

Abstract
Myelotoxicity during thiopurine therapy is enhanced in patients, who because of single nucleotide polymorphisms have decreased activity of the enzyme thiopurine methyltransferase (TPMT) and thus more thiopurine converted into 6-thioguanine nucleotides. Of 601 children with acute lymphoblastic leukemia (ALL) who were treated by the NOPHO ALL-92 protocol, 117 had TPMT genotype determined, whereas for 484 patients only erythrocyte TPMT activity was available. The latter were classified as heterozygous, if TPMT activity was <14 IU/ml, or deficient (<1.0 IU/ml). 526 patients had TPMT wild type, 73 were presumed heterozygous, and two were TPMT deficient. Risk of relapse was higher for the 526 TPMT wild type patients than for the remaining 75 patients (18 vs 7%, P = 0.03). In cox multivariate regression analysis, sex (male worse; P = 0.06), age (higher age worse, P = 0.02), and TPMT activity (wild type worse; P = 0.02) were related to risk of relapse. Despite a lower probability of relapse, patients in the low TPMT activity group did not have superior survival (P = 0.82), possibly because of an excess of secondary cancers among these 75 patients (P = 0.07). These data suggest that children with ALL and TPMT wild type might have their cure rate improved, if the pharmacokinetics/-dynamics of TPMT low-activity patients could be mimicked without a concurrent excessive risk of second cancers.
Keywords: acute lymphoblastic leukemia, thiopurine methyltransferase, genetic polymorphisms, relapse rate, second malignant neoplasm
Introduction
The impact of pharmacogenetic variation on treatment response has become a major research area in recent years. However, for childhood acute lymphoblastic leukemia (ALL) clinical studies have yielded divergent results.1 The thiopurines 6-mercaptopurine (6MP) and 6-thioguanine (6TG) are among the oldest and most widely used agents in the treatment of childhood acute lymphoblastic leukemia (ALL).2–4 They primarily exert their cytotoxicity through conversion into 6-thioguanine nucleotides (6TGN) that are incorporated into DNA and cause DNA damage partly by postreplicative mismatch DNA-repair.5–7 Interindividual variations in response to thiopurine therapy are influenced by genetically determined polymorphisms in the activity of the enzyme thiopurine methyltransferase (TPMT).8 TPMT competes with the formation of 6TGN, as it methylates the thiopurines (especially 6MP) and some of their metabolites. The methylated metabolites are relatively non-toxic, although some (for example, methyl-thioinosine monophosphate) can inhibit purine de novo synthesis. Approximately 10 percent of all Caucasian individuals are TPMT heterozygous, with one wild type and one low-activity allele, and one in 300 individuals are TPMT deficient with two low-activity alleles.8 The natural substrate of TPMT is not known, and beyond their reduced tolerance of thiopurines, the TPMT-deficient individuals are otherwise healthy.
Although (i) TPMT is one of the most widely studied pharmacogenetic polymorphisms in childhood ALL, (ii) TPMT pheno- and genotyping are done routinely by several collaborative ALL groups,9 (iii) TPMT low-activity patients have increased availability of 6MP for 6TGN formation,10 higher E-6TGN levels,10 and higher risk of myelosuppression,5,11 treatment interruption,12 and second cancer12,13 and (iv) TPMT genotyping prior to thiopurine therapy of childhood ALL has been claimed to be cost-effective,14 it remains unclear whether children with ALL and low-activity phenotypes have a relapse rate that differs significantly from that of TPMT wild-type patients.
Several studies have explored the prognostic impact of TPMT low-activity geno- and/or phenotypes, but may have lacked sufficient power to show a statistically significant lower relapse rate compared with TPMT normal activity patients.15–18 In a previous study of children treated according to the NOPHO ALL-92 study, we showed that TPMT phenotype was significantly related to the risk of relapse, but only in a complex model that also included gender, white blood cell count (WBC) at diagnosis, the average neutrophil count during maintenance therapy and the randomized recommendation to adjust therapy by red blood cell levels of 6MP and MTX metabolite levels. With longer follow-up and inclusion of additional patients, we show in this study of 601 patients that ‘TPMT status’ has an independent impact on the risk of relapse in both uni- and multivariate analyses.
Patients and methods
Patients
From January 1992 until October 2001, 1703 children 1.0–14.9 years of age were diagnosed with B-cell precursor (pre-B) or T-cell ALL in the Nordic countries (Denmark, Finland, Iceland, Norway, and Sweden). Two patients with Down syndrome received no antileukemic therapy, 10 patients were treated according to non-NOPHO protocols, one patient was treated according to the previous NOPHO ALL-86 protocol, and 45 patients were treated according to the NOPHO ALL-2000 protocol, before it was officially opened. Of the 1645 patients who started therapy according to the NOPHO ALL-92 protocol, TPMT pheno- and/or genotype was available for 609 patients (Table 1), of whom 485 participated in the randomized ALL-92 study of individualized dose adjustments of 6MP and methotrexate (MTX) during maintenance therapy.11 Thus, a large proportion of the patients were included in this study late in their antileukemic therapy. If patients who experienced an event prior to the start of maintenance therapy were excluded, the patients for whom TPMT pheno- and/or genotype were available did not have an event-free survival that differed significantly from that of the remaining patients, and that was also the case, when the risk groups were analysed separately. Two patients, both with a TPMT wild-type genotype, died in first remission before the start of maintenance therapy. Another two patients developed a relapse before the start of maintenance therapy, one with an M2 bone marrow day 29 (TPMT wild type genotype) and one with a WBC of 600 × 109/l at diagnosis (TPMT heterozygous genotype). Finally, four patients, all of whom had a TPMT wild type genotype, were treated with LSA2L2 maintenance therapy blocks that each consisted of four multidrug courses at 2 week intervals with alternating combinations of 6-thioguanine, cyclophosphamide, hydroxyurea, daunorubicin, MTX, cytarabine and VCR.19 The remaining 329 boys and 272 girls with a median age of 4.2 years and a WBC of 6.0 × 109/l constituted the study cohort, and the latter eight patients, who experienced an event prior to maintenance therapy or who received LSA2L2 maintenance therapy, were only included in one relapse analysis to demonstrate that their inclusion did not significantly change the overall results. The 601 patients did not differ significantly from the remaining 716 patients on the NOPHO ALL-92 protocol, who were in first remission at the start of maintenance therapy, with respect to gender (P = 0.19), age (P = 0.31) or WBC at diagnosis (median: 6.0 vs 8.0 × 109/l, P = 0.07), or the fraction of T-ALL (P = 0.71). The ALL-92 protocol was approved by the ethical committee of Copenhagen (no. V.200.2080/91) as well as by the local ethical committees, and participants gave informed consent according to the Helsinki Declaration.
Table 1.
Patient characteristics in relation to thiopurine methyltransferase activitya
| Wild type a | Heterozygous a | Deficient a | P-value | |
|---|---|---|---|---|
| Male/female* | 287/239 | 41/32 | 1/1 | 0.96 |
| Median age (50% range)* | 4.0 (2.8–6.2) | 4.4 (3.0–6.4) | 6.8 (6.5–7.0) | 0.19 |
| Median WBCb (50% range)* | 6 (3–18) | 6 (4–18) | 18 (3–32) | 0.94 |
| B-/T-lineage** | 513/13 | 68/5 | 2/0 | 0.12 |
| Cytogenetics* | P=0.51 | |||
| High-hyperdiploidc or t(12;21)d | 172 | 19 | 1 | |
| Othere | 92 | 18 | 0 | |
| Nonef | 262 | 36 | 1 | |
| SR/IR/HRg | 250/223/53 | 29/32/12 | 1/1/0 | 0.44 |
| Control/TDM/non-randomizedh | 218/207/101 | 26/32/15 | 1/1/0 | 0.85 |
Patients were classified as heterozygous, if they had one low activity allele; if no TPMT genotype was available, they were classified as heterozygous, if TPMT <14 IU/ml, or deficient if TPMT <1.0 IU/ml.
White blood cell count at diagnosis.
Modal chromosome number 51–61).
TEL/AML1 (=ETV6/RUNX1) translocation.
Other chromosomal aberrations.
No aberrations detected (see text).
Standard/intermediate/high risk treatment (defined in text).
485 patients participated in NOPHO ALL-92 randomized study of therapeutic dose adjustment by red blood cell MTX/6MP metabolite levels, TDM=pharmacological dose adjustment group (Schmiegelow 2003).
P>0.10;
P=0.05–0.10.
The clinical characteristics and treatment assignment of the eligible 329 boys and 272 girls are given in Table 1.
Risk group assignment
The risk group assignment was based on age and white blood cell count (WBC) at diagnosis (SR: age 2.0–9.9 years and WBC <10.0 × 109/l; IR: age 1.0–1.9 or ≥10.0 years and/or WBC 10–49.9 × 109/l; higher risk (that is, high risk (HR) or very high risk (VHR)): WBC ≥50.0 × 109/l) and the presence of higher risk features: T-lineage ALL, the presence of CNS or testicular involvement, translocations t(9;22)(q34;q11) or t(4;11) (q21;q23), lymphomatous leukemia or mediastinal lymphoma, and a poor treatment response (M3 BM at day 15 or M2/M3 at day 29).19 In the NOPHO ALL-92 protocol, patients who had higher risk features were assigned to the VHR treatment arm, if they were at least 5 years of age at diagnosis (due to the use of cranial irradiation in that protocol arm) and in addition had (i) T-cell disease with one or more additional HR-features, (ii) CNS leukemia, (iii) lymphomatous leukemia, and/or (iv) higher risk ALL at diagnosis and a day 15 M3 or a day 29 M2/M3 bone marrow. All the remaining patients with higher risk features were assigned to the HR treatment arm. In the NOPHO ALL-92 protocol, patients treated according to the VHR arm received LSA2L2 maintenance therapy, and they were not part of this study, whereas the SR-, IR-, and HR-ALL patients received oral MTX/6MP maintenance therapy.19
Cytogenetics
Only G-band karyotyping was mandatory in the NOPHO ALL-92 protocol. In addition, many patients were explored with high-resolution comparative genomic hybridization, fluorescent in-situ hybridization, and/or reverse transcriptase PCR for selected translocations. All Nordic karyotypes have subsequently been scrutinized centrally by the NOPHO cytogenetic working group. The karyotypes of the patients were described according to ISCN 1995.20 To explore the impact of the TPMT activity on relapse, the patients were divided into three cytogenetic subsets: (1) those with a favorable karyotype (high-hyperdiploidy with a modal chromosome number of 51–61 chromosomes or a t(12;21)[ETV6/RUNX1]-translocation), (2) those with other aberrations, and (3) those who lacked karyotyping (N = 13) or no aberrant karyotype was identified (Table 2).
Table 2.
Probability of relapse in relation to thiopurine methyltransferase activitya
| pRelapse (s.e.) |
TPMT determined
|
P-value | |
|---|---|---|---|
| Wild type | Low-activity | ||
| Overall | 0.18 (0.02) | 0.07 (0.03) | 0.03 |
| Male/female | 0.21 (0.03)/0.14 0.02) | 0.07 (0.04)/0.06 (0.04) | 0.02 |
| Age 1.0–7.0/8.0–15.0 years | 0.16 (0.02)/0.23 (0.04) | 0.05 (0.03)/0.12 (0.08) | 0.02 |
| WBCb <50/≥50×109/l | 0.17 (0.02)/0.22 (0.07) | 0.07 (0.03)/0.0 | 0.03 |
| B-/T-lineage | 0.18 (0.02)/0.16 (0.10) | 0.07 (0.03)/0.0 | 0.03 |
| HeH or t(12;21)/other/nonec | 0.18 (0.03)/0.23 (0.05)/0.15 (0.02) | 0.11 (0.07)/0.0/0.08 (0.05) | 0.02 |
| SR/IR/HRd | 0.14 (0.02)/0.20 (0.03)/0.21 (0.06) | 0.07 (0.05)/0.09 (0.05)/0.0 | 0.02 |
| Pharmacologye/control groupf | 0.22 (0.03)/0.16 (0.03) | 0.06 (0.06)/0.07 (0.05) | 0.03 |
Patients were classified as heterozygous, if they had one low activity allele; if no TPMT genotype was available, they were classified as heterozygous, if TPMT <14 IU/ml, or deficient if TPMT <1.0 IU/ml. The numbers of any relapse involving bone marrow, isolated CNS relapses, isolated testicular relapse, and other relapses for the TPMTWT and TPMTLA patients were 73/9/5/2 and 4/0/1/0, respectively.
White blood cell counts at diagnosis.
HeH=modal chromosome number 51–61, t(12;21)=TEL/AML1=ETV6/RUNX1 translocation, Other=other chromosomal aberrations, none=no aberrations detected (see text).
Standard/intermediate/high risk treatment (defined in text).
Pharmacology group=dose adjustments by red blood cell levels of MTX and 6MP metabolites vs
Control group (see text and reference 11).
Treatment
Based on risk group assignments, patients were treated according to the SR, IR, or HR arm (Figure 1).19 However, for twenty-three patients the assigned treatment did not match completely the risk group criteria: Nineteen patients with morphologically suspected, but uncertain, M3 bone-marrow on day 15 and/or M2/M3 bone-marrow on day 29 had SR-ALL (N = 14) or IR-ALL (N = 5) by all other criteria and were treated according to the SR or IR treatment arms, respectively. Furthermore, three patients who fulfilled the SR-ALL criteria were by decision of the treating physician assigned to the IR-arm, and one patient, who fulfilled the IR-ALL criteria, was, by decision of the treating physician, assigned to the SR-arm. Finally, one patient who fulfilled the VHR criteria received oral MTX/6MP maintenance therapy. Six of these 24 patients, all of whom had a suspected poor response to induction therapy (M3 day 15 N = 1 or M2/M3 day 29 N = 5), developed a relapse.
Figure 1.
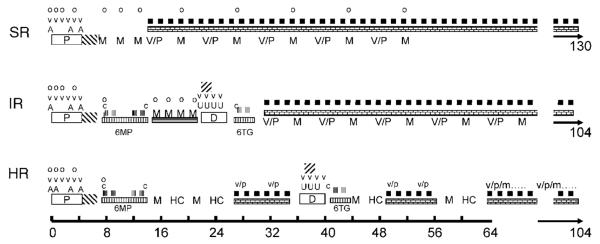
Treatment elements of standard (SR), intermediate (IR), and high risk (HR) ALL according to the NOPHO ALL-92 protocol (see also the text).  =oral prednisolone (60 mg/m2/day). v & V=vincristine (2.0 mg/m2). A=doxorubicin (40 mg/m2 i.v.),
=oral prednisolone (60 mg/m2/day). v & V=vincristine (2.0 mg/m2). A=doxorubicin (40 mg/m2 i.v.),  =Erwinia asparaginase (30 000 IU/m2 per day for 10 days). o=intrathecal MTX by age. M=high-dose MTX (5 g/m2/24 h or 8 g/m2 (HR only)). V/P=vincristine (2.0 mg/m2 × 1)/prednisolone (60 mg/m2/day for 7 days) reinductions.
=Erwinia asparaginase (30 000 IU/m2 per day for 10 days). o=intrathecal MTX by age. M=high-dose MTX (5 g/m2/24 h or 8 g/m2 (HR only)). V/P=vincristine (2.0 mg/m2 × 1)/prednisolone (60 mg/m2/day for 7 days) reinductions.  MTX 20 mg/m2/week.
MTX 20 mg/m2/week.  6MP 75 mg/m2/day. c=cyclophosphamide (1 g/m2 i.v.). Low-dose AraC (75 mg/m2).
6MP 75 mg/m2/day. c=cyclophosphamide (1 g/m2 i.v.). Low-dose AraC (75 mg/m2).  6MP or 6TG 60 mg/m2/day. U=daunorubicin (30 mg/m2),
6MP or 6TG 60 mg/m2/day. U=daunorubicin (30 mg/m2),  =Erwinia asparaginase (30.000 IU/m2 two times weekly for 2 weeks). HC=high dose AraC (2 g/m2 two times daily for 3 days). v/p=vincristine (2.0 mg/m2 × 1)/prednisolone (40 mg/m2/day for 7 days) reinductions without or with (/m) methotrexate i.t. in age-adjusted doses.
=Erwinia asparaginase (30.000 IU/m2 two times weekly for 2 weeks). HC=high dose AraC (2 g/m2 two times daily for 3 days). v/p=vincristine (2.0 mg/m2 × 1)/prednisolone (40 mg/m2/day for 7 days) reinductions without or with (/m) methotrexate i.t. in age-adjusted doses.
As induction therapy, all patients received prednisolone (60 mg/m2/day on days 1–36, then tapered), weekly vincristine (VCR, 2.0 mg/m2 times six), doxorubicin (40 mg/m2 3 (SR and IR) or four doses (HR)), Erwinia asparaginase (30 000 IU/m2 daily on days 37–46), and intrathecal (i.t.) MTX on four occasions.
Consolidation therapy for SR-ALL included three courses of high-dose MTX (HD-MTX) 5 g/m2/24 h with i.t. MTX and Leucovorin rescue, whereas patients with IR-, or HR-ALL received alternating series of (a) i.v. cyclophosphamide (total cumulative dose: 3 g/m2) with low-dose cytarabine (AraC) and either oral 6MP or oral 6TG; (b-1) IR: oral 6MP (25 mg/m2/day) with four courses of HD-MTX 5 g/m2/24 h with i.t. MTX and Leucovorin rescue at 2-week intervals; (b-2) HR: HD-MTX 8 g/m2/24 h with i.t. MTX and Leucovorin rescue times four, alternating with high-dose AraC (12 g/m2) times four with two 2-month intervening periods of oral weekly MTX and daily 6MP with two VCR/prednisolone reinductions per period; (c) 4 weeks of delayed intensification with dexamethasone (10 mg/m2/day for 3 weeks, then tapered), weekly VCR (2.0 mg/m2 four times), weekly anthracycline (30 mg/m2/day doxorubicin three times (HR) or daunorubicin four times (IR)), and Erwinia asparaginase (30.000 IU/m2 four times). 19CNS irradiation was not given to any of the patients.
6MP/MTX maintenance therapy was initiated at treatment weeks 13 (SR), 32 (IR), or 63 (HR) and continued until 2 (IR and HR) or 2½ years (SR) after diagnosis. The starting dose of 6MP was 75 mg/m2/day and of MTX was 20 mg/m2/week. During the first year of maintenance therapy, patients with SR- or IR-ALL received alternate pulses at 4-week intervals of (i) VCR (2.0 mg/m2 once) and prednisolone (60 mg/m2/day for 1 week) and (ii) HD-MTX 5 g/m2/24 h with i.t. MTX and Leucovorin rescue until five courses of HD-MTX had been given. Every 8 weeks throughout maintenance therapy, HR patients received reinductions of VCR (1.5 mg/m2 once) and prednisolone (40 mg/m2/day for 5 days) with i.t. MTX. Between 1992 and 1996, 538 patients with SR-, IR-, or HR-ALL, of whom 485 were included in the present study, were entered into the randomized ALL-92 maintenance therapy trial. It explored the prognostic impact of pharmacologically based monitoring and dose adjustments of oral 6MP/MTX maintenance therapy by erythrocyte levels of 6TGN and MTX.11 As part of that study, and in addition to almost 10 000 measurements of red blood cell 6MP and MTX metabolite levels, 28 580 data sets of MTX and 6MP doses as well as parameters for myelo- and hepatotoxicity were registered.11
TPMT
In total, TPMT genotype data (presence of TPMT low-activity polymorphisms G460A and/or A719G) were available for 117 patients, whereas for 484 patients only erythrocyte TPMT activity was available, and it was measured 1–5 times during maintenance therapy, as described earlier.21 The TPMT pheno- and genotype were not revealed to the physicians, when the patients were on therapy. For patients with more than one TPMT activity measurement, an arithmetic mean TPMT activity was calculated. All TPMT phenotype assays were performed at least 8 weeks after the most recent blood transfusion. The antimode of the TPMT activity distribution was 14 IU/ml.13 The 62 patients below that level, and for whom no TPMT genotype was available, were classified as TPMT presumed heterozygous (N = 60; median TPMT-activity: 10.4, range: 6.2–13.9 IU/ml) or TPMT deficient (0.01 and 0.58 IU/ml, respectively). Among the 39 patients for whom both TPMT geno- and phenotyping was available, all nine patients who were TPMT heterozygous by genotyping had TPMT activity <14 IU/ml. However, also four out of thirty patients (13%), who had the wild-type genotype, had TPMT activity <14 IU/ml (range: 9.0–13.4), but were classified by their genotype as TPMT wild type. In total, 526 patients were classified as TPMT wild type, 73 as heterozygous, and two as TPMT deficient. In the following analyses the latter two subsets were grouped together as TPMT low activity (TPMTLA), which included all patients with a heterozygous genotype as well as the presumed heterozygous patients (TPMT activity <14 IU/ml, no genotype available), and the remainder as TPMT wild type (TPMTWT). The fraction of patients classified as TPMTLA was the same for the 485 patients, who participated in the randomized maintenance therapy study (12.4%) and the 116 who did not (12.9%).
Statistics
Survival analyses were performed with a basic time scale defined by the date of diagnosis. Patients who died in first remission (N = 3) or developed second malignant neoplasms (N = 13) were censored at the time of these events in the analyses of risk factors for a leukemic relapse. Cox proportional hazard regression analyses were performed with the likelihood ratio test for differences in outcome.22,23 Non-parametric methods were applied to compare the distribution of parameters between subgroups.24 The Kaplan–Meier method was applied for estimation of remission duration and for the generation of survival curves.25 Subgroups were compared with the log-rank test,26 stratified where needed. Two-sided P-values <0.05 were regarded as significant.
Results
After a median follow-up of the 491 patients who did not experience an event of 12.4 years (50% range: 10.7–13.8 years), 94 patients had developed a relapse 0.9–12.0 years from diagnosis (median: 3.7 years), with a cumulative incidence of relapse of 0.16. Of the 94 relapses, 77 involved the bone-marrow, and 17 involved the CNS, of which nine were isolated. The overall cumulative risk of CNS relapse was 0.03±0.01. Three patients, all being TPMTWT, died in first remission. Thirteen patients developed a second malignant neoplasm, four of whom were TPMTLA patients (P = 0.07), of which 11 died, including all four TPMTLA patients. Owing to this excess of second malignant neoplasms the overall survival did not differ between the TPMTLA and the TPMTWT patients (0.90 for both groups).
The risk of relapse was significantly lower for the 75 TPMTLA patients than for the 526 TPMTWT patients (7±3 vs 18±2%; P = 0.03) (Figure 2). If the TPMT group was assigned by TPMT activity first and then by TPMT genotype, or if the four patients who were classified as TPMTWT due to their TPMT genotype were excluded, the differences in relapse rate were still statistically significant. The difference in relapse rate was even more pronounced, if the 24 patients with protocol violations in risk and treatment assignment were excluded from the analysis (4±2 vs 17±2%; P = 0.007). In addition, if the eight patients, who received LSA2L2 maintenance therapy or who had an event before initiation of MTX/6MP maintenance therapy were included, the difference in relapse still remained significant (8±3 vs 18±2%; P = 0.045). Finally, if only patients with TPMT phenotyping were included, the TPMT presumed heterozygous patients (TPMT activity <14 IU/ml) had a significantly lower relapse risk than the TPMT presumed wild type (TPMT activity ≥14 IU/ml) (6±3 vs 19±2%; P = 0.02).
Figure 2.
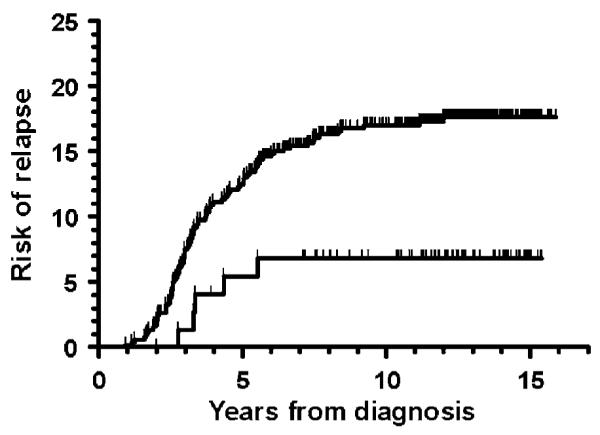
Risk of relapse with respect to the TPMT activity. Lower curve: TPMT low-activity patients (TPMT heterozygous (N=73) or TPMT deficient (N=2), relapse risk: 7±3%). Upper curve: TPMT wild-type patients (N=526, relapse risk: 18±2%); P=0.03.
The relapse rate for patients classified as TPMT wild type was higher than for patients with low TPMT activity across all analysed ALL subtypes, which included sex, age groups, WBC groups, B-lineage versus T-lineage ALL, and karyotype subsets (Table 2). Thus, in the survival analyses the differences in relapse rate remained significant when the TPMT high and low-activity groups were compared with the analyses stratified for these parameters. That was also the case when the survival analyses were stratified by whether the patients were randomized to the control arm (N = 245) or the pharmacology arm (N = 240) in the NOPHO ALL-92 maintenance therapy study11 (Table 2).
We used Cox multivariate regression analysis to test the effect on relapse risk of sex, WBC at diagnosis, age at diagnosis, karyotype subgroup, immune phenotype, and TPMT group (TPMTLA = 1 vs TPMTWT = 2). Sex, age and TPMT group were significantly related to the risk of relapse (overall P-value of the Cox model: 0.002). Owing to major differences in the distribution of clinical characteristics among the patients, the Cox regression analysis was repeated after stratification by immunophenotype (T- vs non-T cell ALL), which did not significantly change the variables in the Cox model or their coefficients. This was also the case when Cox regression analysis was stratified by karyotype groups (favorable karyotypes (see cytogenetics section) vs other aberrations vs normal/missing). Thus, the best-fit model included as covariates sex (male = 1, female 2; B (regression coefficient) = −0.41, P = 0.06), age at diagnosis (B = 0.069, P = 0.02), and TPMT group (B = 1.06, P = 0.02). None of the other covariates reached a significance level <0.10 in any of the backward steps. To explore the impact of the actual TPMT activity measured for those 523 patients for whom this measure was available, we repeated the Cox analysis with stratification for the TPMT group (TPMTLA or TPMTWT). In that analysis sex (B = −0.41, P = 0.07), age at diagnosis (B = 0.088, P = 0.003), and the TPMT activity (B = 0.076, P = 0.05) were related to risk of relapse (overall P-value of the Cox model: 0.002).
Compared to patients who were classified as TPMTWT, those who belonged to the TPMTLA group received lower average 6MP doses (Median: 52.1 vs 60.1 mg/m2/day, P<0.001) and average MTX doses (Median: 14.1 vs 15.8 mg/m2/week, P = 0.001) during maintenance therapy; they achieved lower absolute neutrophil counts (ANC, median: 1.86 vs 1.98 × 109/l; P = 0.12); and they achieved higher erythrocyte 6TGN levels (Median: 338 vs 161 nmol/mmol hemoglobin; P<0.001) but lower erythrocyte MTX polyglutamate levels (Median: 5.2 vs 5.7 nmol/mmol hemoglobin; P = 0.009). For the 484 TPMT phenotyped patients, TPMT activity was related to the average erythrocyte 6TGN level achieved during maintenance (rS = −0.58, P<0.001), and this correlation was more pro nounced for those with TPMT activity below <14 IU/ml (rS = −0.53, P<0.001), than for the TPMT presumed wild type patients (rS = −0.13, P = 0.009). Overall, the patient who developed a relapse did not have higher erythrocyte 6TGN levels than those who stayed in remission (P = 0.31), and the 35 patients with 6TGN levels above 338 nmol/mmol = hemoglobin (median of the TPMTLA patients) did not have a relapse rate that differed significantly from the remaining patients (P = 0.35). The median ANC during maintenance therapy for the 481 patients for whom this information was available, was 1.96 × 109/l. Among the TPMTWT patients, those with an average ANC below 1.96 × 109/l had a significantly lower risk of relapse than those with higher ANC levels (0.13±0.02 vs 0.23±0.03; P = 0.01) (Figure 3).
Figure 3.
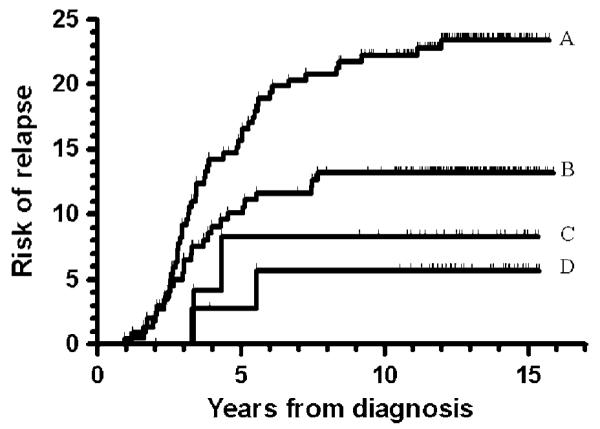
Risk of relapse with respect to the TPMT activity and the average absolute neutrophil counts (ANC) during maintenance therapy. The median for all patients is 1.96 × 109/l. (a) TPMT wild type and ANC ≥1.96 × 109/l, relapse risk: 23±3%; (b) TPMT wild type and ANC <1.96 × 109/l, relapse risk: 13±2%; (c) TPMT low activity and ANC ≥1.96 × 109/l, relapse risk: 9±6%; (d) TPMT low activity and ANC <1.96 × 109/l, relapse risk: 6±4%; P=0.001 (trend analysis, log-rank Mantel–Cox linear trend analysis).
Discussion
Increased understanding of interindividual variation in the disposition of anticancer drugs and the sequencing of the human genome have raised expectations for individualized therapy based on the pharmacogenetic classification of patients,27 not least since high-throughput technology has facilitated exploration of the pharmacogenomic fraction of the millions of single nucleotide polymorphisms (SNP) that occur at a frequency of more than 1%.28 One of the most explored polymorphisms in childhood ALL therapy involves TPMT (E.C. 2.1.1.67), an enzyme that methylates 6MP, 6TG, and some of their metabolites. Of these, 6-methyl-thioinosine 5′-monophosphate can inhibit purine de novo synthesis, which may enhance the incorporation of 6TGN into DNA.29
Although several previous studies have indicated that TPMT heterozygous patients have a reduced risk of relapse, they have lacked sufficient power to be able to demonstrate significant differences in cure rates between wild type and low-activity patients.16–18 Thus, in a recent report from the St Jude Childrens Research Hospital the hematologic relapse of the TPMTLA and TPMTWT patients were very similar to the relapse rates of this study (6.7 and 13.2%, respectively).30 The pharmacodynamics behind the superior outcome of the TPMT low-activity patients in this study may involve several treatment phases in which thiopurines are given together with other anticancer agents: (i) together with cyclophosphamide and low dose AraC during consolidation (6MP, 60 mg/m2) and delayed intensification therapy (6TG, 60 mg/m2) for IR- and HR-ALL, (ii) during consolidation therapy (6MP 25 mg/m2) together with four courses at 2-week intervals of HD-MTX (5 g/m2/24 h) for IR-ALL; (iii) during maintenance therapy together with low-dose oral MTX for all risk groups; and (iv) during the early phase of maintenance for SR- and IR-ALL, where five reinductions with HD-MTX 5 g/m2/24 h are given at 8-week intervals on a backbone of 6MP (75 mg/m2).
This study may have underestimated the superior outcome of the TPMTLA patients treated by the NOPHO ALL-92 protocol. Of the five relapses among these patients, two patients had a suspected poor response to induction therapy with a day 15 M3 bone marrow (N = 1) or a day 29 M2/M3 bone marrow (N = 1), but were by decision of the local physician treated according to the SR- and IR-protocols, respectively. In addition, one patient may not have had a TPMT low-activity allele, as no TPMT genotype was available, the TPMT activity was 13.9 IU/ml, and the mean erythrocyte 6TGN level was only 145 nmol/mml Hb, even though the average 6MP dose was 88.5 mg/m2.
Consolidation therapy with 6MP or 6TG (60 mg/m2) together with cyclophosphamide and low-dose AraC19 was in NOPHO ALL-92 given to IR- and HR-ALL patients. This drug combination is very efficacious in reducing the level of residual leukemia, which improves cure rates,31 and the reduction of residual leukemia was more pronounced for TPMT low activity compared with wild type patients. Thus, in a BFM study of treatment response to a combination of 6MP, cyclophosphamide and low-dose AraC, 55 patients heterozygous for allelic variants of TPMT, conferring on average a 50% reduction in enzyme activity, had a significantly lower rate of MRD positivity (9.1%) at the end of the this consolidation phase compared with 755 patients who had homozygous wild type alleles (22.8%) (P = 0.02).32 However, in this study the difference in outcome was as pronounced for SR-ALL as for IR- and HR-ALL, even though the SR-ALL patients did not receive this kind of consolidation therapy.
Several in vitro33 and in vivo studies have demonstrated the synergistic effect of 6MP and MTX. In the NOPHO ALL-88 study of oral 6MP/MTX maintenance therapy, the most significant pharmacological parameter which predicted the risk of relapse was the product of the red blood cell levels of MTX-polyglutamates and 6TGN.34 In addition, the most significant parameter to predict the rise in WBC levels after cessation of oral 6MP/MTX maintenance therapy was also the product of red blood cell levels of MTX-polyglutamates and 6TGN.35 Furthermore, several studies have demonstrated that the toxicity of HD-MTX is influenced by the treatment intensity of 6MP given concurrently. First, if HD-MTX (5.0 g/m2/24 h) is given without 6MP, the subsequent myelotoxicity is very limited.36 Second, as the dose of 6MP is increased, so is the degree of myelotoxicity, even if the HD-MTX dose is unchanged, and individual adjustments of the 6MP dose can reduce the degree of bone-marrow suppression.36,37 Third, patients with TPMT deficiency are at especially high risk for myelosuppression when 6MP is coadministered with HD-MTX.38 Fourth, it is possible that i.t. MTX, which may result in significant systemic MTX exposure and intracellular concentrations,39 could have influenced the relation between TPMT genotype and post-consolidation residual leukemia in the study reported by Stanulla et al.32 As HD-MTX in the NOPHO ALL-92 protocol was given together with 6MP, both during the consolidation phase (6MP: 25 mg/m2; IR-ALL) and during maintenance therapy (6MP: 75 mg/m2; SR- and IR-ALL), it cannot be determined which 6MP dosages and treatment phases were responsible for the reduced relapse rate among TPMT low-activity patients.
The present study indicates that even among TPMT wild type patients the variation in the TPMT phenotype, that is, enzyme activity, may influence the risk of relapse. This observation emphasizes that, although the genotype of a given polymorphic enzyme may be predictive for efficacy or toxicity, even within the same genotype, patients may differ in their response. Thus, genotyping should, in general, be seen as a supplement to phenotyping rather than a substitute.
The vast majority of the patients in this study had non-B ALL with a WBC at diagnosis less than 50 × 109/l, so this study does not allow conclusions to be drawn with respect to patients with higher risk criteria, including T-lineage ALL. This reflects the fact that most patients had their TPMT pheno- or genotype determined after they initiated maintenance therapy, that is, as part of the randomized NOPHO ALL-92 study, which included only 60 patients with HR-ALL. Of interest, none of the five cases of T-ALL and none of the 12 cases of HR-ALL who had TPMT low activity developed a relapse.
This study indicates that childhood ALL may be far more chemosensitive than what is shown by the event free survival curves of contemporary protocols, and that individualization of therapy to mimic the pharmacokinetics and -dynamics of TPMT low-activity patients might significantly increase the cure rates of childhood ALL. One feature of TPMT heterozygosity is the higher intracellular levels of 6TGN.40 Two European and one US randomized trials have compared 6TG and 6MP in MTX/thiopurine maintenance therapy with 6MP, of which the British and German studies have been published.41,42 Only the British study41 showed a reduced relapse rate in the 6TG arm, which however was counteracted by a 10% risk of veno-occlusive disease (VOD) as also reported by others.43 As an alternative, the use of non-steroidal anti-inflammatory agents that may inhibit TPMT has been proposed,44 and 5-aminosalicylic acid, an inhibitor of TPMT45 has been shown to increase E-6TGN levels during 6-mercaptopurine therapy.46,47 Prospective studies are needed to explore the efficacy of such drug–drug interactions.
Owing to their higher risk myelosuppression15 and supposed risk of second malignant neoplasms,12,13 NOPHO have since 2001 in their ALL-2000 and ALL-2008 protocols adjusted the initial 6MP dosage in maintenance therapy according to the TPMT genotypes (75 mg/m2 for wild type patients, 50 mg/m2 for heterozygous patients, and 10 mg/m2 for TPMT-deficient patients). Subsequently, the 6MP dose is adjusted to the same WBC target. It is yet to early to determine whether this strategy has changed the relapse rate for the TPMT heterozygous/deficient patients.
In conclusion, pharmacogenomic exploration is more than an attempt to understand the pharmacological background for leukemic relapse and serious adverse events, such as second cancers. It may make it possible to identify patients with pharmacogenetically determined reduced relapse rates, and thus could help to set a target for cure rates when drug metabolism is optimal. In this respect, it may also indicate which drugs, treatment phases, and metabolic pathways are of greatest importance, and it may help to identify amendments to therapy or use of alternate drugs that could improve cure rates. Therefore, ‘pharmacogenetic exploration’ should be integrated into the research arms of collaborative protocols.
Acknowledgements
This study has received financial support from The Childhood Cancer Foundation, Denmark; The University Hospital Rigshospitalet; The Children’s Cancer Foundation of Sweden (Grant no.: 53/91, 62/94, 72/96, 98/59, 04/002), The Danish Cancer Society (Grant no.: 91-048, 92-017, 93-017, 95-100-28), The Lundbeck Foundation (Grant no.: 38/99), Novo Nordic Foundation, Home Secretary Research Grant for Individualized Therapy, Danish Research Council for Health and Disease, Michael Goldschmidt Holding A/S, The Nordic Cancer Union (Grant no.: 56-9257, 56-100-03-9102), and the United States National Institutes of Health (Grant No.: R01-GM28157, U01 GM61388). Kjeld Schmiegelow holds the Childhood Cancer Foundation Research Professorship in Pediatric Oncology. We thank all the Nordic pediatric oncology centers that have supported this study with blood sampling and detailed registration of the treatment data. Kjeld Schmiegelow designed the study and wrote the paper. Richard Weinshilboum performed most of the TPMT analyses, whereas Kjeld Schmiegelow performed the rest of the pharmacological analyses. Erik Forestier scrutinized the karyotypes of all patients. Jon Kristensen, Stefan Söderhäll, Kim Vettenranta, and Finn Wesenberg were responsible for providing the clinical data and follow-up data for patients from Iceland, Sweden, Finland, and Norway, respectively. All authors commented and approved the final paper.
References
- 1.Davidsen ML, Dalhoff K, Schmiegelow K. Pharmacogenetics influence treatment efficacy in childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2008 doi: 10.1097/MPH.0b013e3181868570. (in press) [DOI] [PubMed] [Google Scholar]
- 2.Burchenal JH, Murphy ML, Ellison RR, Sykes MP, Tan TC, Leone LA, et al. Clinical evaluation of a new antimetabolite, 6-mercaptopurine, in the treatment of leukemia and allied diseases. Blood. 1953;8:965–987. [PubMed] [Google Scholar]
- 3.Schmiegelow K, Gustafsson G. Acute lymphoblastic leukemia. In: Voute PA, Barrett A, Stevens MCG, Caron M, editors. Cancer in Children. 5th edn Oxford University Press; Oxford: 2005. pp. 138–170. [Google Scholar]
- 4.Coulthard S, Hogarth L. The thiopurines: an update. Invest New Drugs. 2005;23:523–532. doi: 10.1007/s10637-005-4020-8. [DOI] [PubMed] [Google Scholar]
- 5.Lennard L, Welch JC, Lilleyman JS. Thiopurine drugs in the treatment of childhood leukaemia: the influence of inherited thiopurine methyltransferase activity on drug metabolism and cytotoxicity. Br J Clin Pharmacol. 1997;44:455–461. doi: 10.1046/j.1365-2125.1997.t01-1-00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karran P, Attard N. Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat Rev Cancer. 2008;8:24–36. doi: 10.1038/nrc2292. [DOI] [PubMed] [Google Scholar]
- 7.Waters TR, Swann PF. Cytotoxic mechanism of 6-thioguanine: hMutSalpha, the human mismatch binding heterodimer, binds to DNA containing S6-methylthioguanine. Biochemistry. 1997;36:2501–2506. doi: 10.1021/bi9621573. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: insights, challenges and future directions. Oncogene. 2006;25:1629–1638. doi: 10.1038/sj.onc.1209372. [DOI] [PubMed] [Google Scholar]
- 9.Arico M, Baruchel A, Bertrand Y, Biondi A, Conter V, Eden T, et al. The seventh international childhood acute lymphoblastic leukemia workshop report: Palermo, Italy, January 29–30, 2005. Leukemia. 2005;19:1145–1152. doi: 10.1038/sj.leu.2403783. [DOI] [PubMed] [Google Scholar]
- 10.Lennard L, Welch JC, Lilleyman JS. Thiopurine drugs in the treatment of childhood leukaemia: the influence of inherited thiopurine methyltransferase activity on drug metabolism and cytotoxicity. Br J Clin Pharmacol. 1997;44:455–461. doi: 10.1046/j.1365-2125.1997.t01-1-00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmiegelow K, Bjork O, Glomstein A, Gustafsson G, Keiding N, Kristinsson J, et al. Intensification of mercaptopurine/methotrexate maintenance chemotherapy may increase the risk of relapse for some children with acute lymphoblastic leukemia. J Clin Oncol. 2003;21:1332–1339. doi: 10.1200/JCO.2003.04.039. [DOI] [PubMed] [Google Scholar]
- 12.Relling MV, Rubnitz JE, Rivera GK, Boyett JM, Hancock ML, Felix CA, et al. High incidence of secondary brain tumours after radiotherapy and antimetabolites. Lancet. 1999;354:34–39. doi: 10.1016/S0140-6736(98)11079-6. [DOI] [PubMed] [Google Scholar]
- 13.Thomsen JB, Schroder H, Kristinsson J, Madsen B, Szumlanski C, Weinshilboum R, et al. Possible carcinogenic effect of 6-mercaptopurine on bone marrow stem cells: relation to thiopurine metabolism. Cancer. 1999;86:1080–1086. doi: 10.1002/(sici)1097-0142(19990915)86:6<1080::aid-cncr26>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 14.van den Akker-van Marle ME, Gurwitz D, Detmar SB, Enzing CM, Hopkins MM, Gutierrez de ME, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics. 2006;7:783–792. doi: 10.2217/14622416.7.5.783. [DOI] [PubMed] [Google Scholar]
- 15.Relling MV, Hancock ML, Rivera GK, Sandlund JT, Ribeiro RC, Krynetski EY, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91:2001–2008. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 16.Lennard L, Lilleyman JS, Van Loon J, Weinshilboum RM. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336:225–229. doi: 10.1016/0140-6736(90)91745-v. [DOI] [PubMed] [Google Scholar]
- 17.Bostrom B, Erdmann G. Cellular pharmacology of 6-mercaptopurine in acute lymphoblastic leukemia. Am J Pediatr Hematol Oncol. 1993;15:80–86. [PubMed] [Google Scholar]
- 18.Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood. 1999;93:2817–2823. [PubMed] [Google Scholar]
- 19.Gustafsson G, Schmiegelow K, Forestier E, Clausen N, Glomstein A, Jonmundsson G, et al. Improving outcome through two decades in childhood ALL in the Nordic countries: the impact of high-dose methotrexate in the reduction of CNS irradiation. Nordic Society of Pediatric Haematology and Oncology (NOPHO) Leukemia. 2000;14:2267–2275. doi: 10.1038/sj.leu.2401961. [DOI] [PubMed] [Google Scholar]
- 20.Mitelman F. An International System for human Cytogenetic Nomenclature. S Karger; Basel: 1995. [Google Scholar]
- 21.Weinshilboum RM, Raymond FA, Pazmino PA. Human erythrocyte thiopurine methyltransferase: radiochemical microassay and biochemical properties. Clin Chim Acta. 1978;85:323–333. doi: 10.1016/0009-8981(78)90311-x. [DOI] [PubMed] [Google Scholar]
- 22.Cox DR. Regression models and life-tables (with discussion) J R Stat Soc (B) 1972;34:187–220. [Google Scholar]
- 23.Andersen PK, Borgan Ø , Gill RD, Keiding N. Statistical Models Based on Counting Processes. Springer-Verlag; New York: 1993. [Google Scholar]
- 24.Siegel S, Castellan NJ. Nonparametric Statistics for the Behavioral Sciences. McGrawHill; Singapore: 1988. [Google Scholar]
- 25.Kaplan EJ, Meier P. Non-parametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 26.Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother. 1966;50:163–170. [PubMed] [Google Scholar]
- 27.Eichelbaum M, Ingelman-Sundberg M, Evans WE. Pharmacogenomics and individualized drug therapy. Annu Rev Med. 2006;57:119–137. doi: 10.1146/annurev.med.56.082103.104724. [DOI] [PubMed] [Google Scholar]
- 28.Grant SF, Hakonarson H. Recent development in pharmacogenomics: from candidate genes to genome-wide association studies. Expert Rev Mol Diagn. 2007;7:371–393. doi: 10.1586/14737159.7.4.371. [DOI] [PubMed] [Google Scholar]
- 29.Lennard L. The clinical pharmacology of 6-mercaptopurine. Eur J Clin Pharmacol. 1992;43:329–339. doi: 10.1007/BF02220605. [DOI] [PubMed] [Google Scholar]
- 30.Relling MV, Pui CH, Cheng C, Evans WE. Thiopurine methyltransferase in acute lymphoblastic leukemia. Blood. 2006;107:843–844. doi: 10.1182/blood-2005-08-3379. [DOI] [PubMed] [Google Scholar]
- 31.van Dongen JJ, Seriu T, Panzer-Grumayer ER, Biondi A, Pongers-Willemse MJ, Corral L, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352:1731–1738. doi: 10.1016/S0140-6736(98)04058-6. [DOI] [PubMed] [Google Scholar]
- 32.Stanulla M, Schaeffeler E, Flohr T, Cario G, Schrauder A, Zimmermann M, et al. Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA. 2005;293:1485–1489. doi: 10.1001/jama.293.12.1485. [DOI] [PubMed] [Google Scholar]
- 33.Bokkerink JP, Damen FJ, Hulscher MW, Bakker MA, De Abreu RA. Biochemical evidence for synergistic combination treatment with methotrexate and 6-mercaptopurine in acute lymphoblastic leukemia. Hamatol Bluttransfus. 1990;33:110–117. doi: 10.1007/978-3-642-74643-7_20. [DOI] [PubMed] [Google Scholar]
- 34.Schmiegelow K, Schroder H, Gustafsson G, Kristinsson J, Glomstein A, Salmi T, et al. Risk of relapse in childhood acute lymphoblastic leukemia is related to RBC methotrexate and mercaptopurine metabolites during maintenance chemotherapy. Nordic Society for Pediatric Hematology and Oncology. J Clin Oncol. 1995;13:345–351. doi: 10.1200/JCO.1995.13.2.345. [DOI] [PubMed] [Google Scholar]
- 35.Schmiegelow K, Ifversen M. Myelotoxicity, pharmacokinetics, and relapse rate with methotrexate/6-mercaptopurine maintenance therapy of childhood acute lymphoblastic leukemia. Pediatr Hematol Oncol. 1996;13:433–441. doi: 10.3109/08880019609030855. [DOI] [PubMed] [Google Scholar]
- 36.Schmiegelow K, Bretton-Meyer U. 6-mercaptopurine dosage and pharmacokinetics influence the degree of bone-marrow toxicity following high-dose methotrexate in children with acute lymphoblastic leukemia. Leukemia. 2001;15:74–79. doi: 10.1038/sj.leu.2401986. [DOI] [PubMed] [Google Scholar]
- 37.Nygaard U, Schmiegelow K. Dose reduction of coadministered 6-mercaptopurine decreases myelotoxicity following high-dose methotrexate in childhood leukemia. Leukemia. 2003;17:1344–1348. doi: 10.1038/sj.leu.2402990. [DOI] [PubMed] [Google Scholar]
- 38.Andersen JB, Szumlanski C, Weinshilboum RM, Schmiegelow K. Pharmacokinetics, dose adjustments, and 6-mercaptopurine/methotrexate drug interactions in two patients with thiopurine methyltransferase deficiency. Acta Paediatr. 1998;87:108–111. doi: 10.1080/08035259850158001. [DOI] [PubMed] [Google Scholar]
- 39.Bostrom BC, Erdmann GR, Kamen BA. Systemic methotrexate exposure is greater after intrathecal than after oral administration. J Pediatr Hematol Oncol. 2003;25:114–117. doi: 10.1097/00043426-200302000-00006. [DOI] [PubMed] [Google Scholar]
- 40.Lancaster DL, Lennard L, Rowland K, Vora AJ, Lilleyman JS. Thioguanine versus mercaptopurine for therapy of childhood lymphoblastic leukaemia: a comparison of haematological toxicity and drug metabolite concentrations. Br J Haematol. 1998;102:439–443. doi: 10.1046/j.1365-2141.1998.00812.x. [DOI] [PubMed] [Google Scholar]
- 41.Vora A, Mitchell CD, Lennard L, Eden TO, Kinsey SE, Lilleyman J, et al. Toxicity and efficacy of 6-thioguanine versus 6-mercaptopurine in childhood lymphoblastic leukaemia: a randomised trial. Lancet. 2006;368:1339–1348. doi: 10.1016/S0140-6736(06)69558-5. [DOI] [PubMed] [Google Scholar]
- 42.Harms DO, Gobel U, Spaar HJ, Graubner UB, Jorch N, Gutjahr P, et al. Thioguanine offers no advantage over mercaptopurine in maintenance treatment of childhood ALL: results of the randomized trial COALL-92. Blood. 2003;102:2736–2740. doi: 10.1182/blood-2002-08-2372. [DOI] [PubMed] [Google Scholar]
- 43.Jacobs SS, Stork LC, Bostrom BC, Hutchinson R, Holcenberg J, Reaman GH, et al. Substitution of oral and intravenous thioguanine for mercaptopurine in a treatment regimen for children with standard risk acute lymphoblastic leukemia: a collaborative Children’s Oncology Group/National Cancer Institute pilot trial (CCG-1942) Pediatr Blood Cancer. 2007;49:250–255. doi: 10.1002/pbc.20964. [DOI] [PubMed] [Google Scholar]
- 44.Oselin K, Anier K. Inhibition of human thiopurine S-methyltransferase by various nonsteroidal anti-inflammatory drugs in vitro: a mechanism for possible drug interactions. Drug Metab Dispos. 2007;35:1452–1454. doi: 10.1124/dmd.107.016287. [DOI] [PubMed] [Google Scholar]
- 45.Szumlanski CL, Weinshilboum RM. Sulphasalazine inhibition of thiopurine methyltransferase: possible mechanism for interaction with 6-mercaptopurine and azathioprine. Br J Clin Pharmacol. 1995;39:456–459. doi: 10.1111/j.1365-2125.1995.tb04478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hande S, Wilson-Rich N, Bousvaros A, Zholudev A, Maurer R, Banks P, et al. 5-aminosalicylate therapy is associated with higher 6-thioguanine levels in adults and children with inflammatory bowel disease in remission on 6-mercaptopurine or azathioprine. Inflamm Bowel Dis. 2006;12:251–257. doi: 10.1097/01.MIB.0000206544.05661.9f. [DOI] [PubMed] [Google Scholar]
- 47.Gilissen LP, Bierau J, Derijks LJ, Bos LP, Hooymans PM, van GA, et al. The pharmacokinetic effect of discontinuation of mesalazine on mercaptopurine metabolite levels in inflammatory bowel disease patients. Aliment Pharmacol Ther. 2005;22:605–611. doi: 10.1111/j.1365-2036.2005.02630.x. [DOI] [PubMed] [Google Scholar]