

Abstract
Anti-apoptotic Bcl-2 family members can contribute to tumorigenesis and may convey resistance to anti-cancer regimens. Therefore, they are important targets for novel therapeutics, particularly Bcl-2 homology (BH)3 mimetics. Bcl-B (BCL-2-like protein-10) is a relatively understudied member of the Bcl-2 protein family. Its physiological function is unknown, but it has been proven to have an anti-apoptotic activity and to act as a tumor promoter in mice. In human, high Bcl-B protein expression levels correlate with poor prognosis in various carcinomas and predict treatment resistance in acute myeloid leukemia. We here report that protein expression level and anti-apoptotic activity of Bcl-B are dictated by its ubiquitination. We demonstrate that Bcl-B is polyubiquitinated at steady state, in a unique loop between the BH1 and BH2 domains. Mutagenesis identified lysine (K)128 as an acceptor site for polyubiquitin chains, and K119 and K120, but not K181, as potential ubiquitination sites. Mass spectrometry confirmed K128 as a ubiquitination site and defined the polyubiquitin chains as K48-linked, which was confirmed by linkage-specific antibodies. Accordingly, Bcl-B proved to be an instable protein that is subject to ubiquitin-dependent proteasomal degradation at steady state. At equal mRNA expression, protein expression of a lysineless, nonubiquitinated Bcl-B mutant was fivefold higher than that of wild-type Bcl-B, demonstrating that ubiquitination is a key determinant for Bcl-B protein expression levels. Ubiquitination controlled the anti-apoptotic capacity of Bcl-B, in response to a variety of conventional and novel anti-cancer drugs. Certain anti-cancer drugs, known to reduce Mcl-1 protein levels, likewise downregulated Bcl-B. Together, these data demonstrate that polyubiquitination and proteasomal turnover dictate the expression level and anti-apoptotic capacity of Bcl-B.
Keywords: apoptosis, Bcl-2 family, Bcl-B, ubiquitination
Introduction
The Bcl-2 protein family is characterized by Bcl-2 homology (BH) domains that mediate the collaborative or competitive interactions between the different members. The family encompasses the six anti-apoptotic proteins Bcl-2, Bcl-B, Bcl-w, Bcl-xL, Bfl-1 and Mcl-1, many proapoptotic BH3 domain-only proteins and the effector proteins Bax and Bak.1 In response to apoptotic stimuli, the BH3-only proteins relocate to the mitochondria, where they activate Bax and Bak to form large homo-multimeric pores in the mitochondrial outer membrane. Through these pores, mediators are released that enable the activity of initiator- and effector caspases. The anti-apoptotic Bcl-2 proteins generally localize to the mitochondrial membrane. Here they inhibit Bax/Bak multimerization and apoptotic execution by sequestering the BH3 domain of BH3-only proteins, or activated Bax and Bak into a hydrophobic groove formed by their BH1–3 domains.1
It is thought that tumor cells are generally ‘addicted' to overexpression of anti-apoptotic Bcl-2 proteins. Tumor cells encounter many apoptotic stimuli during their growth, such as DNA damage and hypoxia. These stressful conditions will induce proapoptotic BH3-only proteins, and Bax/Bak may reach an activated state. Therefore, tumor cell survival depends on high levels of anti-apoptotic Bcl-2 family members to keep the activity of the proapoptotic Bcl-2 proteins in check.2 The anti-apoptotic Bcl-2 proteins thus present an Achilles' heel of the tumor cells. This notion has led to the development of BH3 mimetics: compounds that specifically bind to anti-apoptotic Bcl-2 proteins, and thereby neutralize their function.
In mouse models of lymphoma, overexpression of anti-apoptotic Bcl-2 proteins can contribute to tumorigenesis,3 as well as resistance to conventional DNA-damaging anti-cancer regimens.4 In human cancer, overexpression of Bcl-2 proper is the driver of follicular lymphoma and a potential mediator of resistance to radio- and chemotherapy.5, 6 Overexpression of other Bcl-2 family members is likewise associated with poor prognosis and potential treatment resistance in hematopoietic malignancies.7, 8 Bcl-2 family protein overexpression may also be a driver of solid tumor types, given the favorable response of small cell lung carcinoma to the BH3 mimetic ABT-263.9
As Bcl-2 family proteins become oncogenic and can confer treatment resistance upon overexpression, it is important to understand how their protein expression levels are regulated. We here present a key mechanism that dictates protein expression level and anti-apoptotic activity of Bcl-B (BCL-2-like protein-10). Bcl-B is the least studied anti-apoptotic Bcl-2 family protein. It was identified in human on the basis of homology with mouse Boo/Diva and chicken Nr-13,10, 11, 12 and clusters phylogenetically with Mcl-1 and Bfl-1.10 It is reportedly expressed in normal plasma cells and various normal epithelial tissues.13 Although its physiological function is largely unknown, data indicate that Bcl-B inhibits apoptosis and can promote tumorigenesis; Bcl-B inhibited Bax-induced apoptosis12, 14, 15 and accelerated Eμ-Myc-driven leukemogenesis in mice.3 Furthermore, it was found overexpressed in breast-, prostate-, gastric-, colorectal- and small cell lung carcinoma.13 In addition, Bcl-B expression levels are predictive for resistance to azacitidine in acute myeloid leukemia patients.16
We here define ubiquitin acceptor sites in Bcl-B and demonstrate its polyubiquitination on lysine K128. This targets Bcl-B for proteasomal degradation, thus limiting its steady-state expression levels. Ubiquitination of Bcl-B also determines its capacity to confer resistance to conventional and novel anti-cancer agents, including the BH3 mimetic ABT-737. Our data imply that ubiquitination controls Bcl-B expression levels, and thereby its anti-apoptotic and oncogenic potential. This notion opens up novel avenues for therapeutic targeting of Bcl-B in tumors that are reliant on Bcl-B overexpression.
Results
Bcl-B is ubiquitinated on specific lysine residues
To determine whether Bcl-B is a substrate for ubiquitination, we expressed N-terminally hemagglutinin (HA)-tagged wild-type (WT) Bcl-B together with FLAG-tagged ubiquitin in HEK 293 T cells. Cells were lysed under denaturing conditions to break all noncovalent protein interactions, and Bcl-B was isolated by anti-HA immunoprecipitation (IP). Immunoblotting with anti-HA monoclonal antibody (mAb) identified Bcl-B at about 25 kDa. The anti-FLAG mAb identified a range of ubiquitinated protein species in the WT Bcl-B isolate (Figure 1a), most likely representing Bcl-B carrying one, two, three or more ubiquitin moieties.
Figure 1.

Bcl-B is ubiquitinated on lysine residues. (a) HEK 293 T cells were transfected to express WT or lysine mutant (K/R) N-terminally HA-tagged Bcl-B, or empty vector, together with FLAG-tagged ubiquitin. Bcl-B was isolated from denatured cell lysates by anti-HA IP, followed by immunoblotting of the precipitates. HA-tagged Bcl-B and FLAG-ubiquitin were detected by probing with, respectively, anti-HA and anti-FLAG antibodies. (b) As in a, now with expression of C-terminally HA-tagged Bcl-B in WT and lysineless K/R mutant form. The two Bcl-B proteins species detected in this case result from alternative use of ATG start codons.10 (c) Bcl-B protein structure was modeled using M4T software. Depicted is a cartoon representation of the model structure, with the N terminus facing downwards. Lysine residues with side chain are represented in black. (d) As in a, now with expression of N-terminally HA-tagged Bcl-B in the form of WT, lysineless K/R mutant and various indicated single-, double- or triple K/R point mutants. Data shown are representative of at least three independent experiments.
Ubiquitin is typically linked to a free amino group present on either lysine residues or the free amino terminus of the substrate. To test whether Bcl-B ubiquitination was lysine-dependent, all four lysine (K) residues of Bcl-B were substituted by arginines (R), generating a lysineless Bcl-B K/R mutant. The HA-tagged Bcl-B K/R mutant was expressed together with FLAG-ubiquitin, and its ubiquitination status was examined by IP and immunoblotting. In this case, ubiquitin was completely lost from Bcl-B (Figure 1a), demonstrating that HA-Bcl-B is directly ubiquitinated on at least one of its four lysine residues.
To exclude that the HA-tag prevented a physiologically relevant ubiquitination on the N terminus of Bcl-B, WT Bcl-B and its K/R mutant were HA-tagged at the carboxy terminus. Next, their ubiquitination status was analyzed as described above. Also in this case, WT Bcl-B was clearly ubiquitinated and ubiquitination was completely lost upon mutation of all four lysines (Figure 1b). We conclude therefore that lysine residues, but not the N terminus of Bcl-B are targeted for ubiquitination.
To gain insight in the localization of the four lysine residues, we used a model of the Bcl-B structure that we had generated by M4T.17 The model revealed that all four lysines are exposed and potentially accessible to ubiquitin ligases (Figure 1c). To identify which of the lysines served as ubiquitin acceptor site, we made N-terminally HA-tagged Bcl-B variants with either one, two or three of the four lysines mutated into arginines. Neither combined double mutation of K119 and K120, nor single mutation of K128 or K181 resulted in a major loss of ubiquitin from Bcl-B (Figure 1d). However, upon combined triple mutation of lysines K119, K120 and K128, all ubiquitin was lost (Figure 1d), demonstrating that these residues, but not the remaining K181 residue, can act as ubiquitin acceptor sites in Bcl-B. Interestingly, multiple ubiquitinated protein species were found in the isolate of the Bcl-B K119, 120R double mutant (Figure 1d). As this mutant has only the K128 residue left as a possible ubiquitin acceptor site, we conclude that Bcl-B can be polyubiquitinated on K128. The collective data indicate that Bcl-B can be ubiquitinated on K119, K120 and/or K128, but not on K181.
Bcl-B carries K48-linked polyubiquitin chains on K128
Ubiquitin ligases can ubiquitinate the substrate on other lysine residues when the primary acceptor site has been lost by mutation. Therefore, we performed mass spectrometry (MS) to identify the primary ubiquitin acceptor site(s) in Bcl-B. Trypsin will cleave ubiquitin C-terminally of R74, leaving the ubiquitin-derived GG-peptide linked to the ubiquitination target residue of the substrate (mass increment 114 Da). On the basis of this mass difference, a GG-modified tryptic peptide can be distinguished from a nonmodifed peptide by MS analysis (reviewed in Kirkpatrick et al.18).
To obtain purified ubiquitin-conjugated Bcl-B, HEK 293 T cells were transfected with HA-Bcl-B, followed by lysis under denaturing conditions. Subsequently, HA-Bcl-B was isolated by IP as detailed in Materials and methods, and the precipitate was separated by SDS–polyacrylamide gel electroporesis. A small sample was run separately to define Bcl-B by immunoblotting (not shown). This analysis revealed that the preparation contained Coomassie-stainable amounts of nonmodified Bcl-B (band I; Figure 2a) and slower migrating protein species, possibly representing Bcl-B carrying one, two or three ubiquitin molecules (bands II, III, IV; Figure 2a).
Figure 2.

Bcl-B is ubiquitinated on K128 by K48-linked chains. (a) HEK 293 T cells were transfected to express N-terminally HA-tagged Bcl-B and subsequently lysed under denaturing conditions. HA-Bcl-B was purified by sequential anti-HA IP using two anti-HA mAb clones, as described in the Materials and methods section. The isolate was analyzed by SDS–polyacrylamide gel electrophoresis (SDS–PAGE) followed by Coomassie SimplyBlue staining. The asterisk indicates the heavy chain band of the antibody used for IP. (b) Purified protein in the respective bands identified in a was digested in gel with trypsin, and liberated peptides were analyzed by nanoLC-MS/MS. Depicted is the tandem mass spectrum of fragmented Bcl-B peptide, containing GG-modified K128. The amino acid sequence of the tryptic peptide with identified y and b ions and their masses is shown. The asterisk indicates the identified b4 ion minus NH3. (c) Same as b, but showing the tandem mass spectrum of fragmented ubiquitin peptide, containing GG-modified K48. The asterisk indicates the identified y6 ion minus NH3. (d) Anti-HA IP samples, isolated as in a from HEK 293 T cells expressing HA-Bcl-B or empty vector (EV), were separated by SDS–PAGE. On the same gel, 100 ng of synthetic K48- or K63-linked di-ubiquitin were loaded (di-Ubi). After blotting, the samples were probed with anti-HA to detect Bcl-B, with anti-ubiquitin P4D1 to detect all ubiquitin species (α-Ubi,) and with the K48- or K63-linkage-specific ubiquitin antibodies (α-K48, α-K63). Dotted lines indicate ubiquitinated species of Bcl-B as detected in α-Ubi blot, specifying the presence of one (Ubi), two, three, four or more ubiquitins, based on the molecular mass. Asterisk indicates some cross-reactivity with the light chain of the antibody used for IP and comigrating nonubiquitinated Bcl-B that were visible by Ponceau S staining. Data shown are representative of multiple independent experiments.
The indicated bands were excised from the gel and subjected to trypsin treatment, after which the extracted peptides were analyzed by MS. This confirmed that bands I, II, III and IV contained Bcl-B. Moreover, ubiquitin was found in bands II, III and IV. The tryptic peptide of Bcl-B that contained K119 and K120 was not found, but in bands II, II and IV, Bcl-B peptide with a GG signature on K128 was identified (Figure 2b). Thus, MS analysis provided the proof that residue K128 in Bcl-B is a primary ubiquitin acceptor site.
Ubiquitin itself has seven lysines that can be ubiquitinated, allowing the formation of different di-ubiquitin or polyubiquitin chains. The functional significance of the ubiquitination process is largely dependent on the chain-type formed. For example, K48-linked polyubiquitin chains generally target a substrate for proteasomal degradation.19 To identify the linkage type of the ubiquitin chains on Bcl-B, the MS data were analyzed for the occurrence of GG-modified ubiquitin peptides. This revealed ubiquitin peptides that were GG-modified on K48 (Figure 2c), whereas no ubiquitin peptides were found that were GG-modified on other lysine residues. GG-modified ubiquitin was only present in bands III and IV that contained Bcl-B carrying more than one ubiquitin. Therefore, these ubiquitin peptides most likely originated from the oligo-ubiquitin species that were conjugated to Bcl-B.
To further examine the ubiquitin chains on Bcl-B, we made use of antibodies that specifically recognize either K48- or K63-linked ubiquitin.20 Bcl-B or control samples were generated as described for MS and were analyzed by immunoblotting with anti-ubiquitin and the K48- and K63-linkage-specific antibodies. To validate these antibodies, we simultaneously probed synthetic K48- or K63-linked di-ubiquitin (Figure 2d). This verified the specificity of the linkage-specific antibodies, but also indicated that their reactivity was relatively weak as compared with reactivity of the anti-ubiquitin antibody (lanes 1, 2, 5, 10). The anti-ubiquitin antibody specifically detected oligo- and polyubiquitinated Bcl-B (lanes 3, 4). The K48-linkage-specific antibody also specifically reacted with Bcl-B carrying 2, 3, 4 or more ubiquitins, but not—as expected—with monoubiquitinated Bcl-B (lanes 7, 8). This confirmed the presence of K48-linked polyubiquitin on Bcl-B. The K63-linkage-specific antibody detected some bands, but these were also present in the control sample and/or did not align with the Bcl-B-ubiquitin conjugates defined by the anti-ubiquitin antibody (lanes 11, 12). The collective data indicate that Bcl-B is polyubiquitinated, predominantly and potentially exclusively with K48-linked chains, with K128 acting as a primary ubiquitin acceptor site.
Steady-state protein expression of Bcl-B is regulated by ubiquitination
The relative protein expression levels of pro- and anti-apoptotic Bcl-2 family members can determine whether the cell survives certain insults or stress conditions, or goes into apoptosis. To determine whether ubiquitination affected protein expression of Bcl-B at steady state, we compared protein levels of the WT and K/R mutant in the acute lymphoblastic leukemia cell lines J16 and MOLT-4. This was done under conditions of equal mRNA expression, as differences in protein expression are then most likely due to post-translational effects. The cell lines were transduced with constructs encoding WT or K/R mutant HA-Bcl-B, followed by an internal ribosomal entry site and green fluorescent protein (GFP). Next, the transduced cell populations were sorted on equal GFP expression by flow cytometry. This should yield stable cell lines that have equal mRNA expression of not only GFP, but also of WT or K/R mutant HA-Bcl-B, as one mRNA encoded both proteins. GFP levels in the resulting J16 and MOLT-4 cell lines transduced with WT or K/R mutant Bcl-B were equal, as assessed by flow cytometry (Figure 3a) and immunoblotting (Figure 3b). Under these standardized conditions, the steady-state protein expression level of the Bcl-B K/R mutant was much higher than that of WT Bcl-B (Figure 3b). Quantification of the results from multiple experiments showed that the protein expression level of the Bcl-B K/R mutant was close to fivefold higher than that of WT Bcl-B, in both J16 and MOLT-4 (Figure 3c). These data indicate that steady-state protein expression of Bcl-B is regulated by its ubiquitination.
Figure 3.

Steady-state protein expression of Bcl-B is regulated by ubiquitination. (a) MOLT-4 and J16 cells were retrovirally transduced with an internal ribosomal entry site (IRES)–GFP vector to express N-terminally HA-tagged Bcl-B in WT or lysineless K/R mutant form. In addition, control cells (- in panel b) were created by transduction with empty IRES–GFP vector. Cells were flow cytometrically sorted twice for equal GFP expression. Histograms depict GFP fluorescence intensity of the different cell lines after the second sort. (b) Total cell lysates of the cell lines described in a were analyzed by immunoblotting for protein expression of Bcl-B (anti-α-HA), GFP and actin. The asterisk indicates a background band of unknown nature. Data are representative of three independent experiments. (c) Quantification of Bcl-B protein expression levels, as determined in b. Bcl-B signal intensity was corrected for GFP signal intensity in the same cells and Bcl-B WT expression level was set to 1. Data represent mean +s.d. from three independent experiments.
Ubiquitination dictates the half-life of Bcl-B and targets Bcl-B for proteasomal degradation
To follow the stability and degradation of Bcl-B in detail, MOLT-4 cells expressing WT or K/R mutant HA-Bcl-B in conjunction with GFP were treated with the protein synthesis inhibitor cycloheximide (CHX) for different periods of time. Cell lysates were analyzed by immunoblotting for protein levels of Bcl-B, whereas GFP was monitored as a control for equal protein loading. Within 360 min after inhibition of protein synthesis, almost all WT Bcl-B protein had disappeared (Figure 4a). Prevention of Bcl-B ubiquitination stabilized the protein, as shown by a decreased degradation rate of the Bcl-B K/R mutant compared with WT Bcl-B (Figures 4a and b). These findings were refined using metabolic pulse-chase radiolabeling. MOLT-4 cells expressing WT or K/R mutant HA-Bcl-B were pulsed with [35S]-methione and -cysteine, and the fate of the radiolabeled Bcl-B protein pool was followed throughout a 6-h chase period. As with the CHX assays, we found that WT Bcl-B was more rapidly degraded than the K/R mutant (Figure 4c). The half-life of WT Bcl-B was calculated to be 68 min, whereas Bcl-B K/R had an almost threefold increased half-life of 170 min (Figures 4d and e). These data indicate that Bcl-B is an instable protein, whose half-life is to a significant extent determined by steady-state ubiquitination.
Figure 4.

Ubiquitination targets Bcl-B for proteasomal degradation. (a) MOLT-4 cells stably expressing WT or K/R mutant HA-Bcl-B in an internal ribosomal entry site (IRES)–GFP configuration were treated with CHX (50 μg/ml) for the indicated time periods. Cell lysates were analyzed by immunoblotting for Bcl-B (α-HA) and GFP as a stable protein and loading control (α-GFP). Antibodies were labeled with fluorescent dyes to allow read-out on the Odyssey infrared imager. Data shown are representative of three independent experiments. (b) Quantification of three independent experiments such as shown in a. Data represent mean ±s.d. Bcl-B signal intensity was corrected for GFP signal intensity in the same cells, the 0-min time point was set to 100% and data points were connected by a one-phase decay curve fit. (c) The same cell lines as in a were labeled with [35S]-cysteine and -methionine for 30 min, followed by a chase with nonradioactive amino acids for the indicated time periods. Bcl-B was immunoprecipitated with anti-HA mAb and resolved by SDS–polyacrylamide gel electroporesis. Radioactive signals were quantified by phosphorimaging. Data shown are representative of three independent experiments. (d) Quantification of three independent experiments such as shown in c. Data represent mean ±s.d. Local background was subtracted from the HA-Bcl-B signals, the 0-min time point was set to 100%, and data points were connected by a one-phase decay curve fit. (e) The HA-Bcl-B half-life was calculated from the individual experiments shown in c. The mean half-life +s.d. standard are shown (n=3). Asterisk indicates statistically significant difference (Student's t-test; *P>0.05). (f) J16 cells stably expressing WT or K/R mutant HA-Bcl-B were treated with CHX, either alone or together with the proteasome inhibitors MG132 (MG, 50 μM) or epoxomicin (EPX, 10 μM), or the lysosome inhibitors bafilomycin A1 (BAF, 1 μM), or chloroquine (CHQ, 200 μM). After 8 h of treatment, cell lysates with equal protein content were analyzed by immunoblotting for Bcl-B (α-HA) or actin (α-actin). Data shown are representative of three independent experiments. Numbers above blot indicate signal intensity of HA-Bcl-B, corrected for actin signal, untreated sample of Bcl-B WT or Bcl-B K/R was set to 1.
K48-linked ubiquitin conventionally targets substrates to the proteasome.19 Therefore, our finding that Bcl-B is polyubiquitinated with K48-linked chains suggested that it is subject to proteasomal degradation. To examine the mechanism of Bcl-B degradation, J16 cells were incubated with CHX alone, or together with either proteasome inhibitors (MG132, epoxomycin) or lysosome inhibitors (bafilomycin A1, chloroquine). Proteasome inhibitors greatly reduced the degradation of WT Bcl-B, whereas lysosome inhibitors had no effect (Figure 4f). The protein levels of the Bcl-B K/R mutant did not decrease dramatically upon CHX treatment. Some degradation was observed and this could be blocked to some extent by proteasome inhibitors (Figure 4f). Together, these data indicate that steady-state ubiquitination of Bcl-B promotes its proteasomal degradation and greatly shortens its half-life.
Prevention of Ubiquitination does not affect Bcl-B localization or its ability to interact with BH3-only proteins
Prosurvival Bcl-2 proteins inhibit the apoptotic pathway at the mitochondrial membrane, where they sequester proapoptotic Bcl-2 family members. To assess a possible impact of ubiquitination on Bcl-B function, we first determined whether the localization of Bcl-B was affected by prevention of its ubiquitination. For this purpose, the localization of WT and K/R mutant HA-Bcl-B in U2OS cells was analyzed by confocal laser-scanning microscopy. The N-terminal HA tag did not affect the mitochondrial localization signal, which is present as a hydrophobic helix at the C terminus of Bcl-B.10, 12 Both WT and K/R Bcl-B primarily localized to the mitochondria, as revealed by colocalization with the mitochondrion-selective dye Mitotracker (Figure 5a). Image analysis revealed that WT Bcl-B and the K/R mutant did so to a similar extent (Figure 5b), indicating that the mitochondrial localization of Bcl-B was not affected by deletion of its ubiquitin acceptor sites.
Figure 5.

The lysineless Bcl-B K/R mutant behaves like WT Bcl-B in terms of localization and interactions with BH3-only proteins. (a) Subcellular localization. U2OS cells transfected to express HA-Bcl-B and Life-Act-GFP were stained to detect nuclei (DAPI, 4',6-diamidino-2-phenylindole), mitochondria (Mitotracker Deep Red) and Bcl-B (anti-HA), and examined by confocal laser-scanning microscopy. Representative cells of multiple independent experiments are shown. (b) Colocalization of Mitotracker and HA-Bcl-B signal of experiments such as shown in a was quantified using the Intensity Correlation Analysis plugin and ImageJ software. A total of 80 or 96 cells were analyzed for Bcl-B WT and Bcl-B K/R, respectively. (c) Interaction with BH3-only proteins. HEK 293 T cells were transfected to express WT or K/R mutant HA-Bcl-B, together with Myc-tagged Bik, Bim, Puma or Noxa. Cells were lysed in CHAPS buffer and Bcl-B was isolated by IP with anti-HA mAb. Total cell lysates (TCL) and precipitates (IP) were analyzed by immunoblotting with anti-α-HA or α-Myc antibody. Data shown are representative of multiple independent experiments.
We subsequently examined whether prevention of Bcl-B ubiquitination influenced its ability to interact with proapoptotic BH3-only proteins. HEK 293 T cells were transfected to express WT or K/R mutant HA-Bcl-B, together with Myc-tagged BH3-only proteins. Bim, Bik, Puma and Noxa interacted to a similar extent with WT and K/R mutant Bcl-B, as assessed by anti-HA IP, followed by anti-Myc immunoblotting (Figure 5c). This indicated that deletion of its ubiquitin acceptor sites did not affect the ability of Bcl-B to sequester these proapoptotic BH3-only proteins. In summary, mutation of internal lysines and consequent prevention of ubiquitination did not affect Bcl-B localization or interaction with its proapoptotic relatives.
Bcl-B ubiquitination regulates its capacity to protect cells against anti-cancer therapeutics
Reportedly, Bcl-B overexpression can protect cells against apoptosis, but only a few proapoptotic agents have been tested.12, 21, 22 We aimed to extend this to clinically relevant anti-cancer regimens and to test whether Bcl-B ubiquitination affected its anti-apoptotic capacity. For this purpose, we compared the ability of WT Bcl-B and its nonubiquitinatable K/R mutant to protect cells from a range of conventional and targeted anti-cancer drugs with diverse modes of action. J16 and MOLT-4 cells were employed, that have a mutant and WT p53 status, respectively. We used the cell lines expressing GFP only, WT HA-Bcl-B or its K/R mutant at equal mRNA levels (described in Figure 3). The cells were treated with a dose range of the toposiomerase inhibitor etoposide and the microtubule destabilizer vincristine as conventional agents, and the BH3 mimetic ABT-737,23 the cyclin-dependent kinase inhibitor roscovitine and the death receptor agonist tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) as novel targeted agents. For TRAIL, only J16 was included, as MOLT-4 cells are not sensitive to TRAIL-induced apoptosis. Cell death was monitored after 48 h by propidium iodide uptake. It was apoptotic in all cases, as determined by inhibition with a pan-caspase inhibitor and by nuclear fragmentation (results not shown). In both cell lines, WT Bcl-B provided very little protection to any of the drugs (Figure 6). However, as compared with WT Bcl-B, the Bcl-B K/R mutant provided significant protection, particularly to ABT-737 in both J16 and MOLT-4 cells, but also to etoposide and vincristine in both cell types, and to roscovitine and TRAIL in J16 (Figure 6).
Figure 6.

Ubiquitination of Bcl-B affects its potential to inhibit cell death. J16 (left panels) and MOLT-4 (right panels) cell lines expressing empty vector (EV), WT Bcl-B or K/R mutant Bcl-B (Figure 3a) were treated with the targeted anti-cancer drugs ABT-737, roscovitine or TRAIL, or the conventional anti-cancer drugs etoposide or vincristine, at the indicated dose range. After 24 h, dead cells were identified by flow cytometric analysis of propidium iodide uptake. Data represent mean +s.d. from three independent experiments. Asterisks indicate statistically significant difference (Student's t-test; *P<0.05, **P<0.01, ***P<0.001).
To exclude that the HA-tag impacted on the anti-apoptotic function of Bcl-B, we also tested J16 cells expressing untagged versions of Bcl-B WT and K/R, side by side with J16 cells expressing HA-tagged Bcl-B. This revealed that untagged and HA-tagged Bcl-B provided cells with similar levels of protection against etoposide and ABT-737 (Supplementary Figure S1). Moreover, also for untagged Bcl-B, the K/R mutant provided significantly more protection to both stimuli than the WT protein (Supplementary Figure S1). We conclude that ubiquitination of Bcl-B regulates its steady-state expression level and, thereby, its capacity to protect cells against a diverse array of conventional and targeted anti-cancer drugs.
Roscovitine, etoposide and vincristine downregulate Bcl-B protein levels, as they do for Mcl-1
It has been reported that certain anti-cancer agents reduce protein expression levels of the anti-apoptotic Bcl-2 protein Mcl-1, and thus facilitate tumor cell killing.24, 25, 26 Roscovitine lowers Mcl-1 levels by blocking transcription,24 but vincristine and etoposide promote Mcl-1 degradation by activating the E3 ligases SCFFBW7 (Wertz et al.25) and Mule,26 respectively. To investigate whether such modes of regulation also apply to Bcl-B, we treated J16 cells stably expressing WT Bcl-B with roscovitine, etoposide or vincristine in a time course. Samples were subjected to immunoblotting for endogenous Mcl-1, actin and HA-Bcl-B. Treatment with roscovitine and etoposide rapidly reduced Mcl-1 protein levels (Figure 7), as previously reported.24, 26 Interestingly, Bcl-B protein levels were reduced to a similar extent in response to these two drugs. Vincristine also reduced Bcl-B and Mcl-1 levels, but with slower kinetics. This is consistent with a mutation in the Mcl-1 ligase SCFFBW7 that has been reported for Jurkat cells,27 from which the J16 cell line is derived. Thus, Bcl-B might be subject to the same modes of regulation as Mcl-1. The data imply that treatment outcome may be determined by steady-state Bcl-B turnover, but also by drug-induced Bcl-B degradation.
Figure 7.
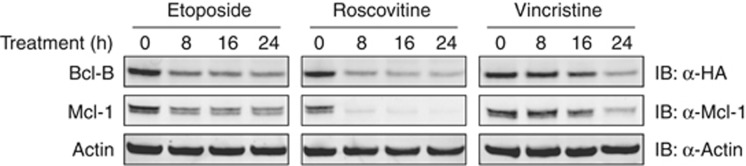
Certain anti-cancer agents reduce Bcl-B and Mcl-1 protein levels. J16 cells stably expressing HA-tagged Bcl-B were incubated for indicated periods with etoposide (5 μg/ml), roscovitine (100 μM) or vincristine (5 ng/ml). Lysates were subjected to immunoblotting for Bcl-B (anti-HA) and endogenous Mcl-1. Actin levels served as a control for equal loading. Data shown are representative for at least two independent experiments.
Discussion
Overexpression of anti-apoptotic Bcl-2 family members allows tumor cells to survive the many prodeath signals they encounter during their rapid growth under unfavorable circumstances. In addition, it can provide them with resistance against anti-cancer drugs. In this study, we show that the capacity of Bcl-B to protect cells against apoptosis is controlled by its ubiquitin-mediated proteasomal degradation.
IP experiments and MS data revealed that Bcl-B was polyubiquitinated on internal lysines. Mutations of the lysines into argines showed that three out of the four lysines of Bcl-B can serve as ubiquitin acceptor sites and that K128 is polyubiquitinated. A crystal structure of Bcl-B was recently reported,28 but it does not include the regions in the molecule that contain the lysines. Our homology-based model closely matches this structure and indicates that the defined ubiquitin acceptor site K128 and the potential acceptor sites K119 and K120 reside in the previously identified unstructured loop between the BH1 and BH2 domains,10, 29 specifically between α-helices 5 and 6 (Figure 1c). This suggests that these three lysines are highly exposed and, therefore, can be efficiently targeted for ubiquitination. We found that K181 is not ubiquitinated, possibly because its location is too distant from the active site of the ubiquitination machinery. Alternatively, it might be inaccessible because of its proximity to the trans-membrane domain. Interestingly, the unstructured loop containing K119, K120 and K128 is not found in any other human anti-apoptotic Bcl-2 family protein.29 The loop region is also highly variable within vertebrate species in both sequence and length. However, the conservation within primates is high, possibly pointing at a recent evolutionary role for Bcl-B ubiquitination.29
Lysineless, nonubiquitinatable Bcl-B was more stable than WT Bcl-B, demonstrating that ubiquitination targets Bcl-B for degradation. Accordingly, we found that Bcl-B is degraded by the proteasome. However, preventing ubiquitination of Bcl-B did not completely prevent its proteasomal degradation. Possibly, Bcl-B can also be degraded by an ubiquitin-independent proteasomal pathway, as has been shown for the Bcl-2 family members Mcl-1 and Bim.30, 31 Recently, Beverly et al.32 also described that lysineless Bcl-B is more stable that WT Bcl-B. This correlated with improved oncogenic potential, as the mutant more potently accelerated Eμ-Myc-driven leukemogenesis than WT Bcl-B.
In contrast to our data, these authors conclude that in HEK 293 T cells, ectopically expressed Bcl-B is exclusively monoubiquitinated on multiple lysine residues, based on coexpression with lysineless HA-tagged ubiquitin. We cannot exclude that multiple monoubiquitin species can be appended on to a single Bcl-B molecule, but our data using a Bcl-B mutant with only one acceptor lysine residue clearly indicate that Bcl-B is polyubiquitinated on K128. Furthermore, both MS analysis and probing with validated linkage-specific antibody revealed that WT Bcl-B carries K48-linked polyubiquitin chains. Moreover, we show that Bcl-B is targeted for proteasomal degradation in agreement with the canonical function of K48-linked polyubiquitin chains.19 We therefore conclude that polyubiquitination of Bcl-B drives its rapid degradation.
We demonstrate here that Bcl-B can protect tumor cells against various novel and conventional anti-cancer treatments, provided that it is expressed at high-enough levels. In T-leukemic cells, WT Bcl-B provided resistance against the BH3 mimetic ABT-737, but not against roscovitine, etoposide, vincristine and TRAIL. Upon expression at the same mRNA levels, protein levels of the nonubiquitinatable Bcl-B K/R mutant were higher than those of WT Bcl-B, due to its decreased proteasomal turnover. In contrast to WT Bcl-B, Bcl-B K/R provided resistance against all tested anti-cancer treatments, in both p53-WT and p53-mutant leukemic cells. As Bcl-B K/R behaves like WT Bcl-B in terms of BH3-only protein binding and localization, this demonstrates that ubiquitination controls the anti-apoptotic activity of Bcl-B. This implies that loss of Bcl-B ubiquitination in cancer cells, which may occur by defects in its ubiquitination machinery or overexpression of its deubiquitinating enzyme(s), may promote resistance to diverse therapies.
Interestingly, roscovitine, etoposide and, to a lesser extent, vincristine reduced Bcl-B protein levels. Etoposide and vincristine are known to induce degradation of Mcl-1.25, 26 As these drugs reduced Bcl-B and Mcl-1 protein levels with similar kinetics, degradation of both proteins may be regulated by the same mechanism. Roscovitine, on the other hand, can block Mcl-1 transcription. Thereby, it mediated a rapid drop in Mcl-1 protein levels, given the inherent instability of Mcl-1.24 On the basis of this principle, translation inhibitors are now under clinical evaluation to kill chronic myeloid leukemia cells33 that depend on high Mcl-1 protein levels.34 Our data suggests that these various anti-cancer drugs could be useful in tumor types that depend on Bcl-B for survival or treatment resistance.
These findings also have implications for cancer therapy with BH3 mimetic drugs. ABT-737 and its clinically applied orally available analog ABT-263 are selective for certain anti-apoptotic Bcl-2 proteins. In the cellular context, ABT-737 targets Bcl-2, Bcl-xL and Bcl-w with different efficacy,22, 35 but not Bfl-1, Mcl-136, 37 and Bcl-B.22 Given that expression levels of Bfl-1,38 Mcl-139 and Bcl-B are determined by ubiquitin-dependent proteasomal degradation, stimulation of their degradation may alleviate ABT-737 resistance, as already demonstrated for Mcl-1.40
Our data imply that in cancer diagnostics, Bcl-B expression should be monitored at the protein rather than at the mRNA level, for which a flow cytometric assay was recently reported.16 Elevated Bcl-B protein expression indeed correlated with resistance to azacytidine in myeloid leukemia in that study. In addition, it is important to find the machinery that determines the ubiquitination status of Bcl-B. For Mcl-1, two E3 ligases (MULE, SCFFBW7) and a deubiquitinating enzyme (USP9X) have been identified.25, 39, 41, 42 Interestingly, high expression of USP9X resulted in high Mcl-1 levels and correlated with poor prognosis of multiple myeloma patients.42 Conversely, loss of the E3 ligase SCFFBW7 made T-ALL cells resistant to ABT-737, by increasing Mcl-1 levels.41 Similarly, ligases or deubiquitinating enzymes of Bcl-B could represent prognostic markers or even druggable targets.
Materials and methods
Constructs
The cDNA encoding full-length human Bcl-B (ImaGenes, Berlin, Germany) was cloned into modified versions of pEGFP-N or pEGFP-C, in which the enhanced GFP coding sequence was replaced by a double HA-tag sequence. The N-terminal HA-tag was modified to contain a serine and aspartate in front of the HA-tag, to make it a target for N-terminal acetyltransferase A.43 Point mutants of Bcl-B were obtained by site-directed mutagenesis PCR. For retroviral transduction, N-terminally HA-tagged WT Bcl-B cDNA and its K/R mutant were cloned into pMSCV-IRES-GFP. In addition, pMSCV constructs were created encoding untagged WT and K/R mutant Bcl-B. The plasmids pcDNA3-FLAG-Ubiquitin44 and Liveact-GFP45 have been described. The cDNAs encoding full-length human Bik, Bim, Puma and Noxa were obtained from Geneservice Ltd (Cambridge, UK) and cloned into pCDNA3 with a double Myc-tag at the N terminus.
Cell lines, gene transfection and transduction
The T-lymphoblastic leukemia cell lines J16 and MOLT-4 were cultured in Iscove's modified Dulbecco's medium and the osteosarcoma cell line U2OS, the human embryonic kidney cell line HEK 293T and its derivative Phoenix-Ampho were cultured in Dulbecco's modified Eagle's medium. Transfections of cDNA were carried out in serum-free medium with polyethyleneimine at a DNA:polyethyleneimine ratio of 1:3 (w/w). Retroviral particles were produced in the Phoenix-Ampho cell line. Retroviral transduction of J16 and MOLT-4 cells was performed as described.46 Upon integration of the retroviral construct in the host cell genome, Bcl-B and GFP proteins are expressed from the same mRNA, because of the presence of a ribosomal entry site. Transduced cells expressing WT or K/R mutant Bcl-B were sorted on equal GFP signal using a FACSAria (BD Biosciences, San Jose, CA, USA). The resulting stable cell lines used for analysis are polyclonal, representing thousands of cells of the J16 or MOLT-4 cell lines used for transduction.
Western blotting
SDS–polyacrylamide gel electroporesis was performed as described,44 and blotting was performed using the Trans-Blot Turbo system (Bio-Rad, Hercules, CA, USA). Primary antibodies used were as follows: rabbit anti-GFP polyclonal,47 mouse anti-Actin mAb MAB1501R (Millipore, Billerica, MA, USA), rabbit polyclonal anti-Mcl-1 S-19 (Santa Cruz, Dallas, TX, USA), rabbit anti-Actin mAb D6A8 (Cell Signaling, Danvers, MA, USA), rabbit anti-Ubiquitin mAb Apu3 (K63-specific) and Apu2 (K48-specific) (Millipore), horseradish peroxidase-conjugated anti-Flag mAb M2 (Sigma-Aldrich, St Louis, MO, USA), mouse anti-Ubiquitin mAb P4D1 (Santa Cruz), and fluorochrome-conjugated anti-HA mAb 12CA5, anti-Myc mAb 9E10 (both purified in house) and anti-FLAG mAb M2 (Sigma-Aldrich). Fluorochromes DY-682 (HA) or DY-800 (Myc, FLAG) were from Dyomics (Jena, Germany). Fluorescently labeled secondary antibodies were from LI-COR (Lincoln, NE, USA). Fluorescence signals were visualized and quantified on the Odyssey Imaging System (LI-COR), and chemiluminescence signals (Pierce Biotechnology, Rockford, IL, USA) by the ChemiDoc imaging system (Bio-Rad) or exposure to film, GE Healthcare Life Sciences, Buckinghamshire, UK.
Immunoprecipitation
For analysis of Bcl-B ubiquitination, HEK 293 T cells were collected 24 h after transfection and lysed in SDS buffer (50 mM Tris–HCl, pH 8.0, 1% SDS, 0.5 mM EDTA, 10 mM dithiothreitol) 10 min, 95 °C. Next, nine volumes of NP-40 buffer were added and lysates were cleared by centrifugation at 17 000 g for 10 min at 4 °C. Equal amounts of protein were incubated with anti-HA mAb 12CA5 and Protein G Sepharose beads (GE Healthcare Life Sciences). For analysis of Bcl-B interactions, transfected HEK 293T cells were incubated with the pan-caspase inhibitor Q-VD-OPH (10 μM). IPs were performed in Chaps buffer directly (Noxa) or after fixing with formaldehyde as described (Bik, Bim, Puma).22, 48 For MS, HEK 293T cells were collected 24 h after transfection in phosphate-buffered saline (PBS) with 2 mM N-ethylmaleimide and lysed in SDS buffer. After quenching with NP-40, IP was performed with anti-HA mAb 12CA5, bound protein was eluted from the Protein G beads with SDS buffer, after which a second IP in excess NP-40 buffer was performed with anti-HA 3F10 affinity matrix (Roche, Basel, Switzerland). This eluate was separated by SDS–polyacrylamide gel electroporesis and the gel was Coomassie-stained with SimplyBlue SafeStain (Invitrogen, Carlsbad, CA, USA).
Mass spectrometry
For MS analysis, selected bands were cut from the gel and reduced with dithiothreitol. To avoid false-positive interpretation of ubiquitination,49 the N-methylated form of iodoacetamide was used as alkylation reagent instead of standard iodoacetamide. Trypsin digestion was performed using the Proteineer DP digestion robot (Bruker, Bremen, Germany). The tryptic peptides were extracted from the gel, lyophilized, dissolved in 95/3/0.1 v/v/v water/acetonitril/formic acid and subsequently analyzed by online nano high-performance liquid chromatography MS/MS. An 1100 HPLC system (Agilent Technologies, Santa Clara, CA, USA)50 coupled to a 7-T LTQ-FT Ultra mass spectrometer (Thermo Electron, Waltham, MA, USA) was used, essentially as described51 and detailed in Supplementary Materials and methods.
Assessment of protein stability
J16 or MOLT-4 cell lines expressing HA-Bcl-B were treated with CHX (50 μg/ml) for the indicated periods of time. Cells were lysed in NP-40-buffer and subjected to western blotting. For metabolic pulse-chase labeling, MOLT-4 cells were starved for 1 h in methionine- and cysteine-free medium, followed by a 30-min pulse with medium with [35S]-methionine and -cysteine (1 mCi/ml total, Perkin Elmer, Waltham, MA, USA). Subsequently, the medium was removed and replaced with complete medium containing 5 mM additional unlabeled methionine and cysteine. At the indicated time points, a cell sample was withdrawn, and cells were washed, lysed in NP-40 buffer and subjected to anti-HA IP and SDS–polyacrylamide gel electroporesis. Imaging and quantification were performed on a phosphorimager (Fujifilm, Tokyo, Japan).
Confocal laser-scanning microscopy
At 24 h after transfection, U2OS cells were stained with Mitrotracker Deep Red (Invitrogen), fixed with 4% PFA in PBS for 20 min, quenched with 125 mM glycine in PBS and permeabilized with 0.1% Triton-X 100 in PBS. Cells were incubated with 1% bovine serum albumin in PBS for 30 min to block nonspecific antibody binding, after which rat anti-HA mAb 3F10 (1:100, Roche) was added, followed by Alexa-594-conjugated goat anti-rat IgG (Invitrogen) and DAPI (4',6-diamidino-2-phenylindole). Next, cells were mounted onto slides using Fluor-gel (Electron Microscopy Sciences, Hatfield, PA, USA). Images were taken on a TCS SP5 microscope (Leica, Wetzlar, Germany) and colocalization was quantified using the Intensity Correlation Analysis plugin and ImageJ software (NIH, Bethesda, MD, USA).
Reagents
Stock solutions were prepared in dimethylsulfoxide for etoposide, epoxomicin, CHX, chloroquine diphosphate (Sigma-Aldrich), Q-VD-OPH (SM Biochemicals, Yorba Linda, CA, USA), ABT-737 (Chemietek, Indianapolis, IN, USA), MG132 (Calbiochem, Darmstadt, Germany), r-roscovitine (Cayman Chemicals, Ann Arbor, MI, USA) and bafilomycin A1 (Santa Cruz). Vincristine in solution for injection was from Faulding Pharmaceuticals Plc (Warwickshire, UK). Soluble recombinant Isoleucine Zippered TRAIL46 and di-ubiquitin molecules52 have previously been described.
Cell death assays
To assess cell death, cells were plated in round-bottom 96-well plates, at 25 000 cells/well in 100 μl Iscove's modified Dulbecco's medium. Stimuli were given in 100 μl Iscove's modified Dulbecco's medium, keeping the solvent constant, and cells were subsequently placed in the incubator. After 48 h, cells were washed with PBS and subsequently stained with propidium iodide (1 μg/ml) in PBS with bovine serum albumin for 5 min, to monitor dead cells. Cells were analyzed on a FACSArray (BD Biosciences), equipped with a plate loader.
Acknowledgments
We thank Lennert Janssen, Drs Ricky Johnstone, Henning Walczak and Huib Ovaa for kindly providing reagents, and Drs Abdel Aouacheria and Mark Hinds for advise on the Bcl-B protein structure. This work was supported by grant ECHO 700.57.008 from the Netherlands Organization for Scientific Research and grant NKI 2008-4110 from the Dutch Cancer Society, awarded to JB, and GMCJ was supported by the Netherlands Proteomics Center.
Glossary
- BH
Bcl-2 homology
- CHX
cycloheximide
- DUB
deubiquitinating enzyme
- IP
immunoprecipitation
- mAb, monoclonal antibody; WT
wild-type.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
Supplementary Material
References
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo M, Moore VDG, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Beverly LJ, Varmus HE. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene. 2009;28:1274–1279. doi: 10.1038/onc.2008.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, Rosenthal CT, Lowe SW. Genetic analysis of chemoresistance in primary murine lymphomas. Nat Med. 2000;6:1029–1035. doi: 10.1038/79542. [DOI] [PubMed] [Google Scholar]
- Knoops L, Haas R, de Kemp S, Majoor D, Broeks A, Eldering E, et al. In vivo p53 response and immune reaction underlie highly effective low-dose radiotherapy in follicular lymphoma. Blood. 2007;110:1116–1122. doi: 10.1182/blood-2007-01-067579. [DOI] [PubMed] [Google Scholar]
- Tothova E, Fricova M, Stecova N, Kafkova A, Elbertova A. High expression of Bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Neoplasma. 2002;49:141–144. [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Pepper C, Lin TT, Pratt G, Hewamana S, Brennan P, Hiller L, et al. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood. 2008;112:3807–3817. doi: 10.1182/blood-2008-05-157131. [DOI] [PubMed] [Google Scholar]
- Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouacheria A, Arnaud E, Venet S, Lalle P, Gouy M, Rigal D, et al. Nrh, a human homologue of Nr-13 associates with Bcl-Xs and is an inhibitor of apoptosis. Oncogene. 2001;20:5846–5855. doi: 10.1038/sj.onc.1204740. [DOI] [PubMed] [Google Scholar]
- Lee R, Chen J, Matthews CP, McDougall JK, Neiman PE. Characterization of NR13-related human cell death regulator, Boo/Diva, in normal and cancer tissues. Biochim Biophys Acta. 2001;1520:187–194. doi: 10.1016/s0167-4781(01)00268-8. [DOI] [PubMed] [Google Scholar]
- Ke N, Godzik A, Reed JC. Bcl-B, a novel Bcl-2 family member that differentially binds and regulates Bax and Bak. J Biol Chem. 2001;276:12481–12484. doi: 10.1074/jbc.C000871200. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Kitada S, Winter JN, Variakojis D, Lichtenstein A, Zhai D, et al. Bcl-B expression in human epithelial and nonepithelial malignancies. Clin Cancer Res. 2008;14:3011–3021. doi: 10.1158/1078-0432.CCR-07-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai D, Jin C, Huang Z, Satterthwait AC, Reed JC. Differential regulation of Bax and Bak by anti-apoptotic Bcl-2 family proteins Bcl-B and Mcl-1. J Biol Chem. 2008;283:9580–9586. doi: 10.1074/jbc.M708426200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai D, Ke N, Zhang H, Ladror U, Joseph M, Eichinger A, et al. Characterization of the anti-apoptotic mechanism of Bcl-B. Biochem J. 2003;376:229–236. doi: 10.1042/BJ20030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluzeau T, Robert G, Mounier N, Karsenti JM, Dufies M, Puissant A, et al. BCL2L10 is a predictive factor for resistance to Azacitidine in MDS and AML patients. Oncotarget. 2012;3:490–501. doi: 10.18632/oncotarget.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Fuentes N, Madrid-Aliste CJ, Rai BK, Fajardo JE, Fiser A. M4T: a comparative protein structure modeling server. Nucleic Acids Res. 2007;35:W363–W368. doi: 10.1093/nar/gkm341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick DS, Denison C, Gygi SP. Weighing in on ubiquitin: the expanding role of mass-spectrometry-based proteomics. Nat Cell Biol. 2005;7:750–757. doi: 10.1038/ncb0805-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- Zhang H, Holzgreve W, De Geyter C. Bcl2-L-10, a novel anti-apoptotic member of the Bcl-2 family, blocks apoptosis in the mitochondria death pathway but not in the death receptor pathway. Hum Mol Genet. 2001;10:2329–2339. doi: 10.1093/hmg/10.21.2329. [DOI] [PubMed] [Google Scholar]
- Rooswinkel RW, van de Kooij B, Verheij M, Borst J. Bcl-2 is a better ABT-737 target than Bcl-xL or Bcl-w and only Noxa overcomes resistance mediated by Mcl-1, Bfl-1, or Bcl-B. Cell Death Dis. 3:e366. doi: 10.1038/cddis.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, et al. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005;65:5399–5407. doi: 10.1158/0008-5472.CAN-05-0233. [DOI] [PubMed] [Google Scholar]
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- Hao Z, Duncan GS, Su YW, Li WY, Silvester J, Hong C, et al. The E3 ubiquitin ligase Mule acts through the ATM-p53 axis to maintain B lymphocyte homeostasis. J Exp Med. 2012;209:173–186. doi: 10.1084/jem.20111363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautureau GJ, Yabal M, Yang H, Huang DC, Kvansakul M, Hinds MG. The restricted binding repertoire of Bcl-B leaves Bim as the universal BH3-only prosurvival Bcl-2 protein antagonist. Cell Death Dis. 2012;3:e443. doi: 10.1038/cddis.2012.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemin Y, Cornut-Thibaut A, Gillet G, Penin F, Aouacheria A. Characterization of unique signature sequences in the divergent maternal protein Bcl2l10. Mol Biol Evol. 2011;28:3271–3283. doi: 10.1093/molbev/msr152. [DOI] [PubMed] [Google Scholar]
- Stewart DP, Koss B, Bathina M, Perciavalle RM, Bisanz K, Opferman JT. Ubiquitin-independent degradation of antiapoptotic MCL-1. Mol Cell Biol. 2010;30:3099–3110. doi: 10.1128/MCB.01266-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins CM, Tsvetkov P, Johnson M, Joyce CL, Lamb CA, Bryant NJ, et al. BIMEL, an intrinsically disordered protein, is degraded by 20S proteasomes in the absence of poly-ubiquitylation. J Cell Sci. 2011;124:969–977. doi: 10.1242/jcs.058438. [DOI] [PubMed] [Google Scholar]
- Beverly LJ, Lockwood WW, Shah PP, Erdjument-Bromage H, H Varmus. Ubiquitination, localization, and stability of an anti-apoptotic BCL2-like protein, BCL2L10/BCLb, are regulated by Ubiquilin1. Proc Natl Acad Sci USA. 2012;109:E119–E126. doi: 10.1073/pnas.1119167109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemunaitis J, Mita A, Stephenson J, Mita MM, Sarantopoulos J, Padmanabhan-Iyer S, et al. Pharmacokinetic study of omacetaxine mepesuccinate administered subcutaneously to patients with advanced solid and hematologic tumors. Cancer Chemother Pharmacol. 2013;71:35–41. doi: 10.1007/s00280-012-1963-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aichberger KJ, Mayerhofer M, Krauth MT, Skvara H, Florian S, Sonneck K, et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005;105:3303–3311. doi: 10.1182/blood-2004-02-0749. [DOI] [PubMed] [Google Scholar]
- Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, et al. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood. 2012;119:5807–5816. doi: 10.1182/blood-2011-12-400929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitecross KF, Alsop AE, Cluse LA, Wiegmans A, Banks KM, Coomans C, et al. Defining the target specificity of ABT-737 and synergistic antitumor activities in combination with histone deacetylase inhibitors. Blood. 2009;113:1982–1991. doi: 10.1182/blood-2008-05-156851. [DOI] [PubMed] [Google Scholar]
- Fan G, Simmons MJ, Ge S, Dutta-Simmons J, Kucharczak J, Ron Y, et al. Defective ubiquitin-mediated degradation of antiapoptotic Bfl-1 predisposes to lymphoma. Blood. 2010;115:3559–3569. doi: 10.1182/blood-2009-08-236760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–107. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- Goetze S, Qeli E, Mosimann C, Staes A, Gerrits B, Roschitzki B, et al. Identification and functional characterization of N-terminally acetylated proteins in Drosophila melanogaster. PLoS Biol. 2009;7:e1000236. doi: 10.1371/journal.pbio.1000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SWG, de Vries E, Maas C, Keller AM, D'Santos CS, Borst J. Apoptosis induction by Bid requires unconventional ubiquitination and degradation of its N-terminal fragment. J Cell Biol. 2007;179:1453–1466. doi: 10.1083/jcb.200707063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbrugge I, de Vries E, SWG Tait, EHJ Wissink, Walczak H, Verheij M, et al. Ionizing radiation modulates the TRAIL death-inducing signaling complex, allowing bypass of the mitochondrial apoptosis pathway. Oncogene. 2008;27:574–584. doi: 10.1038/sj.onc.1210696. [DOI] [PubMed] [Google Scholar]
- Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, et al. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7–RILP–p150Glued and late endosome positioning. J Cell Biol. 2009;185:1209–1225. doi: 10.1083/jcb.200811005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland BW, Toews J, Kast J. Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J Mass Spectrom. 2008;43:699–715. doi: 10.1002/jms.1415. [DOI] [PubMed] [Google Scholar]
- Nielsen ML, Vermeulen M, Bonaldi T, Cox J, Moroder L, Mann M. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat Methods. 2008;5:459–460. doi: 10.1038/nmeth0608-459. [DOI] [PubMed] [Google Scholar]
- Meiring HD, van der Heeft E, ten Hove GJ, de Jong APJM. Nanoscale LC–MS(n): technical design and applications to peptide and protein analysis. J Sep Sci. 2002;25:557–568. [Google Scholar]
- van Lummel M, van Veelen PA, Zaldumbide A, de Ru A, Janssen GMC, Moustakas AK, et al. Type 1 diabetes-associated HLA-DQ8 transdimer accommodates a unique peptide repertoire. J Biol Chem. 2012;287:9514–9524. doi: 10.1074/jbc.M111.313940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Oualid F, Merkx R, Ekkebus R, Hameed DS, Smit JJ, de Jong A, et al. Chemical synthesis of ubiquitin, ubiquitin-based probes, and diubiquitin. Angew Chem Int Ed Engl. 2010;49:10149–10153. doi: 10.1002/anie.201005995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.