

Abstract
In hepatitis C virus (HCV) infection, there is accumulating data suggesting the presence of cellular immune responses to HCV in exposed but seemingly uninfected populations. Some studies have suggested cross-reactive antigens rather than prior HCV exposure as the main reason for the immune responses. In this study we address this question by analyzing the immune response of chimpanzees that have been sequentially exposed to increasing doses of HCV virions. The level of viremia, as well as the immune responses to HCV at different times after virus inoculation, were examined. Our data indicate that HCV infective doses as low as 1–10 RNA (+) virions induce detectable cellular immune responses in chimpanzees without consistently detectable viremia or persistent seroconversion. However, increasing the infective doses of HCV to 100 RNA (+) virions overcame the low-inoculum-induced immune response and produced high-level viremia followed by seroconversion.
Keywords: Hepatitis C virus, HCV, Immune response, IFN-γ, Cellular immunity, Protection, Exposure
Introduction
Hepatitis C virus (HCV) is one of the major health problems in the world. The prevalence of HCV in adult populations of most developed countries is 1–2% but infection rates are much higher in high-risk groups and in most developing countries. WHO estimates that 3% of the world's population has been infected with HCV and that >170 million persons are chronic carriers. About 20% of those with chronic HCV infections develop cirrhosis and/or hepatocellular carcinoma (HCC). The annual incidence of HCC is 2–4% in HCV carriers who have been infected for 20–30 years (Alter et al., 1999; Seeff et al., 2001).
Infection of chimpanzees with HCV results in patterns of infection closely resembling those in man. In general, the course of infection in chimpanzees with HCV can be summarized as follows: HCV-RNA is usually detected in serum 3–7 days after infection and is rapidly amplified to levels of >106 copies/ml. Mild ALT elevation may occur in weeks 2–3; however, the main peak of ALT elevation does not usually occur until 5–10 weeks after infection. In our experience HCV RNA usually remains detectable in self-limited infection for 9–39 weeks (X = 18.6) (Abe et al., 1992). In chronic infection, which occurs in 50–80% of humans, and about 50% of chimpanzees, HCV RNA remains detectable for life (Prince and Brotman, 1994). However, neither cirrhosis nor hepatocellular carcinoma has been reported as a complication of HCV infection in chimpanzees.
HCV antigen-specific IFN-γ secreting T cells have been observed in the peripheral blood and the intrahepatic lymphocytes (IHLs) during HCV infection (Anthony et al., 2001; Cooper et al., 1999; Lechner et al., 2000; Rice and Walker, 1995; Schirren et al., 2000; Shata et al., 2002). A multispecific strong CD8+ response may control HCV replication to some extent, yet the cellular immune response appears to be unable to clear the virus in most HCV infections (Hiroishi et al., 1997; Rehermann et al., 1996; Wong et al., 1998). However, when the cellular responses from humans and chimpanzees that clear HCV infection were analyzed, it has been found that viral clearance was associated with an early and broad CTL response to multiple HCV epitopes (Cooper et al., 1999; Nelson et al., 1997; Shata et al., 2002). Moreover, it was shown that chimpanzees that cleared HCV infection are partially resistant to challenge with HCV (Bassett et al., 2001; Major et al., 2002). Similarly, a strong and persistent T cell response was associated with viral clearance in patients acutely infected with HCV, and in HCV-seronegative humans with exposure to HCV (Bronowicki et al., 1997; Koziel et al., 1997; Lechner et al., 2000; Takaki et al., 2000).
In HIV infection, high-risk individuals with repeated exposure to HIV, but without disease, seroconversion, or viremia, have been shown to have strong cellular immune responses against HIV. It has been suggested that the strong cellular immune responses in these individuals protected them from detectable HIV infection, or modified the course of disease following a fully infectious challenge. In the present study we examined the threshold of the infective doses of HCV that could induce cellular immune responses to HCV without detectable viremia, or seroconversion. We also examine the impact of low-inoculum-induced immune responses on establishment of persistent versus resolved infection following higher dose challenge.
Results
Titration of the infective doses and induction of anti-HCV humoral responses
To determine the infectious titer (CID50), plasma of the chronically HCV-infected chimpanzee (chimpanzee 317), taken 2–4 weeks after inoculation and before the appearance of anti-HCV, was diluted to have approximately 1, 10, and 100 RNA (+) virions/ml. Two virgin chimpanzees, chimpanzee 251 and chimpanzee 325, were challenged with progressively higher doses of RNA (+) virions as described under Materials and methods. Sera, PBLs, and liver biopsies were collected from these chimpanzees at different time points after challenge and were subjected to immunological and virological studies. As shown in Fig. 1A and B, HCV RNA could not be detected by real-time PCR in the sera of either chimpanzee except after challenge with 100 RNA (+). However, transient, very low level viremia in chimpanzee 325 was detected three independent times when tested by a transcription-mediated amplification (TMA) assay for HCV, which has a sensitivity of 14 copies/ml (50% detection limit) (Fig. 1B). Viral replication was detected by quantitative PCR after challenge with 100 RNA (+) virions, but was self-limited in chimpanzee 251, lasting about 12 weeks. In contrast, in chimpanzee 325, viremia initially peaked, then dropped, then rebounded again, and subsequently persisted for more than 50 weeks at a low set point (<105 copies/ml). After challenge with 10 RNA (+) virions, despite the absence of detectable viremia, borderline short-lived increases in the level of anti-HCV antibodies were observed in both chimpanzees (Fig. 1A and B). The anti-HCV levels were boosted dramatically after challenge with 100 RNA (+) virions. In chimpanzee 251, the level of anti-HCV dropped after 30 weeks to borderline and remained low thereafter. By comparison, the level of anti-HCV in chimpanzee 325 dropped initially during the first 12 weeks with decrease in viral load but rebounded to a high level after reappearance of viremia and persisted thereafter. The levels of liver enzymes (AST and ALT) were not significantly changed after challenging with different doses of HCV (data not shown).
Fig. 1.
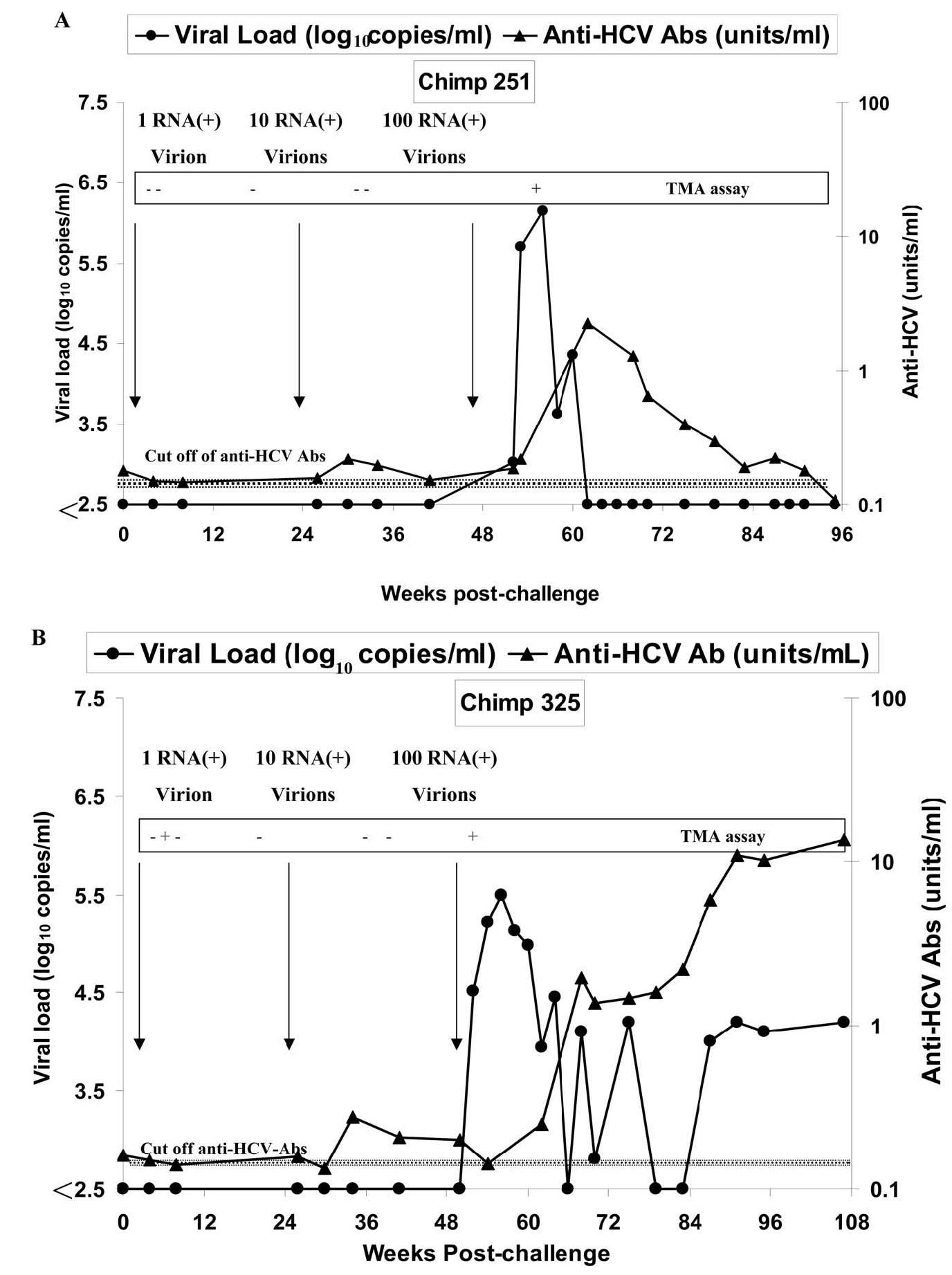
Viral load and anti-HCV level after challenge with different doses of HCV. Sera from chimpanzees 251 (A) and chimpanzee 325 (B) were tested for anti-HCV (filled triangle) and viral load (filled circle) at different time points after challenge with HCV, using quantitative ELISA and PCR, respectively. The lower detection limit (LDL) of quantitative PCR is 250 copies/ml, and for qualitative TMA assay is 14 copies/ml at 50% detection limit. The lower detection limit of anti-HCV antibodies is 0.2 units/ml.
HCV-specific cellular immune responses in peripheral blood
Cryopreserved peripheral blood lymphocytes (PBLs) from chimpanzees 251 and 325, at different time points after infection, were examined using IFN-γ ELISPOT assays. To measure the presence of HCV-specific activated effector T cells, crypreserved PBLs were thawed and used directly without expansion in the IFN-γ ELISPOT assay (ex vivo ELISPOT). Cells were stimulated for 18 h with autologous target cells infected with HCV (NS3, C-through NS3, or NS3 through NS5)-recombinant vaccinia virus. As shown in Fig. 2A and B, an increase in the number of IFN-γ-secreting cells (ISCs) from chimpanzees 251 and 325 was seen even after challenge with an estimated single RNA (+) virion (285 and 195 ISCs/106 cells, respectively). The ISC number increased after challenge with a dose of 10, but not significantly after challenge with 100 RNA (+) molecules. We observed that the responses to NS3-vaccinia constructs were not always in agreement with the responses to C-NS3 or NS3-NS5-vaccinia constructs, although all the constructs contain the NS3 region. The reason for this discrepancy is not known, but it may reflect differences in the processing of dominant NS3 epitopes among these constructs, as well as changes in the dominant epitopes due to mutation.
Fig. 2.
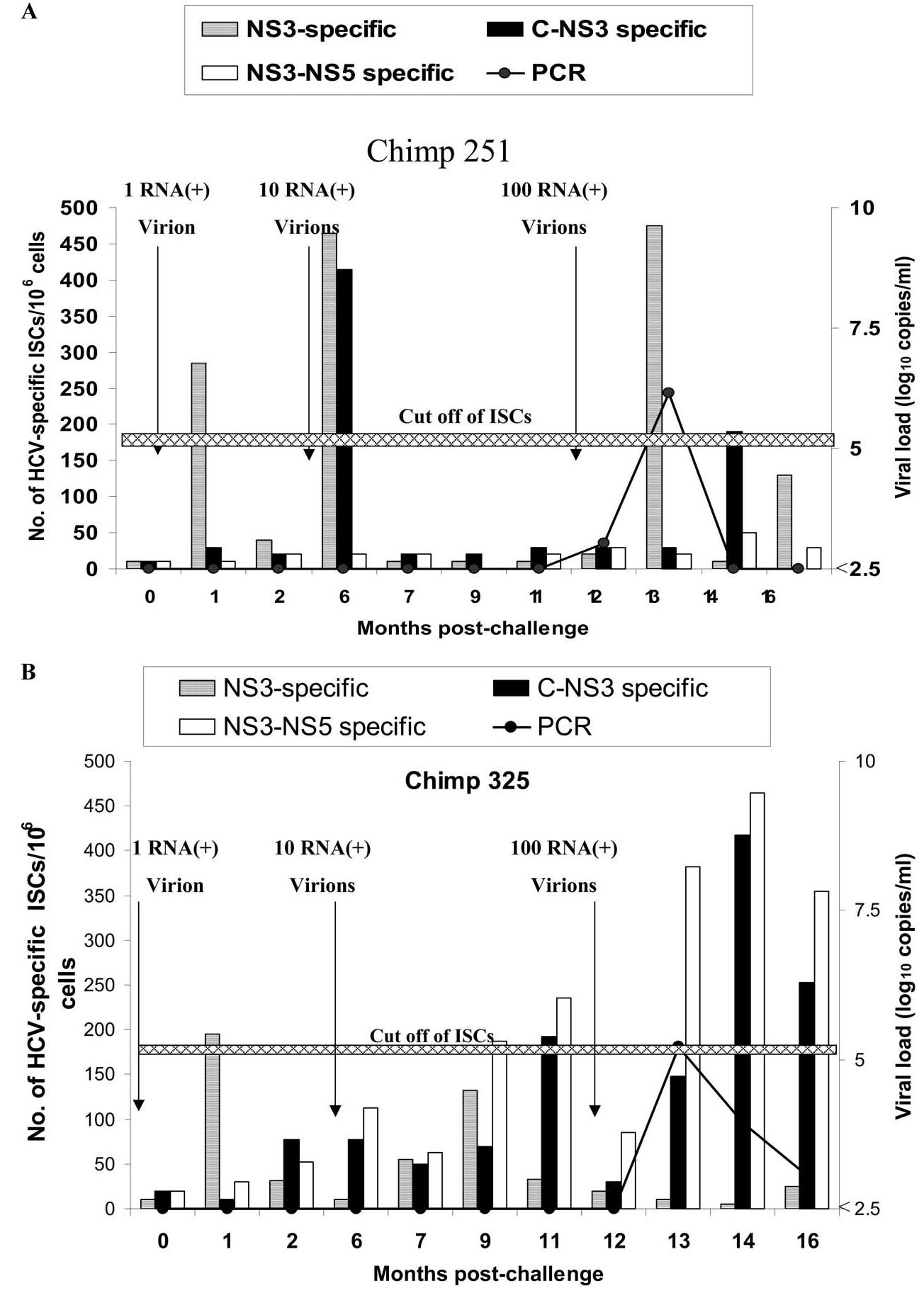
Direct ex vivo IFN-γ ELISPOT for chimpanzees 251 and 325 after infection with different doses. PBLs from chimpanzee 251 (A) and chimpanzee 325 (B) were tested for anti-HCV cellular response using IFN-γ ELISPOT assay as described under Materials and methods. Cells from time 0 (before challenge) were infected with recombinant HCV-C-NS3-vaccinia (filled column), HCV-NS3-NS5 (unfilled column), or HCV-NS3 (hatched column) at an m.o.i. of 10:1 and used as stimulator cells. PBLs from different time points after challenge were incubated with the stimulator cells, and the number of IFN-γ-secreting cells (ISCs) was measured 18 h after in vitro stimulation. IFN-γ ELISPOT was done at all time points indicated in the graph. Sera from chimpanzees 251 and 325 were tested for viral load (line) using quantitative PCR as described in Fig. 1. The data represent average of two experiments. The cutoff value of ISCs (horizontal line) was calculated as described under Materials and methods as approximately 185.5 ISCs/106. The average numbers of direct ex vivo ISCs in the presence of parent vaccinia were 78 ± 54 ISCs/106 cells.
To measure the presence of HCV-specific memory T cells, crypreserved PBLs from chimpanzees 251 and 325 were thawed and expanded in vitro with autologous target cells infected with ALVAC-HCV (core through NS3) for 6 days. Expanded cells were assessed for HCV specificity using IFN-γ ELISPOT assay (in vitro expanded ELISPOT). As shown in Fig. 3A and B, memory HCV-specific T cells from chimpanzees 251 and 325 were detected even after challenge with 1 RNA (+) molecule (2125 and 1525 ISCs/106 cells, respectively). The pool of memory T cells in both chimpanzees did not increase significantly after challenge with higher doses (10 and 100 RNA), which is in agreement with another article (Blattman et al., 2000). To find if there is a qualitative difference between memory T cells in chimpanzees 251 and 325, the levels of IL-4 (data not shown) and TNF-α were measured in the supernatant of the in vitro expanded C-NS3 stimulated T cells. As shown in Fig. 4A and B, the levels of TNF-α in the supernatant of memory T cells after challenge with 10 and 100 RNA (+) molecules were two- to threefold higher in chimp 251 (287 and 366.5 pg/ml, respectively) in comparison to chimp 325 (118 and 146 pg/ml, respectively). However, the levels of IL-4 in the supernatant of the stimulated cells from the two chimpanzees were below the detection level (data not shown).
Fig. 3.
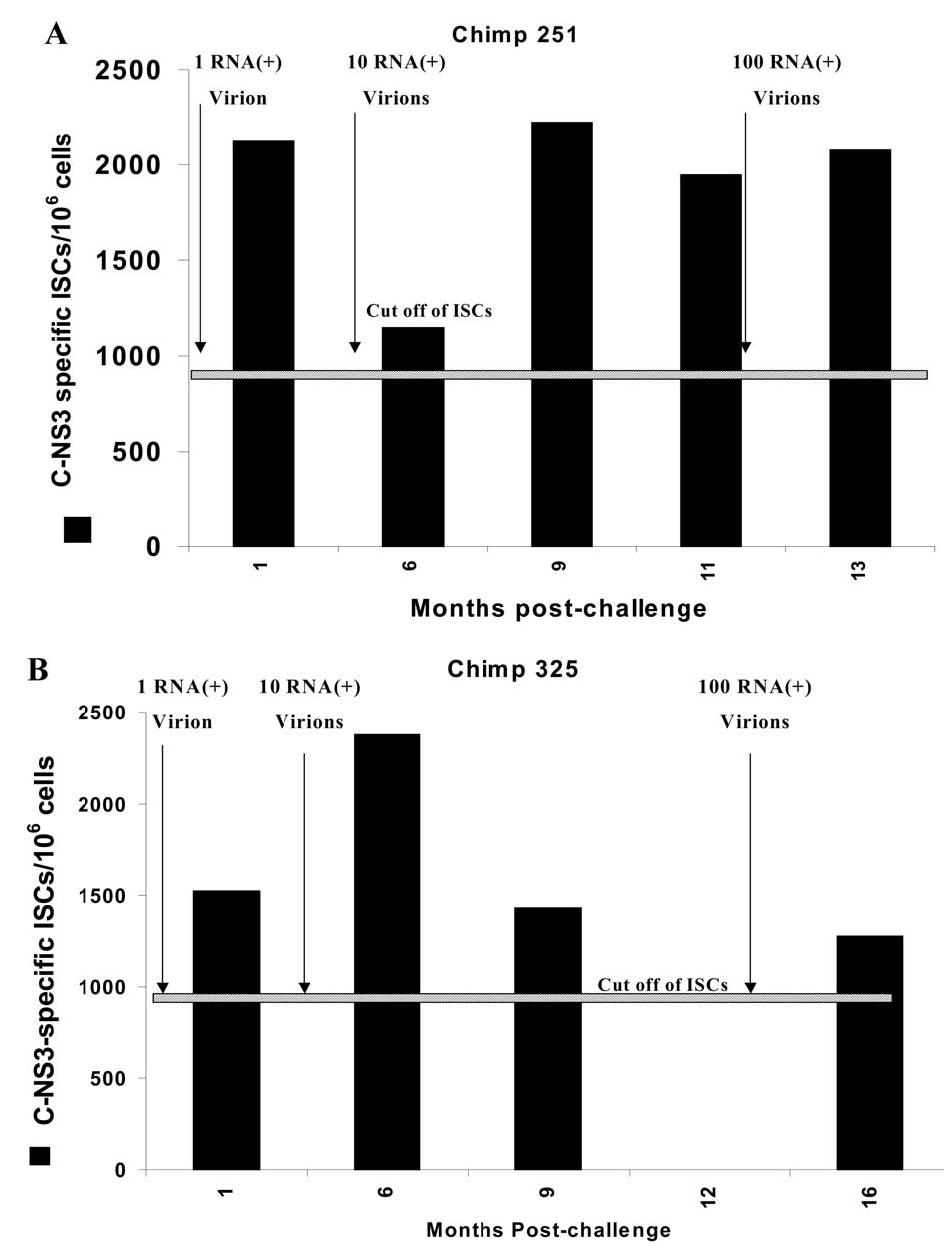
Expanded ELISPOT for chimpanzees 251 and 325 after infection with different doses. Expanded PBLs from chimpanzee 251 (A) and chimpanzee 325 (B) were tested for anti-HCV cellular response using IFN-γ ELISPOT assay as described under Materials and methods. Briefly autologous PBLs cells (at time 0) were infected for 1 h with recombinant HCV-C-NS3-Alvac (filled column) at an m.o.i. of 10:1 and used as stimulator cells. Autologous PBLs from different time points after challenge were incubated with the stimulator cells for 1 week at 37°C and 5% CO2. On day 7, IFN-γ ELISPOT assay was done as described in Fig. 2. The numbers of HCV-specific ISCs/106 cells were plotted after subtracting the background (parental vaccinia virus alone). The cutoff value of ISCs (horizontal line) was calculated as described under Materials and methods as approximately 870.3 ISCs/106 cells.
Fig. 4.
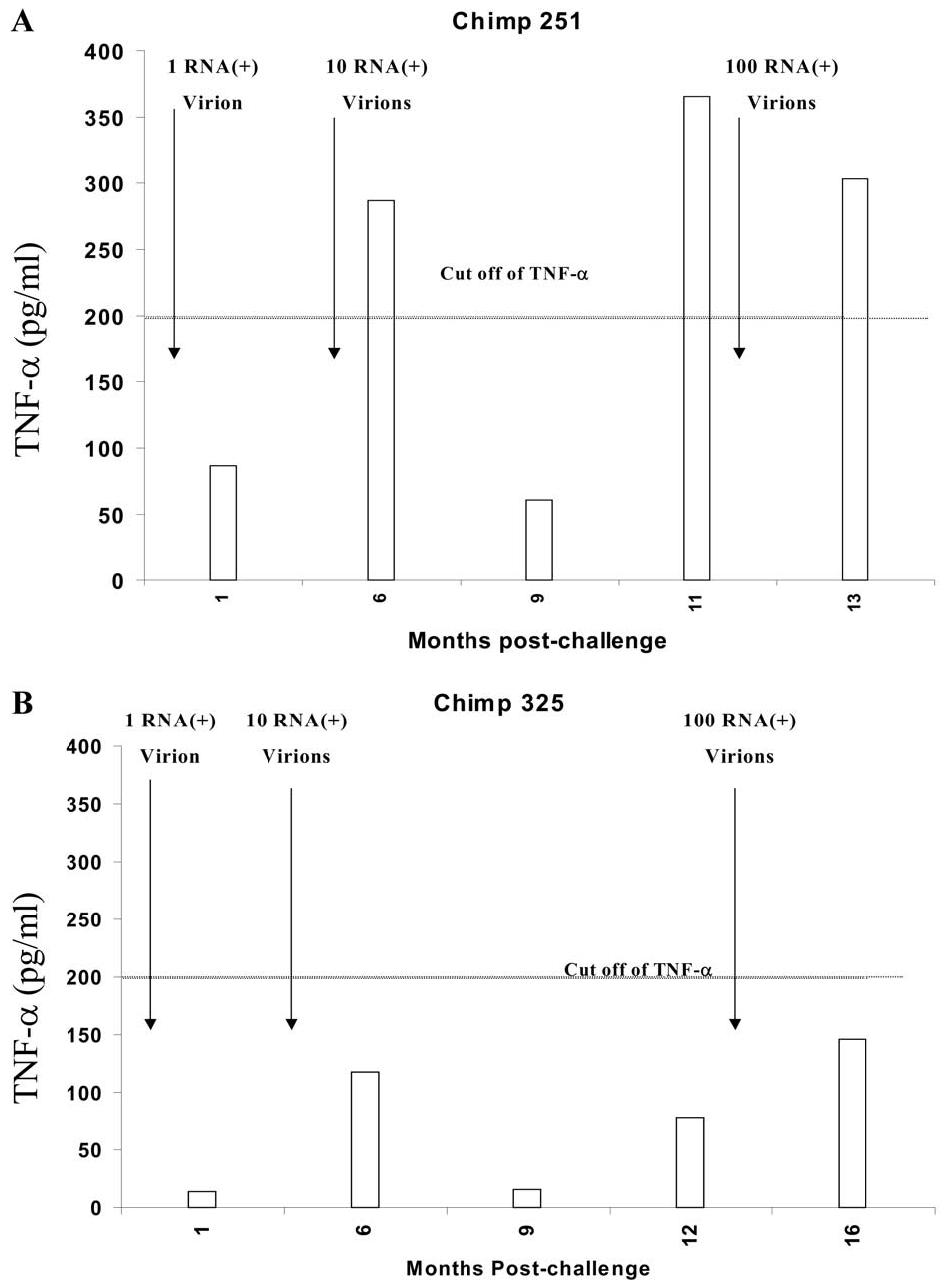
TNF-α secretion from expanded ELISPOT for chimpanzees 251 and 325 after infection with different doses. The levels of TNF-α in the supernatant expanded PBLs from chimpanzee 251 (A) and chimpanzee 325 (B) were measured as previously described. TNF-α was expressed as pg/ml after subtracting the background (TNF-α secreted by vaccinia vector alone). The cutoff value was calculated as the level of TNF-α in the presence of vaccinia vector + 2 SD and was approximately 200 pg/ml.
Responses to Class 1 restricted epitopes of HCV-NS3 protein
To identify responses to class I restricted epitopes of HCV-NS3 protein of genotype 1b, 312 (10-mer) overlapping peptides for NS3 genotype 1b were synthesized with two amino acid offsets. Due to the limiting number of cells for the assay (2 × 105 cells/well are needed for each peptide), the peptides were used as pools of 10 peptides (32 pools were used). The peptide pools contained individual peptides at a concentration of 4 nM, corresponding to the optimum concentration for peptides with moderate binding affinity to class I (Wentworth et al., 1996). The pooled peptides were added for 18 h to the PBMCs from chimpanzees 251 and 325 at different time points after challenge, and the peptide-specific cells were evaluated using direct exvivo IFN-γ secreting cells ELISPOT assay. As shown in Fig. 5, there were multiple borderline NS3-reacting ISCs for chimpanzee 251 even before challenge with HCV (Fig. 5A). This may reflect cross-reactive antigens. One to two months postchallenge with one RNA (+) virion, the responses to NS3-specific epitopes were boosted significantly to previous recognized regions (such as pools 25, 41, 49, 153, 161, and 297) (Fig. 5B). Moreover, new regions of NS3 were recognized (such as pools 105, 113, 209, 241, 249, and 305). Conversely, the response to some previously recognized epitopes were decreased in other NS3 regions (such as pools 57, 65, 145, 169, 185, and 265). However, 3 months postchallenge with one RNA (+), the NS3-specific responses dropped dramatically in all the regions (data not shown). Rechallenging chimpanzee 251 with 10 RNA (+) molecules restricted the response to limited, previously recognized NS3 regions (pools 161 and 185) (Fig. 5C). Rechallenging chimpanzee 251 with 100 RNA (+) molecules stimulated a broad spectrum of NS3-specific ISCs (Fig. 5D). Some ISC responses were to previously recognized regions (such as pools 49, 57, and 241). Other ISC responses were to newly recognized NS3 regions (such as pools 73, 81, 121, 193, 217, and 289). The change in the breadth of HCV epitope recognition was previously reported by other laboratories and may be related to the high mutation rate of HCV (Erickson et al., 2001; Wang et al., 2002). In chimpanzee 325, no preexisting cross-reactive NS3 epitopes were recognized before challenge (Fig. 6A) and only a borderline response to a few epitopes could be detected 1–2 months postchallenge (Fig. 6B). The responses were absent or weaker in general than that of chimpanzees 251 at all time points (Figs. 5 and 6).
Fig. 5.
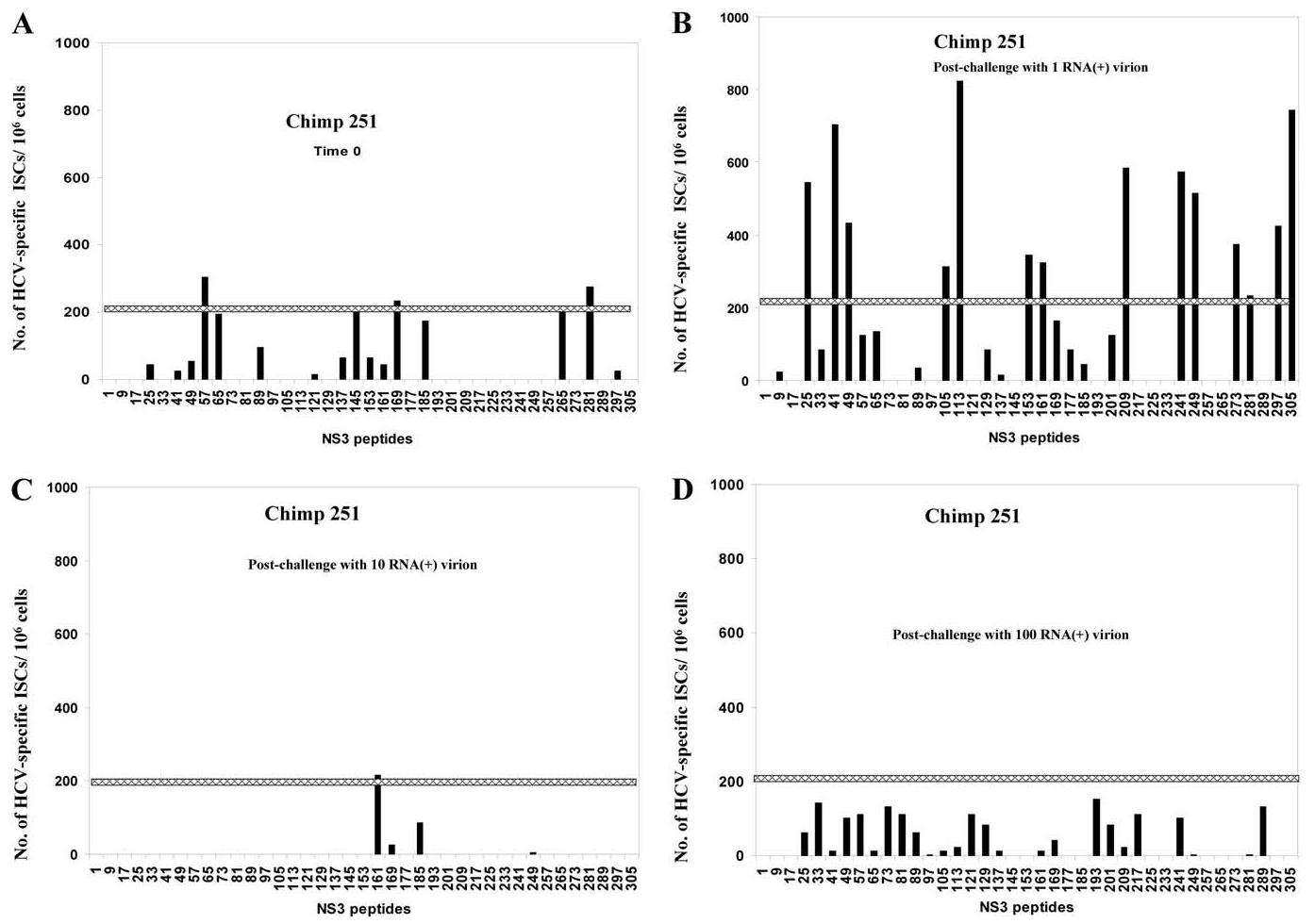
NS3-epitope mapping of effector cells from chimpanzees 251 after challenge with 1-100 RNA (+) virion molecules. PBLs from chimpanzee 251 were tested for cellular response to pools of 10-mer NS3 overlapping peptides using IFN-γ ELISPOT assay as described under Materials and methods. PBLs at time 0 (A), after challenge with an estimated one RNA (+) molecule (B), or after challenge with 10 (C), and 100 (D) RNA (+) molecules, were incubated with pools of ten 10-mer overlapping peptides of NS3 (HCV genotype 1b) at a concentration of 4 nM for each peptide. The numbers of IFN-γ-secreting cells (ISCs) were measured 18 h after in vitro stimulation. The numbers of HCV-specific ISCs/106 cells were plotted after subtracting the background (media alone). The cutoff value of ISCs (horizontal line) was calculated as described under Materials and methods and estimated to be approximately 217 ISCs/106.
Fig. 6.
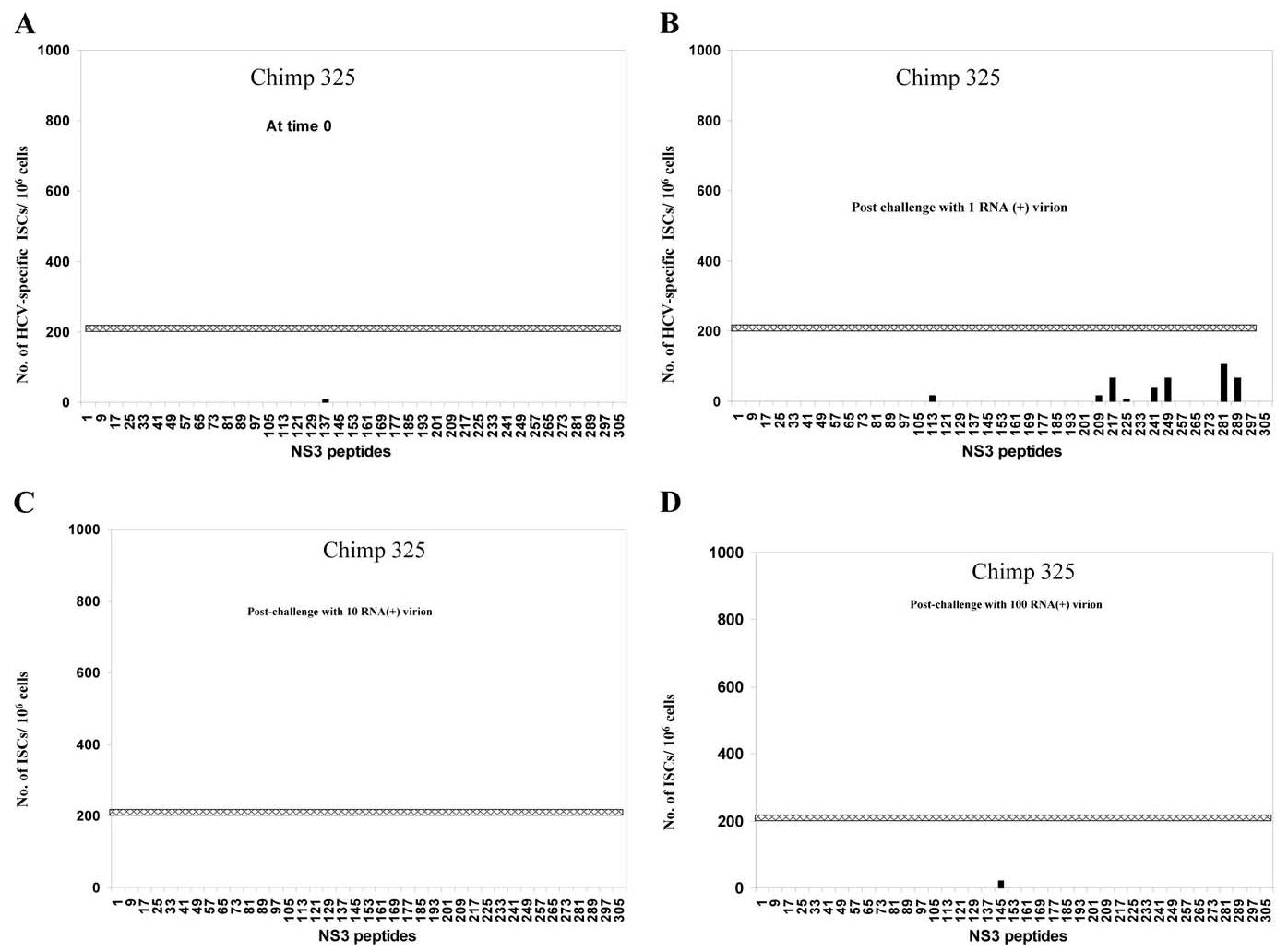
NS3-epitope mapping of effector cells from chimpanzees 325 after challenge with 1-100 RNA (+) virion molecules. PBLs from chimpanzee 325 were tested for cellular response to pools of 10-mer NS3 overlapping peptides using IFN-γ ELISPOT assay as described under Materials and methods. PBLs at time 0 (A), and after challenge with an estimated one RNA (+) molecule (B), or after challenge with 10 (C), and 100 (D) RNA (+) molecules, were incubated with pools of ten 10-mer overlapping peptides of NS3 (HCV genotype 1b) at a concentration of 4 nM for each peptide. The numbers of IFN-γ-secreting cells (ISCs) were measured 18 h after in vitro stimulation. The numbers of HCV-specific ISCs/106 cells were plotted after subtracting the background (media alone). The cutoff value of ISCs (horizontal line) was calculated as described under Materials and methods and estimated to be approximately 217 ISCs/106.
HCV-specific cellular immune response of intrahepatic lymphocytes (IHLs)
To identify T cell response of IHLs after challenge with different doses of HCV, IHLs were prepared as described under Materials and methods, and the number of HCV-specific cells was evaluated using direct ex vivo IFN-γ-secreting-cells ELISPOT assay. As shown in Fig. 7A, there were significant increases in the number of HCV-specific IFN-γ-secreting cells 1 month postchallenge with 1 RNA (+) virion in chimpanzee 251. However, in chimpanzee 325, very weak HCV-specific IHL responses were seen only after challenge with 10 RNA (+) virions and persisted thereafter (Fig. 7B). It is clear that both the IHL and the PBL responses in chimpanzee 251 are stronger than those seen in chimpanzees 325 even after challenge with 100 RNA (+) molecules and the appearance of viremia.
Fig. 7.
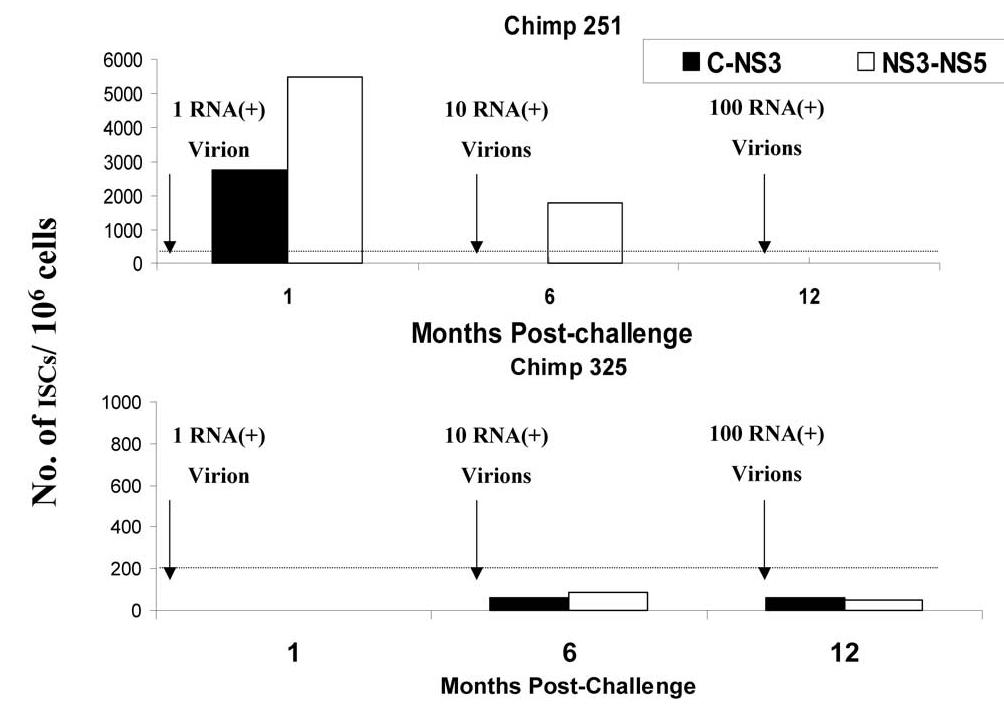
Response of intrahepatic lymphocytes (IHLs) from chimpanzees 251 and 325 to HCV after challenge with different doses. IHLs prepared as described under Materials and methods, from chimpanzee 251 (A) or chimpanzee 325 (B), were tested for anti-HCV cellular response using direct ex vivo IFN-γ ELISPOT assay. Autologous PBLs from time 0 (before challenge) were infected with recombinant HCV-C-NS3-vaccinia (filled column), HCV-NS3-NS5-vaccinia (unfilled column), at an m.o.i. of 10:1 and used as stimulator cells. IHLs at 1, 6, and 12 months postchallenge were incubated with stimulator cells at 37°C and 5% CO2. The numbers of direct ex vivo IFN-γ-secreting cells (ISCs) were measured 18 h after in vitro stimulation. The numbers of HCV-specific ISCs/106 cells were plotted after subtracting the background (parental vaccinia virus alone). The cutoff value of direct ex vivo ISCs (horizontal line) was calculated as described under Materials and methods as approximately 185.5 ISCs/106. The data represent the average of two experiments.
Proliferation responses to HCV proteins
To identify proliferation responses to HCV proteins after challenge with different doses of HCV RNA, PBMCs from chimpanzees 251 and 325 collected at different time points before and after challenge with HCV were examined in T cell proliferation assays using 3H-thymidine incorporation. No significant T cell proliferation (stimulation index > 3) was seen at all time points in chimp 325. However, in chimp 251, blastogenesis was seen only at week 59 (8 weeks postchallenge with 100 RNA (+) molecules); the stimulation index was 3.32, which declined later (data not shown). This low-level stimulation with HCV proteins was not related to the viability of the thawed cells, since the PHA stimulation index was >50 at all time points.
Discussion
It has been shown that about 20–50% of HCV infections in humans are self-limited. Although cellular immune responses clearly play a significant role in controlling viral multiplication (Cooper et al., 1999; Nelson et al., 1997), the mechanisms by which those individuals are able to clear the virus remain controversial (Cerny and Chisari, 1994, 1999; Farci et al., 1992; Lai et al., 1994b; Prince et al., 1992). Likewise, some high-risk individuals, such as the household contacts of HCV-infected patients (Brettler et al., 1992; Meisel et al., 1995) or up to 20% of injection drug abusers consistently exposed to the virus through needle sharing (Thomas et al., 1995), do not develop apparent infection despite repeated exposure to HCV. The reasons for the resistance to infection are not clear, but development of cellular immune responses to HCV has been reported in some seronegative healthy individuals who were either sexual partners of HCV-infected patients, or laboratory personnel with possible occupational exposure to the virus (Bronowicki et al., 1997; Koziel et al., 1997; Scognamiglio et al., 1999).
This study addressed the two following important issues of HCV infection: (1) The threshold of viral exposure that elicits immune responses without detectable viremia and (2) the nature of protective immunity in HCV. The chimpanzee is an excellent model for the study of HCV infection. Analogous to humans, about 50% of chimpanzees clear the virus. Immune exposed uninfected chimpanzees have not been previously investigated. However, it has been shown that chimpanzees with self-limited HCV infection were relatively resistant to rechallenge with HCV infection (Bassett et al., 2001; Major et al., 2002; Nascimbeni et al., 2003). In this study, we examined the threshold dose of HCV that was able to elicit specific cellular immune responses. Two “virgin” (not experimentally infected with HCV) chimpanzees were injected with increasing doses of HCV at 6-month intervals, while both corresponding immune responses and viral load were monitored. Our data indicated that low-dose exposure to 1 or 10 RNA (+) virions could induce HCV-specific cellular immune responses without quantifiable persistent viremia or seroconversion. The levels of induced immune responses were significantly different between the two chimpanzees. Chimpanzee 251, which was able to clear the infection even after high-dose challenge, had early stronger cellular responses in comparison to chimpanzee 325, which failed to clear the infection and subsequently became chronically infected. Ex vivo IFN-γ ELISPOT assays were done to detect HCV-specific effector cells. Borderline, but detectable, ex vivo IFN-γ cellular immune responses were observed after the low-dose challenges. These may represent responses to aborted or low-level viral replication. Attempts to detect viral RNA from sera or liver biopsies after 1–10 RNA (+) virions challenges using quantitative PCR were unsuccessful (data not shown). Despite this failure, low viral hepatic multiplication could not be excluded completely due to the small fragments of liver tissue analyzed from the liver biopsies, as well as the low frequency of infected hepatocytes in HCV, specifically in low-dose infections (Gosalvez et al., 1998). Moreover, using the Genprobe TMA assay which is more sensitive (14 copies/ml) (Giachetti et al., 2002), we confirmed a low level of viral replication after 1 RNA (+) molecule in chimp 325 (Fig. 1B). Higher doses of infection ≥ 100 RNA (+) virions induced detectable viremia and seroconversion as well as ex vivo IFN-γ cellular responses in both chimpanzees, but with variable intensities; the responses were, in general, stronger in chimpanzee 251 than in chimpanzee 325. In expanding the HCV-specific memory cells in vitro, HCV-specific IFN-γ memory cells could be detected in both chimpanzees with a range of 1500–2500 ISCs/106 cells even after challenge with low doses, 1–10 RNA (+) molecules. The level of HCV-specific memory T cells did not increase after challenge with 100 RNA (+) molecules. Remarkably, on expanding the T cells in vitro using target cells infected with ALVAC-HCV construct, the number of ISCs in response to vector alone (background) was still higher than that without expansion (148 ± 360 and 78.7 ± 54 ISCs/106 cells, respectively). The reason for this increase in the background is not clear, but it may represent an anamnestic response to cross-reactive pox viruses or cross-reactive antigens (Brehm et al., 2002; Kim et al., 2002; Welsh and Selin, 2002). However, memory-specific HCV responses were still significantly higher than the background with a cutoff value of 870.3 ISCs/106 cells. We also noticed that the numbers of ISCs in expanded in vitro ELISPOT assay is not always in agreement with that of ex vivo ELISPOT assay, probably because they measured two different subpopulations of Agspecific T cells, namely memory and effector T cells, respectively. Besides, the numbers of memory Ag-specific T cells are influenced by the in vitro condition for expansion.
On dissecting the fine specificity of the immune response to HCV antigens using pools (10 peptides) of NS3 10-mer overlapping peptides, detectable levels of the IFN-γ responses to some of the pool peptides were mainly observed in chimpanzee 251 prechallenge and postchallenges with 1, 10, and 100 RNA (+) virions. It is clear that chimpanzee 251 had a low-level preexisting immune response to cross-reacting antigens to NS3 that is absent in chimpanzee 325. The importance of the preexisting cross-reactive cellular immune response in the outcome of infection has been suggested by others (Brehm et al., 2002; Kim et al., 2002; Wedemeyer et al., 2001; Welsh and Selin, 2002). Consistent with the generalized weak response in chimpanzee 325, the level of the IFN-γ responses to the pool of NS3 peptides were very limited or absent even after challenges with 100 RNA (+) virions.
A discrepancy between the numbers of ISCs generated by vaccinia-NS3 and NS3 peptides is observed. The reasons for this discrepancy are not clear but comparison between the two assays might be difficult for the following reasons: (1) by using vaccinia-NS3, the target cells have to process the NS3 before presenting to T cells. Besides, there is a possible competition among NS3, vaccinia and endogenous epitopes to MHC class I binding. In the case of ELISPOT assay using overlapping NS3 peptides, the peptides bind MHC class 1 without processing and without competition with vaccinia peptides. (2) Stimulation with overlapping peptides may underestimate the number of epitopes recognized because they were 10-mer overlapping with offset of 2 Amino Acids (AA). Theoretically, they may represent only 50% of the possible recognizable epitopes. (3) We have used 10-mer peptide, which has been suggested as the average optimum length of class I restricted peptides (range from 8–13 AA), but this size may or may not be the optimum length for the dominant NS3 peptides. (4) Due to limitation of the available cells, peptides were used at one concentration (4 μM), which represents the response to moderate affinity peptides. Therefore, we could underestimate low avidity responses. Additionally, peptides were used as pools of 10. Some peptides in the pool may act as antagonists to the dominant epitopes without actual stimulation of T cells and may lead to underestimation of the responses.
Since measuring the immune response in PBMCs may not reflect the local immune responses in the liver, we monitored the HCV-specific immune response in IHLs. Due to the low number of cells isolated from liver biopsies, functional analysis of IHLs was limited. Detectable levels of HCV-specific IFN-γ responses were seen after 1 and 6 months postchallenge in chimpanzee 251. Nevertheless, no HCV-specific IFN-γ responses were detected after rechallenge with 100 RNA, unlike those detected in the PBMCs. The reasons for this are not clear but this may reflect the low yield of IHLs with few HCV-specific lymphocytes in the liver biopsies. Similar to cellular responses in peripheral blood mononuclear cells (PBMCs) the IHL responses of chimpanzee 325, in comparison to chimpanzee 251, were absent or weak, even after challenge with 100 RNA (+) virions.
When proliferation to either HCV antigens or HCV peptides (data not shown) was evaluated using 3H-thymidine uptake, no significant proliferation (stimulation index, S.I., > 3) was observed at all time points in both chimpanzees except at one point: 6–8 weeks after challenge with 100 RNA (+) molecules in chimpanzee 251. Viability of the frozen PBMCs was not the reason for the weak proliferation observed, since stimulation index in response to PHA was >50 in all time points tested (data not shown), but weak responses to HCV Ags have been reported by others (Lauer et al., 2002; Rico et al., 2002; Valdez et al., 2000).
One of the striking observations made from this study was that chimpanzee 251 had preexisting cross-reacting cellular immune responses to NS3 peptides of HCV. This chimpanzee cleared the virus more rapidly after rechallenge with 100 RNA (+) virions of HCV in comparison to chimpanzee 325 that had no preexisting immune responses. It is clear from our data that the outcome of infection as well as the duration required for viral clearance are directly related to the level of the immune responses induced by viral challenge. For example, chimpanzee 251, who had strong effector HCV-specific immune response, cleared the virus within 12 weeks. By contrast, chimpanzee 325, who had weaker immune responses, failed to clear the virus. However, the number of IFN-γ-secreting memory-specific HCV responses were in general lower in chimpanzee 325 by comparison to chimpanzee 251. To address the possibility of a qualitative difference in the HCV-specific memory T cells, secreted IL-4 and TNF-alpha cytokines were measured. Quantitative analysis of IL-4 levels in the supernatant of HCV-stimulated in vitro expanded PBLs showed no differences (data not shown). However, a significant increase (P < 0.01) in the level of TNF-α secretion by memory T cells of chimpanzee 251 in comparison to chimpanzee 325 has been observed after challenge with 10 and 100 RNA (+) molecules, as shown in Fig. 4A and B. The role of TNF-α in the outcome of HCV infection has been suggested by other studies (Ando et al., 1997; Gruener et al., 2001; Kakumu et al., 1997; Shata et al., 2002; Zhu et al., 1998).
Three important conclusions can be drawn from this study.
First, low doses of HCV infection can induce cellular immune responses without persistently detectable viremia or seroconversion. This conclusion is supported by the following data:
Memory HCV-specific T cells were established after challenge with 1–10 RNA (+) molecules in both chimpanzees and did not increase significantly after rechallenge with higher doses.
Induction of effector HCV-specific IFN-γ-secreting cells in chimpanzees 251 after challenge with 1 and 10 RNA (+) molecules. The weak cellular immune response in chimpanzee 325, even after challenge with 100 RNA (+) molecules, may reflect an inherited weakness in response to HCV.
Significant induction of HCV-specific ISCs in IHLs in chimpanzee 251 even after challenge with 1 RNA (+) molecule. Chimpanzee 325 failed to induce significant numbers of HCV-specific ISCs in IHLs even after challenge with 100 RNA (+) molecules and subsequently established viremia.
Borderline boosting of antibody responses was seen in both chimpanzees after challenge with 10 RNA (+) molecules.
By using the more sensitive Genprobe TMA assay, we were able to detect HCV RNA 4 weeks after challenge with 1 RNA (+) molecule in chimpanzee 325. The failure to detect HCV RNA after low-dose challenges in chimpanzee 251 may reflect the low level of replication in this chimpanzee, the fluctuation of the viremia level, as well as the small sample sizes.
The presence of preexisting NS3-specific ISCs in chimpanzee 251 upon peptide stimulation is of great interest and may reflect cross-reactive antigens (Brehm et al., 2002) or previous unrecognized exposure to HCV. Attempts to identify strong HCV-specific memory T cells in chimp 251 prechallenge were not successful (data not shown). Therefore, we are more inclined toward cross-reactive antigens as an explanation for the presence of a preexisting cellular immune response in this particular chimpanzee.
The second conclusion from out study is the correlation between the outcome of HCV infection and the level of induced cellular immune responses in the host. This is supported by the fact that chimpanzee 251, which resolved the infection, had early stronger cellular immune response in comparison to chimpanzee 325, which became chronically infected with a low set point of viral load. However, these immune responses failed to induce sterile immunity against a challenge > 100 RNA (+) molecules, although they lowered the duration of viremia, and in chimpanzee 251, ultimately resulted in clearance of the infection.
The third conclusion from our study is the probable lack of a role of anti-HCV in controlling viral replication. As shown in Fig. 1, chimpanzee 325 had 1–2 log higher level of anti-HCV in comparison to chimpanzee 251; however, this chimpanzee failed to control viral replication and developed chronic HCV infection. It is interesting to notice that the drop in viral load in chimpanzee 251 is associated with an increase in the anti-HCV. It is not clear if these antibodies have a role in the control of viral replication in this chimpanzee and if there is a quality difference between the anti-HCV in chimpanzee 251 and chimpanzee 325.
Materials and methods
Chimpanzees
Chimpanzees (Pan troglodytes verus) were housed in the New York Blood Center's primate laboratory, Vilab II, at the Liberian Institute for Biomedical Research (LIBR) in Robertsfield, Liberia. To titrate infectivity, two chimpanzees, 251 and 325, were experimentally infected sequentially with HCV genotype 1b at different doses [1, 10, and 100 RNA (+) molecules]. Serum samples, and cryopreserved PBMCs were taken at 1- to 4-week intervals up to 2 years after challenge with HCV. Liver biopsies were taken at 4-week intervals. Intrahepatic lymphocytes were prepared from the liver biopsies as described below and cryopreserved in liquid N2 until analyzed. Serum samples were frozen and stored at −70°C until examined.
Quantitation of HCV RNA
HCV RNA was quantitated using real-time PCR assay with molecular beacon technology using a Perkin–Elmer model 7700, as previously described (Lai et al., 1994a; Lee and Prince, 2001; Takeuchi et al., 1999) with modifications. Briefly, RNA was extracted from plasma, or lysate of liver biopsies using Qiagen HCV RNA kits. PCR and cDNA mixtures were set up with a Beckman Biomek 2000 pipetting station in a laminar flow hood in a room dedicated to PCR setup. This permits quantitation of both strong and weak specimens by comparison with a standard curve derived from serial dilutions of synthetic HCV RNA as performed in this study; the method was sensitive to approximately 100 RNA molecules/ml using a synthetic RNA standard and gives linear results between 102.5 and 107 RNA molecules/ml. This methodology has two major advantages: the ability to quantitate strong and weak specimens from a single dilution, and the fact that the amplified samples are not used for other assays, gel, or ELISA, which can serve as a source for contamination. Quality control is performed routinely by including 4–10 HCV negative control sera, and 2–4 positive sera, during each PCR run, as external negative and positive controls, respectively, to monitor the extraction and amplification efficiencies. Assays in which positive control quantities are outside of the mean ± 2 SD for all assays run (QA curve) are discarded.
Qualitative detection of HCV RNA
Transcription-mediated amplification was done as described (Giachetti et al., 2002; Linnen et al., 2002). The specificity of the TMA-driven assay is ≥99.5% in both normal specimens and plasma containing potentially interfering substances or other blood-borne pathogens. The sensitivity of the assay is 14 copies/ml at 50% detection.
Titrating the infective doses of HCV
Chimpanzee 317 was infected with HCV strain 1b (BK) at a dose of 10 CID50 and became chronically infected. HCV viral load in the acute phase plasma (4 weeks post-inoculation) was quantitatively measured using PCR as described above. This plasma was found to have viral load of 106.32 copies/ml. The plasma was diluted to obtain different concentrations of viral RNA molecules. Chimpanzees 251 and 325 were challenged intravenously initially with 1 ml of plasma from chimpanzee 317 diluted to contain approximately 1 RNA (+) molecule/ml. After 6 months, both chimpanzees were rechallenged with 1 ml of the diluted plasma containing approximately 10 RNA (+) molecules. After another 6 months, they were rechallenged again with 1 ml of the diluted plasma containing approximately 100 RNA (+) molecules. Between each challenge, sera and PBMCs were collected at biweekly intervals, and liver biopsies were obtained monthly.
HCV ELISA
Quantitative measurement of anti-HCV antibody response was performed using Ortho HCV 3.0 ELISA test system (Ortho Diagnostic System, Rariton, NJ), and serial dilution of chimpanzee sera, and positive control sera was performed according to the manufacturer's instructions with some modification. Briefly, dilution of positive control sera with well-defined quantities of anti-HCV antibodies and dilutions of chimpanzee sera were run simultaneously in the Ortho HCV 3.0 ELISA test system. The level of anti-HCV antibodies in the chimpanzee sera was expressed as units in comparison to the positive control serum. One unit of positive control serum is equivalent to the dilution at 50% binding. The results were expressed as units/ml.
Isolation and propagation of peripheral blood lymphocytes
PBMCs drawn at different intervals after infection were purified by Ficoll–Hypaque and then cryopreserved at 1°C/min in the presence of 20% autologous serum and 10% dimethyl sulfoxide (DMSO). Before performing any assay, PBLs were thawed, washed three times, and counted for functional analysis. The effect of freezing and thawing is an issue of considerable importance. Therefore freshly isolated and freeze/thawed PBLs were tested in parallel to establish the effects of cryopreservation. The tested antigens induced a comparable number of spots in ELISPOT assays with both frozen as well as fresh PBLs (data not shown).
Isolation of liver infiltrating lymphocytes
The peripheral blood compartment may not be sufficiently representative of the immune response in the infected animals, since HCV-specific T cells may be preferentially concentrated in HCV-infected liver. Therefore, functional analysis of the intrahepatic lymphocytes was done after challenge with HCV in parallel with the PBLs. One problem of IHL analysis is the low yield of the liver biopsy (approximately 2–5 × 105 cells/1–2 cm of liver biopsy). Therefore, direct IFN-γ ELISPOT assay was done using only 1–3 × 104 cells/well. To isolate IHLs, the liver specimens were cut into small pieces and then treated with collagenase IV (Sigma, 0.5 mg/ml) and DNAse (Sigma, 100 μg/ml) at 37°C for 20–30 min with continuous shaking. Treated samples were passed through a 200-gauge stainless mesh and then resuspended in 1 ml of RPMI 1640 medium. The cells were mixed with 4 ml of 30% (W/V metrizamide in PBS) and centrifuged at 1500 g for 20 min at 4°C in 15–ml conical tubes. Cells at the metrizamide/PBS interface were collected and washed twice with RPMI and then counted (Kawarabayashi et al., 2000). IHLs were functionally examined using IFN-γ ELISPOT assay as described below.
Synthetic HCV peptides
NS3 peptides
A panel of 312 overlapping peptides with a length of 10 amino acids and offset of two, derived from the NS3 region of genotype 1b, according to its sequences (Takamizawa et al., 1991), was synthesized (Mimotopes Pty Ltd., Clayton Victoria, Australia) and pools of 10 successive overlapping peptides were used to study recognition by HCV NS3-specific IFN-γ-secreting cells. Data were presented as the number of ISCs in the presence of pool peptides (10 overlapping 10-mer peptides) after subtracting the cut-off value. The cutoff value of ISCs was calculated for each experiment as the average number of ISCs in response to a control peptide + 2 SD and range from 100 to 200 ISCs/106 cells.
Generation of poxvirus recombinants expressing HCV proteins
Recombinant virus
Hepatitis C genes from HCV-H (genotype 1a) and HCV-BK (genotype 1b) were inserted into two poxviruses; ALVAC, the vaccine strain of canarypox (Perkus et al., 1989), and the L variant of WR, a laboratory strain of vaccinia virus (Panicali et al., 1981; Perkus et al., 1989). Poxvirus recombinants were generated as previously described (Perkus et al., 1989). For construction of poxvirus recombinants expressing an internal segment of HCV (e.g., NS2 or NS3), the poxvirus promoter was immediately followed by an initiation codon, which was added to the native HCV cleavage site. To maximize gene expression, all occurrences of the T5NT poxvirus early transcription termination signal in the HCV sequences were mutagenized without changing the encoding amino acid (Yuen and Moss, 1987). These constructs have been used successfully in our laboratory to infect target cells for in vitro stimulation of HCV-specific CD8+ T cells in 51Cr-release assay (Cohard et al., 1998) as well as in ELISPOT assay (Pancholi et al., 2000; Prince and Shata, 2001; Shata et al., 2002).
IFN-γ ELISPOT assay
ELISPOT assay was done according to the manufacturer's instructions contained in the γ-IFN ELISPOT Kit (MABTECH Cat. No. M34201-A, Sweden) with modifications. Briefly, 96-well nitrocellulose-bottomed Millititer plates (Millipore, Bedford, MA) were coated with murine anti-human IFN-γ mAb at concentration of 15 μg/ml in PBS and incubated at 4°C. After 24 h, the plates were washed and blocked with 10% human AB+ serum (1 h at 37°C). For in vitro expanded ELISPOT assay, autologous PBMCs from the chimpanzee before infection (defined as time 0 PBMCs) were infected with recombinant ALVAC-HCV, or control ALVAC at a multiplicity of infection (m.o.i.) 10:1, and used as stimulator cells. PBMCs at different time points postchallenge were incubated with the stimulator cells at E:T ratio of 10:1 as described (Cohard et al., 1998; Wong et al., 2001). Ten units/ml of recombinant IL-2 (R&D Systems, Minneapolis, MN, USA) were added on the third day. On day 6, expanded T cells were washed three times and counted. To estimate the number of HCV-specific interferon-γ secreting cells, in vitro expanded or ex vivo unexpanded PBMCs were added at different concentrations (104–105 cells/well) in 100 μl volume of complete medium (RPMI-1640 containing 10% AB+ serum). The stimulator cells, autologous PBMCs at time 0, infected with either recombinant vaccinia-HCV or parental vaccinia at m.o.i. of 10:1, were added at a concentration of 105 cells/well in 100 μl volume of complete medium. In the case of peptide stimulation, the unexpanded ex vivo PBMCs were pulsed with the test peptides at a concentration of 4 nM as described (Shata et al., 2002; Tobery et al., 2001). After 18-h incubation at 37°C, the plates were washed five times with washing buffer, PBS containing 0.5% (v/v) Tween 20 (Sigma, St. Louis, MO), using an ELISPOT plate washer (Millipore). Biotinylated anti-human IFN-γ mAb (clone 4S.B3) at a concentration of 1 μg/ml in blocking buffer, PBS containing 0.5% (v/v) Tween 20 and 1% (w/v) bovine serum albumin (Sigma), was added, and the plates were incubated at room temperature for 2 h. Plates were then again washed five times, and streptavidin-HRP (1:1000) in blocking buffer was added and incubated at room temperature for 2 h, followed by washing five times with washing buffer and the addition of substrate 3-amino-9-ethyl carbazole (AEC) reagent in substrate buffer (Sigma). After developing the spots for 10–15 min, the plates were washed with distilled water and air-dried. The number of spots was enumerated using a computerized-assisted ELISPOT image analyzer (Zeiss System, Axioplan 2 imaging), and KS ELISPOT 4.2 program (Zeiss), designed to detect spots using predetermined criteria based on size, shape, and colorimetric density. Analysis of the data was calculated from the equation:
HCV-specific ISCs were plotted after subtracting the number of ISCs induced by parental vaccinia. The average numbers of direct ex vivo, or expanded ISCs induced by parental vaccinia calculated from 16 experiments, were 78.7 ± 54 and 148 ± 360 ISCs/106, respectively. The cutoff level of ISCs is calculated as the average number of ISCs in the presence of parental vaccinia + 2 SD and were 185.5 and 870.3 ISCs/106 cells for the direct ex vivo and expanded ISCs, respectively. To eliminate samples with poor viability, those having PHA responses <2500 ISCs/106 cells, equivalent to the mean − 2 SD of all the samples tested in 18 experiments, were excluded from analysis. The average numbers of (ISCs)/106 cells, after PHA stimulation, in three uninfected chimpanzees and six chronically HCV-infected chimpanzees, were 3783.3 ± 398.2 and 5148.1 ± 1241, respectively.
T cell proliferation (3H-thymidine uptake)
PBMCs, prepared as described above, were added at a concentration of 105 cells per well in 96-well round-bottom tissue culture plate in a volume of 100 μl. The medium consisted of RPMI-1640 with 2 mM glutamine, antibiotics, 5 mM HEPES buffer, and either 10% human AB serum or chimpanzee serum. Cultures were incubated with pooled HCV antigens (c22-3, rNS3, c200, HCV-NS5) kindly provided by Dr. Michael Houghton (Chiron, Emeryville, CA) at 1 μg/ml for each individual protein, as described (Rosen et al., 1999; Tsai et al., 1997), or with PHA (5 μg/ml) (Sigma). The cells were incubated at 37°C in humidified 5% CO2. On the sixth day 50 μl of fresh medium containing 1 μCi of 3H-thymidine was added for each well. After 18 h, the cells were harvested using a Skatron Combi cell harvester (Molecular Devices Corp., CA), and the 3H-thymidine incorporation was counted using a Microbeta Plus 1450 Liquid Scintillation Counter (Perkin–Elmer Life Science, MA). Stimulation index was calculated from the equation:
To eliminate samples with poor cell viability, data were excluded from the analysis if the stimulation index after PHA stimulation was less than 10.
Cytokine measurement in the supernatant of HCV-specific memory cells
The levels of cytokines (IL-4 and TNF-α) in the supernatant from ex vivo expanded C-NS3 specific memory T cells, after in vitro restimulation for 24 h with Vaccinia-CNS3-infected PBLs (at time 0), were measured using Human IL-4 OptEIA kit (BD Biosciences, San Jose, CA, Cat. No. 2678KK) and human-TNF-α OptEIA kit (B.D. Biosciences, Cat. No. 2600KK), according to the manufacturer's instructions. The cutoff level was calculated as the level of cytokine in the presence of parental vaccinia vector alone + 2 SD.
Acknowledgments
All chimpanzee studies were conducted in accordance with Institutional Animal Care and Use Committee-approved protocol No. 202 and the NIH Guide for the Care and Use of Laboratory Animals. This work was supported in part by NIH Grants RO1-AI47349. We acknowledge the dedicated assistance of Musa Konneh, Joseph Thomas, John Zeonuway, George Saycoyah, and Etmonia Davis at the New York Blood Center's primate laboratory, Vilab II, in the conduct of these studies. We also acknowledge Ms. Marie Fuderanan for editorial assistance.
References
- Abe K, Inchauspe G, Shikata T, Prince AM. Three different patterns of hepatitis C virus infection in chimpanzees. Hepatology. 1992;15((4)):690–695. doi: 10.1002/hep.1840150423. [DOI] [PubMed] [Google Scholar]
- Alter MJ, Kruszon-Moran D, Nainan OV, McQuillan GM, Gao F, Moyer LA, Kaslow RA, Margolis HS. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N. Engl. J. Med. 1999;341((8)):556–562. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- Ando K, Hiroishi K, Kaneko T, Moriyama T, Muto Y, Kayagaki N, Yagita H, Okumura K, Imawari M. Perforin, Fas/Fas ligand, and TNF-alpha pathways as specific and bystander killing mechanisms of hepatitis C virus-specific human CTL. J. Immunol. 1997;158((11)):5283–5291. [PubMed] [Google Scholar]
- Anthony DD, Post AB, Valdez H, Peterson DL, Murphy M, Heeger PS. ELISPOT analysis of hepatitis C virus protein-specific IFN-gamma-producing peripheral blood lymphocytes in infected humans with and without cirrhosis. Clin. Immunol. 2001;99((2)):232–240. doi: 10.1006/clim.2001.5018. [DOI] [PubMed] [Google Scholar]
- Bassett SE, Guerra B, Brasky K, Miskovsky E, Houghton M, Klimpel GR, Lanford RE. Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infection. Hepatology. 2001;33((6)):1479–1487. doi: 10.1053/jhep.2001.24371. [DOI] [PubMed] [Google Scholar]
- Blattman JN, Sourdive DJ, Murali-Krishna K, Ahmed R, Altman JD. Evolution of the T cell repertoire during primary, memory, and recall responses to viral infection. J. Immunol. 2000;165((11)):6081–6090. doi: 10.4049/jimmunol.165.11.6081. [DOI] [PubMed] [Google Scholar]
- Brehm MA, Pinto AK, Daniels KA, Schneck JP, Welsh RM, Selin LK. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat. Immunol. 2002;3((7)):627–634. doi: 10.1038/ni806. [DOI] [PubMed] [Google Scholar]
- Brettler DB, Mannucci PM, Gringeri A, Rasko JE, Forsberg AD, Rumi MG, Garsia RJ, Rickard KA, Colombo M. The low risk of hepatitis C virus transmission among sexual partners of hepatitis C-infected hemophilic males: an international, multicenter study. Blood. 1992;80((2)):540–543. [PubMed] [Google Scholar]
- Bronowicki JP, Vetter D, Uhl G, Hudziak H, Uhrlacher A, Vetter JM, Doffoel M. Lymphocyte reactivity to hepatitis C virus (HCV) antigens shows evidence for exposure to HCV in HCV-seronegative spouses of HCV-infected patients. J. Infect. Dis. 1997;176((2)):518–522. doi: 10.1086/517279. [DOI] [PubMed] [Google Scholar]
- Cerny A, Chisari FV. Immunological aspects of HCV infection. Intervirology. 1994;37((2)):119–125. doi: 10.1159/000150366. [DOI] [PubMed] [Google Scholar]
- Cerny A, Chisari FV. Pathogenesis of chronic hepatitis C: immunological features of hepatic injury and viral persistence [see comments] Hepatology. 1999;30((3)):595–601. doi: 10.1002/hep.510300312. [DOI] [PubMed] [Google Scholar]
- Cohard M, Liu Q, Perkus M, Gordon E, Brotman B, Prince AM. Hepatitis C virus-specific CTL responses in PBMC from chimpanzees with chronic hepatitis C: determination of CTL and CTL precursor frequencies using a recombinant canarypox virus (ALVAC) J. Immunol. Methods. 1998;214((1–2)):121–129. doi: 10.1016/s0022-1759(98)00054-4. [DOI] [PubMed] [Google Scholar]
- Cooper S, Erickson AL, Adams EJ, Kansopon J, Weiner AJ, Chien DY, Houghton M, Parham P, Walker CM. Analysis of a successful immune response against hepatitis C virus. Immunity. 1999;10((4)):439–449. doi: 10.1016/s1074-7613(00)80044-8. [DOI] [PubMed] [Google Scholar]
- Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, McKinney D, Sette A, Hughes AL, Walker CM. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 2001;15((6)):883–895. doi: 10.1016/s1074-7613(01)00245-x. [DOI] [PubMed] [Google Scholar]
- Farci P, Alter HJ, Govindarajan S, Wong DC, Engle R, Lesniewski RR, Mushahwar IK, Desai SM, Miller RH, Ogata N, et al. Lack of protective immunity against reinfection with hepatitis C virus. Science. 1992;258((5079)):135–140. doi: 10.1126/science.1279801. [DOI] [PubMed] [Google Scholar]
- Giachetti C, Linnen JM, Kolk DP, Dockter J, Gillotte-Taylor K, Park M, Ho-Sing-Loy M, McCormick MK, Mimms LT, McDonough SH. Highly sensitive multiplex assay for detection of human immunodeficiency virus type 1 and hepatitis C virus RNA. J. Clin. Microbiol. 2002;40((7)):2408–2419. doi: 10.1128/JCM.40.7.2408-2419.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosalvez J, Rodriguez-Inigo E, Ramiro-Diaz JL, Bartolome J, Tomas JF, Oliva H, Carreno V. Relative quantification and mapping of hepatitis C virus by in situ hybridization and digital image analysis. Hepatology. 1998;27((5)):1428–1434. doi: 10.1002/hep.510270534. [DOI] [PubMed] [Google Scholar]
- Gruener NH, Lechner F, Jung MC, Diepolder H, Gerlach T, Lauer G, Walker B, Sullivan J, Phillips R, Pape GR, Klenerman P. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J. Virol. 2001;75((12)):5550–5558. doi: 10.1128/JVI.75.12.5550-5558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroishi K, Kita H, Kojima M, Okamoto H, Moriyama T, Kaneko T, Ishikawa T, Ohnishi S, Aikawa T, Tanaka N, Yazaki Y, Mitamura K, Imawari M. Cytotoxic T lymphocyte response and viral load in hepatitis C virus infection. Hepatology. 1997;25((3)):705–712. doi: 10.1002/hep.510250336. [DOI] [PubMed] [Google Scholar]
- Kakumu S, Okumura A, Ishikawa T, Yano M, Enomoto A, Nishimura H, Yoshioka K, Yoshika Y. Serum levels of IL-10, IL-15 and soluble tumour necrosis factor-alpha (TNF-alpha) receptors in type C chronic liver disease. Clin. Exp. Immunol. 1997;109((3)):458–463. doi: 10.1046/j.1365-2249.1997.4861382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi N, Seki S, Hatsuse K, Ohkawa T, Koike Y, Aihara T, Habu Y, Nakagawa R, Ami K, Hiraide H, Mochizuki H. Decrease of CD56(+)T cells and natural killer cells in cirrhotic livers with hepatitis C may be involved in their susceptibility to hepatocellular carcinoma. Hepatology. 2000;32((5)):962–969. doi: 10.1053/jhep.2000.19362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Brehm MA, Welsh RM, Selin LK. Dynamics of memory T cell proliferation under conditions of heterologous immunity and bystander stimulation. J. Immunol. 2002;169((1)):90–98. doi: 10.4049/jimmunol.169.1.90. [DOI] [PubMed] [Google Scholar]
- Koziel MJ, Wong DK, Dudley D, Houghton M, Walker BD. Hepatitis C virus-specific cytolytic T lymphocyte and T helper cell responses in seronegative persons. J. Infect. Dis. 1997;176((4)):859–866. doi: 10.1086/516546. [DOI] [PubMed] [Google Scholar]
- Lai J, Prince AM, Wolfe L, Andrus L. A simplified method for PCR detection of hepatitis C virus RNA from human serum. PCR Methods Appl. 1994a;3((5)):308–309. doi: 10.1101/gr.3.5.308. [DOI] [PubMed] [Google Scholar]
- Lai ME, Mazzoleni AP, Argiolu F, De Virgilis S, Balestrieri A, Purcell RH, Cao A, Farci P. Hepatitis C virus in multiple episodes of acute hepatitis in polytransfused thalassaemic children. Lancet. 1994b;343((8894)):388–390. doi: 10.1016/s0140-6736(94)91224-6. [DOI] [PubMed] [Google Scholar]
- Lauer GM, Nguyen TN, Day CL, Robbins GK, Flynn T, McGowan K, Rosenberg ES, Lucas M, Klenerman P, Chung RT, Walker BD. Human immunodeficiency virus type 1-hepatitis C virus coinfection: intraindividual comparison of cellular immune responses against two persistent viruses. J. Virol. 2002;76((6)):2817–2826. doi: 10.1128/JVI.76.6.2817-2826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner F, Wong DK, Dunbar PR, Chapman R, Chung RT, Dohrenwend P, Robbins G, Phillips R, Klenerman P, Walker BD. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 2000;191((9)):1499–1512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Prince AM. Automation of nucleic acid extraction for NAT screening of individual blood units. Transfusion. 2001;41((4)):483–487. doi: 10.1046/j.1537-2995.2001.41040483.x. [DOI] [PubMed] [Google Scholar]
- Linnen JM, Gilker JM, Menez A, Vaughn A, Broulik A, Dockter J, Gillotte-Taylor K, Greenbaum K, Kolk DP, Mimms LT, Giachetti C. Sensitive detection of genetic variants of HIV-1 and HCV with an HIV-1/HCV assay based on transcription-mediated amplification. J. Virol. Methods. 2002;102((1–2)):139–155. doi: 10.1016/s0166-0934(02)00012-5. [DOI] [PubMed] [Google Scholar]
- Major ME, Mihalik K, Puig M, Rehermann B, Nascimbeni M, Rice CM, Feinstone SM. Previously infected and recovered chimpanzees exhibit rapid responses that control hepatitis C virus replication upon rechallenge. J. Virol. 2002;76((13)):6586–6595. doi: 10.1128/JVI.76.13.6586-6595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel H, Reip A, Faltus B, Lu M, Porst H, Wiese M, Roggendorf M, Kruger DH. Transmission of hepatitis C virus to children and husbands by women infected with contaminated anti-D immunoglobulin. Lancet. 1995;345((8959)):1209–1211. doi: 10.1016/s0140-6736(95)91992-9. [DOI] [PubMed] [Google Scholar]
- Nascimbeni M, Mizukoshi E, Bosmann M, Major ME, Mihalik K, Rice CM, Feinstone SM, Rehermann B. Kinetics of CD4+ and CD8+ memory T-cell responses during hepatitis C virus rechallenge of previously recovered chimpanzees. J. Virol. 2003;77((8)):4781–4793. doi: 10.1128/JVI.77.8.4781-4793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DR, Marousis CG, Davis GL, Rice CM, Wong J, Houghton M, Lau JY. The role of hepatitis C virus-specific cytotoxic T lymphocytes in chronic hepatitis C. J. Immunol. 1997;158((3)):1473–1481. [PubMed] [Google Scholar]
- Pancholi P, Liu Q, Tricoche N, Zhang P, Perkus ME, Prince AM. DNA prime-canarypox boost with polycistronic hepatitis C virus (HCV) genes generates potent immune responses to HCV structural and nonstructural proteins. J. Infect. Dis. 2000;182((1)):18–27. doi: 10.1086/315646. [DOI] [PubMed] [Google Scholar]
- Panicali D, Davis SW, Mercer SR, Paoletti E. Two major DNA variants present in serially propagated stocks of the WR strain of vaccinia virus. J. Virol. 1981;37((3)):1000–1010. doi: 10.1128/jvi.37.3.1000-1010.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkus ME, Limbach K, Paoletti E. Cloning and expression of foreign genes in vaccinia virus, using a host range selection system. J. Virol. 1989;63((9)):3829–3836. doi: 10.1128/jvi.63.9.3829-3836.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince AM, Brotman B. The biology of hepatitis C virus infection. Lessons learned from chimpanzees. Curr. Stud. Hematol. Blood Transfus. 1994;61:195–207. doi: 10.1159/000423276. [DOI] [PubMed] [Google Scholar]
- Prince AM, Brotman B, Huima T, Pascual D, Jaffery M, Inchauspe G. Immunity in hepatitis C infection. J. Infect. Dis. 1992;165((3)):438–443. doi: 10.1093/infdis/165.3.438. [DOI] [PubMed] [Google Scholar]
- Prince AM, Shata MT. Immunoprophylaxis of hepatitis C virus infection. Clin. Liver Dis. 2001;5((4)):1091–1103. doi: 10.1016/s1089-3261(05)70211-7. [DOI] [PubMed] [Google Scholar]
- Rehermann B, Chang KM, McHutchinson J, Kokka R, Houghton M, Rice CM, Chisari FV. Differential cytotoxic T-lymphocyte responsiveness to the hepatitis B and C viruses in chronically infected patients. J. Virol. 1996;70((10)):7092–7102. doi: 10.1128/jvi.70.10.7092-7102.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice CM, Walker CM. Hepatitis C virus-specific T lymphocyte responses. Curr. Opin. Immunol. 1995;7((4)):532–538. doi: 10.1016/0952-7915(95)80099-9. [DOI] [PubMed] [Google Scholar]
- Rico MA, Quiroga JA, Subira D, Garcia E, Castanon S, Sallberg M, Leroux-Roels G, Weiland O, Pardo M, Carreno V. Features of the CD4+ T-cell response in liver and peripheral blood of hepatitis C virus-infected patients with persistently normal and abnormal alanine aminotransferase levels. J. Hepatol. 2002;36((3)):408–416. doi: 10.1016/s0168-8278(01)00281-1. [DOI] [PubMed] [Google Scholar]
- Rosen HR, Hinrichs DJ, Gretch DR, Koziel MJ, Chou S, Houghton M, Rabkin J, Corless CL, Bouwer HG. Association of multispecific CD4(+) response to hepatitis C and severity of recurrence after liver transplantation. Gastroenterology. 1999;117((4)):926–932. doi: 10.1016/s0016-5085(99)70352-5. [DOI] [PubMed] [Google Scholar]
- Schirren CA, Jung MC, Gerlach JT, Worzfeld T, Baretton G, Mamin M, Hubert Gruener N, Houghton M, Pape GR. Liver-derived hepatitis C virus (HCV)-specific CD4(+) T cells recognize multiple HCV epitopes and produce interferon gamma. Hepatology. 2000;32((3)):597–603. doi: 10.1053/jhep.2000.9635. [DOI] [PubMed] [Google Scholar]
- Scognamiglio P, Accapezzato D, Casciaro MA, Cacciani A, Artini M, Bruno G, Chircu ML, Sidney J, Southwood S, Abrignani S, Sette A, Barnaba V. Presence of effector CD8+ T cells in hepatitis C virus-exposed healthy seronegative donors. J. Immunol. 1999;162((11)):6681–6689. [PubMed] [Google Scholar]
- Seeff LB, Hollinger FB, Alter HJ, Wright EC, Cain CM, Buskell ZJ, Ishak KG, Iber FL, Toro D, Samanta A, Koretz RL, Perrillo RP, Goodman ZD, Knodell RG, Gitnick G, Morgan TR, Schiff ER, Lasky S, Stevens C, Vlahcevic RZ, Weinshel E, Tanwandee T, Lin HJ, Barbosa L. Long-term mortality and morbidity of transfusion-associated non-A, non-B, and type C hepatitis: a National Heart, Lung, and Blood Institute collaborative study. Hepatology. 2001;33((2)):455–463. doi: 10.1053/jhep.2001.21905. [DOI] [PubMed] [Google Scholar]
- Shata MT, Anthony DD, Carlson NL, Andrus L, Brotman B, Tricoche N, McCormack P, Prince A. Characterization of the immune response against hepatitis C infection in recovered, and chronically infected chimpanzees. J. Viral Hepat. 2002;9((6)):400–410. doi: 10.1046/j.1365-2893.2002.00373.x. [DOI] [PubMed] [Google Scholar]
- Takaki A, Wiese M, Maertens G, Depla E, Seifert U, Liebetrau A, Miller JL, Manns MP, Rehermann B. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat. Med. 2000;6((5)):578–582. doi: 10.1038/75063. [DOI] [PubMed] [Google Scholar]
- Takamizawa A, Mori C, Fuke I, Manabe S, Murakami S, Fujita J, Onishi E, Andoh T, Yoshida I, Okayama H. Structure and organization of the hepatitis C virus genome isolated from human carriers. J. Virol. 1991;65((3)):1105–1113. doi: 10.1128/jvi.65.3.1105-1113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Katsume A, Tanaka T, Abe A, Inoue K, TsukiyamaKohara K, Kawaguchi R, Tanaka S, Kohara M. Real-time detection system for quantification of hepatitis C virus genome [see comments] Gastroenterology. 1999;116((3)):636–642. doi: 10.1016/s0016-5085(99)70185-x. [DOI] [PubMed] [Google Scholar]
- Thomas DL, Vlahov D, Solomon L, Cohn S, Taylor E, Garfein R, Nelson KE. Correlates of hepatitis C virus infections among injection drug users. Medicine (Baltimore) 1995;74((4)):212–220. doi: 10.1097/00005792-199507000-00005. [DOI] [PubMed] [Google Scholar]
- Tobery TW, Wang S, Wang XM, Neeper MP, Jansen KU, McClements WL, Caulfield MJ. A simple and efficient method for the monitoring of antigen-specific T cell responses using peptide pool arrays in a modified ELISpot assay. J. Immunol. Methods. 2001;254((1–2)):59–66. doi: 10.1016/s0022-1759(01)00397-0. [DOI] [PubMed] [Google Scholar]
- Tsai SL, Liaw YF, Chen MH, Huang CY, Kuo GC. Detection of type 2-like T-helper cells in hepatitis C virus infection: implications for hepatitis C virus chronicity. Hepatology. 1997;25((2)):449–458. doi: 10.1002/hep.510250233. [DOI] [PubMed] [Google Scholar]
- Valdez H, Anthony D, Farukhi F, Patki A, Salkowitz J, Heeger P, Peterson DL, Post AB, Asaad R, Lederman MM. Immune responses to hepatitis C and non-hepatitis C antigens in hepatitis C virus infected and HIV-1 coinfected patients. AIDS. 2000;14((15)):2239–2246. doi: 10.1097/00002030-200010200-00004. [DOI] [PubMed] [Google Scholar]
- Wang H, Bian T, Merrill SJ, Eckels DD. Sequence variation in the gene encoding the nonstructural 3 protein of hepatitis C virus: evidence for immune selection. J. Mol. Evol. 2002;54((4)):465–473. doi: 10.1007/s00239-001-0037-6. [DOI] [PubMed] [Google Scholar]
- Wedemeyer H, Mizukoshi E, Davis AR, Bennink JR, Rehermann B. Cross-reactivity between hepatitis C virus and influenza A virus determinant-specific cytotoxic T cells. J. Virol. 2001;75((23)):11392–11400. doi: 10.1128/JVI.75.23.11392-11400.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh RM, Selin LK. No one is naive: the significance of heterologous T-cell immunity. Nat. Rev. Immunol. 2002;2((6)):417–426. doi: 10.1038/nri820. [DOI] [PubMed] [Google Scholar]
- Wentworth PA, Sette A, Celis E, Sidney J, Southwood S, Crimi C, Stitely S, Keogh E, Wong NC, Livingston B, Alazard D, Vitiello A, Grey HM, Chisari FV, Chesnut RW, Fikes J. Identification of A2-restricted hepatitis C virus-specific cytotoxic T lymphocyte epitopes from conserved regions of the viral genome. Int. Immunol. 1996;8((5)):651–659. doi: 10.1093/intimm/8.5.651. [DOI] [PubMed] [Google Scholar]
- Wong DK, Dudley DD, Afdhal NH, Dienstag J, Rice CM, Wang L, Houghton M, Walker BD, Koziel MJ. Liver-derived CTL in hepatitis C virus infection: breadth and specificity of responses in a cohort of persons with chronic infection. J. Immunol. 1998;160((3)):1479–1488. [PubMed] [Google Scholar]
- Wong DK, Dudley DD, Dohrenwend PB, Lauer GM, Chung RT, Thomas DL, Walker BD. Detection of diverse hepatitis C virus (HCV)-specific cytotoxic T lymphocytes in peripheral blood of infected persons by screening for responses to all translated proteins of HCV. J. Virol. 2001;75((3)):1229–1235. doi: 10.1128/JVI.75.3.1229-1235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen L, Moss B. Oligonucleotide sequence signaling transcriptional termination of vaccinia virus early genes. Proc. Natl. Acad. Sci. USA. 1987;84((18)):6417–6421. doi: 10.1073/pnas.84.18.6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N, Khoshnan A, Schneider R, Matsumoto M, Dennert G, Ware C, Lai MM. Hepatitis C virus core protein binds to the cytoplasmic domain of tumor necrosis factor (TNF) receptor 1 and enhances TNF-induced apoptosis. J. Virol. 1998;72((5)):3691–3697. doi: 10.1128/jvi.72.5.3691-3697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]