;120:1177–1192. doi: 10.1016/j.mod.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Kugler JE, Deng T, Bustin M. The HMGN family of chromatin-binding proteins: dynamic modulators of epigenetic processes. Biochim Biophys Acta. 2012;1819:652–656. doi: 10.1016/j.bbagrm.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugler JE, Horsch M, Huang D, Furusawa T, Rochman M, Garrett L, Becker L, Bohla A, Hölter SM, Prehn C, Rathkolb B, Racz I, Aguilar-Pimentel JA, Adler T, Adamski J, Beckers J, Busch DH, Eickelberg O, Klopstock T, Ollert M, et al. High mobility group N proteins modulate the fidelity of the cellular transcriptional profile in a tissue- and variant-specific manner. J Biol Chem. 2013;288:16690–16703. doi: 10.1074/jbc.M113.463315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Kahn MA, Dinh L, de Vellis J. NT-3-mediated TrkC receptor activation promotes proliferation and cell survival of rodent progenitor oligodendrocyte cells in vitro and in vivo. J Neurosci Res. 1998;54:754–765. doi: 10.1002/(SICI)1097-4547(19981215)54:6<754::AID-JNR3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma'ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26:2438–2444. doi: 10.1093/bioinformatics/btq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau P, Verrier JD, Nielsen JA, Johnson KR, Notterpek L, Hudson LD. Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J Neurosci. 2008;28:11720–11730. doi: 10.1523/JNEUROSCI.1932-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, Bruce JN, Canoll P. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PloS One. 2011;6:e20041. doi: 10.1371/journal.pone.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sandoval J, Doh ST, Cai L, López-Rodas G, Casaccia P. Epigenetic modifiers are necessary but not sufficient for reprogramming non-myelinating cells into myelin gene-expressing cells. PloS One. 2010;5:e13023. doi: 10.1371/journal.pone.0013023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Gao Q, Li P, Zhao Q, Zhang J, Li J, Koseki H, Wong J. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat Commun. 2013;4:1563. doi: 10.1038/ncomms2562. [DOI] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Husstege M, Muggironi M, Liu A, Casaccia-Bonnefil P. Histone deacetylase activity is necessary for oligodendrocyte lineage progression. J Neurosci. 2002;22:10333–10345. doi: 10.1523/JNEUROSCI.22-23-10333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. The EMBO J. 2000;19:662–671. doi: 10.1093/emboj/19.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzio G, Wagener C, Gutierrez MI, Cartwright P, Helin K, Giacca M. E2F family members are differentially regulated by reversible acetylation. J Biol Chem. 2000;275:10887–10892. doi: 10.1074/jbc.275.15.10887. [DOI] [PubMed] [Google Scholar]
- Masaoka A, Gassman NR, Kedar PS, Prasad R, Hou EW, Horton JK, Bustin M, Wilson SH. HMGN1 protein regulates poly(ADP-ribose) polymerase-1 (PARP-1) self-PARylation in mouse fibroblasts. J Biol Chem. 2012;287:27648–27658. doi: 10.1074/jbc.M112.370759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H, Helin K. The E2F transcription factors: key regulators of cell proliferation. Biochim Biophys Acta. 2000;1470:M1–M12. doi: 10.1016/s0304-419x(99)00030-x. [DOI] [PubMed] [Google Scholar]
- Nair SS, Kumar R. Chromatin remodeling in cancer: a gateway to regulate gene transcription. Mol Oncol. 2012;6:611–619. doi: 10.1016/j.molonc.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins JR. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differentiation. 1998;9:585–593. [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- Nygård M, Wahlström GM, Gustafsson MV, Tokumoto YM, Bondesson M. Hormone-dependent repression of the E2F-1 gene by thyroid hormone receptors. Mol Endocrinol. 2003;17:79–92. doi: 10.1210/me.2002-0107. [DOI] [PubMed] [Google Scholar]
- Pasquini JM, Adamo AM. Thyroid hormones and the central nervous system. Dev Neurosci. 1994;16:1–8. doi: 10.1159/000112080. [DOI] [PubMed] [Google Scholar]
- Raff M. Intracellular developmental timers. Cold Spring Harbor Symp Quant Biol. 2007;72:431–435. doi: 10.1101/sqb.2007.72.007. [DOI] [PubMed] [Google Scholar]
- Raff MC, Durand B, Gao FB. Cell number control and timing in animal development: the oligodendrocyte cell lineage. Int J Dev Biol. 1998;42:263–267. [PubMed] [Google Scholar]
- Reeves R. Nuclear functions of the HMG proteins. Biochim Biophys Acta. 2010;1799:3–14. doi: 10.1016/j.bbagrm.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–F177. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, Tajima S, Mitsuya K, Okano M, Koseki H. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- Shen S, Liu A, Li J, Wolubah C, Casaccia-Bonnefil P. Epigenetic memory loss in aging oligodendrocytes in the corpus callosum. Neurobiol Aging. 2008;29:452–463. doi: 10.1016/j.neurobiolaging.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. The ins and outs of RB: coupling gene expression to the cell cycle clock. Trends Cell Biol. 1994;4:15–18. doi: 10.1016/0962-8924(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Sohn J, Natale J, Chew LJ, Belachew S, Cheng Y, Aguirre A, Lytle J, Nait-Oumesmar B, Kerninon C, Kanai-Azuma M, Kanai Y, Gallo V. Identification of Sox17 as a transcription factor that regulates oligodendrocyte development. J Neurosci. 2006;26:9722–9735. doi: 10.1523/JNEUROSCI.1716-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonabend AM, Yun J, Lei L, Leung R, Soderquist C, Crisman C, Gill BJ, Carminucci A, Sisti J, Castelli M, Sims PA, Bruce JN, Canoll P. Murine cell line model of proneural glioma for evaluation of anti-tumor therapies. J Neuro-oncol. 2013;112:375–382. doi: 10.1007/s11060-013-1082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiss VA, Casaccia P. Cell-context specific role of the E2F/Rb pathway in development and disease. Glia. 2010;58:377–390. doi: 10.1002/glia.20933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiss VA, Nguyen T, Dugas J, Ibrahim A, Barres B, Androulakis IP, Casaccia P. Identification of a gene regulatory network necessary for the initiation of oligodendrocyte differentiation. PloS One. 2011;6:e18088. doi: 10.1371/journal.pone.0018088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–816. [PMC free article] [PubMed] [Google Scholar]
- Tamiya T, Mizumatsu S, Ono Y, Abe T, Matsumoto K, Furuta T, Ohmoto T. High cyclin E/low p27Kip1 expression is associated with poor prognosis in astrocytomas. Acta Neuropathol. 2001;101:334–340. doi: 10.1007/s004010000261. [DOI] [PubMed] [Google Scholar]
- Tang XM, Beesley JS, Grinspan JB, Seth P, Kamholz J, Cambi F. Cell cycle arrest induced by ectopic expression of p27 is not sufficient to promote oligodendrocyte differentiation. J Cell Biochem. 1999;76:270–279. doi: 10.1002/(sici)1097-4644(20000201)76:2<270::aid-jcb10>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Tikoo R, Osterhout DJ, Casaccia-Bonnefil P, Seth P, Koff A, Chao MV. Ectopic expression of p27Kip1 in oligodendrocyte progenitor cells results in cell-cycle growth arrest. J Neurobiol. 1998;36:431–440. doi: 10.1002/(SICI)1097-4695(19980905)36:3<431::AID-NEU10>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Tokumoto YM, Durand B, Raff MC. An analysis of the early events when oligodendrocyte precursor cells are triggered to differentiate by thyroid hormone, retinoic acid, or PDGF withdrawal. Dev Biol. 1999;213:327–339. doi: 10.1006/dbio.1999.9397. [DOI] [PubMed] [Google Scholar]
- Tokumoto YM, Apperly JA, Gao FB, Raff MC. Posttranscriptional regulation of p18 and p27 Cdk inhibitor proteins and the timing of oligodendrocyte differentiation. Dev Biol. 2002;245:224–234. doi: 10.1006/dbio.2002.0626. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Maekawa M. Methylation of DNA in cancer. Adv Clin Chem. 2010;52:145–167. doi: 10.1016/S0065-2423(10)52006-7. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Wells J, Boyd KE, Fry CJ, Bartley SM, Farnham PJ. Target gene specificity of E2F and pocket protein family members in living cells. Mol Cell Biol. 2000;20:5797–5807. doi: 10.1128/MCB.20.16.5797-5807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Hernandez M, Shen S, Sabo JK, Kelkar D, Wang J, O'Leary R, Phillips GR, Cate HS, Casaccia P. Differential modulation of the oligodendrocyte transcriptome by sonic hedgehog and bone morphogenetic protein 4 via opposing effects on histone acetylation. J Neurosci. 2012;32:6651–6664. doi: 10.1523/JNEUROSCI.4876-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Bieda M, Jin VX, Rabinovich A, Oberley MJ, Green R, Farnham PJ. A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res. 2007;17:1550–1561. doi: 10.1101/gr.6783507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zezula J, Casaccia-Bonnefil P, Ezhevsky SA, Osterhout DJ, Levine JM, Dowdy SF, Chao MV, Koff A. p21cip1 is required for the differentiation of oligodendrocytes independently of cell cycle withdrawal. EMBO Rep. 2001;2:27–34. doi: 10.1093/embo-reports/kve008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Gao Q, Li P, Liu X, Jia Y, Wu W, Li J, Dong S, Koseki H, Wong J. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 2011;21:1723–1739. doi: 10.1038/cr.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]