

Abstract
Background:
Hereditary tyrosinemia type 1 (HT1) is a rare autosomal recessive inborn error of metabolism caused by deficiency of fumarylacetoacetate hydrolase enzyme. This disease manifests with severe liver and kidney impairment and is associated with an increased risk of liver cancer. The aim of this study was to evaluate clinical, laboratory, imaging, and histopathologic characteristics in the children with HT1 who had referred for liver transplantation.
Methods:
The present retrospective study was conducted on 45 children with HT1 who had referred to Organ Transplantation Center affiliated to Shiraz University of Medical Sciences between March 2005 and March 2010.
Results:
There were 64.4% boys and 35.6% girls with mean age of 3.75±1.28 year (ranges from 2 months to 13 years). The most first clinical presentation was hepatic (80%) and the most prevalent physical findings were hepatomegaly (57.8%), splenomegaly (51.1%), ascites (42.2%), and jaundice (37.9%). The most relevant laboratory parameters were the high serum succinylacetone, alpha-fetoprotein, and tyrosine levels. The most common findings in the patient's abdominal ultrasonography were multiple hepatic nodules (75.6%) and inhomogeneous parenchymal echogenicity of liver (48.9%), while hyper and hypo attenuated nodules (60%) and non-homogeneous pattern of liver parenchyma (53.3%) were the most prevalent findings in abdominal computed tomography scan. In the histopathology of the liver, the most important finding was cirrhosis in all the patients. In this study, 14 patients (31.1%) received Nitisinone (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyklohexanedione; NTBC).
Conclusions:
This study described clinical and laboratory findings in the children with HT1 who had referred for liver transplantation because of end-stage liver disease from all over country, which indicates delay in diagnosis and treatment of this disease. Considering the results of this study, newborn screening for this disease is highly suggested.
Keywords: Liver transplantation, newborn screening, succinylacetone, tyrosinemia
INTRODUCTION
Hereditary tyrosinemia type 1 (HT1) is an autosomal recessive inborn error of metabolism caused by deficiency of fumarylacetoacetate hydrolase (FAH). The FAH enzyme is expressed mainly in the liver (90%) and the kidney (10%).[1] The prevalence of HT1 is low throughout the world and is estimated to be no greater than 1 in 100,000 live births with several exceptions. However, the prevalence of the disease is high in some regions of Finland and Quebec (1:63,000 and 1:2,000, respectively) due to founder allele effect.[2] The marked elevation of plasma tyrosine and increased urinary excretion of succinylacetone (SA), maleylacetoacetate, and fumarylacetoacetate cause liver impairment and renal-tubular dysfunction.[3] SA is a potent inhibitor of the δ-aminolevulinic acid dehydratase step in porphyrin synthesis, causing the porphyria-like neurological crises.[4]
HT1 has various clinical manifestations and the affected individual can present at any age from the neonatal period to adulthood. There is a considerable variability of presentation even between the members of the same family.[5,6] The reason of milder phenotype is probably self-induced correction of the fumarylacetoacetase defect in hepatocytes.[7,8,9,10,11] Clinically, HT1 may be classified based on the age at the onset of the symptoms, which broadly correlates with disease severity: An acute form manifests before the age of 6 months with acute liver failure; a subacute form presents between 6 months and 1 year of age with liver disease, failure to thrive, coagulopathy, hepatosplenomegaly, rickets, and hypotonia; and a chronic form presents after the first year of life with chronic liver disease, renal disease, rickets, cardiomyopathy and/or porphyria-like syndrome.[12,13,14,15]
Increased urinary excretion of SA and δ-aminolevulinic acid together with elevated plasma tyrosine level are pathognomonic of HT1.[16,17]
Historically, HT1 was treated with a tyrosine and phenylalanine restricted diet, with or without liver transplantation. In 1992, a new drug Orfadin®, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3- cyklohexanedione (NTBC, Nitisinone), which is a potent inhibitor of 4-hydroxyphenylpyruvate dioxygenase was introduced; it has revolutionized the treatment of HT1 and is now the mainstay of therapy.[18,19,20]
The aim of this study is to present a detailed clinical and laboratory features of 45 Iranian children with HT1 who had referred for liver transplantation.
METHODS
The medical records of all the children less than 14 year who had been diagnosed and treated as HT1 and referred to Organ Transplantation Center of Nemazee Hospital affiliated to Shiraz University of Medical Sciences between March 2005 and March 2010 were reviewed, retrospectively. The diagnosis was established according to the clinical manifestations, high serum alpha-fetoprotein (AFP) levels, and positive serum SA.
Overall, 45 children and adolescents with diagnosis of HT1 were enrolled in to the present study.
The clinical signs and symptoms of the patients at the times of presentation and referral to this center were retrieved from their medical files. Furthermore, the paraclinical tests, including serum levels of alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase (AP), total and direct bilirubin, plasma proteins, serum calcium and phosphor, AFP, serum tyrosine level, and serum SA were extracted from their medical files. Tyrosine in plasma was analyzed by ion exchange chromatography on automatic amino acids analyzer 400 (Ingo's, Czech Republic) with ninhydrin detection at 440 nm and at 570 nm for the amino acids with secondary amino group. Amino acids were identified by the retention time. Moreover, AFP concentration was measured by a UniCel DxI 800 Access immunoanalyzer (Beckman Coulter, Inc., Fullerton, CA). Besides, the results of abdominal ultrasonography and computed tomography scan, electrocardiograms and echocardiographic findings, and histopathologic examinations of the liver biopsies that were carried out as a part of pre-transplant evaluations were reviewed in all patients.
RESULTS
Among the 45 patients, 29 were male (64.4%) and 16 (35.6%) were female. The mean age of the patients was 17.8 ± 10.3 months (range, 1 month to 10 years) at the first clinical presentation and 3.75 ± 1.28 years (range, 2 months to 13 years) at the time of diagnosis. The first presentations of the disease were hepatic disorders in 36 children (80%), rickets in 4 (8.9%), neurologic in 3 (6.7%), and renal in 2 patients (4.4%). The most common clinical presentations were jaundice (n = 17; 37.9%), vomiting (n = 12; 26.7%), irritability (n = 8; 17.8%), anorexia (n = 8; 17.8%), polyuria (n = 8; 17.8%), weakness (n = 7; 15.6%), and convulsion (n = 4; 8.9%). Other clinical presentations are shown in Table 1. The patients’ families had no history of the disease. The physical examination of the patients at the time of referral to this center revealed hepatomegaly in 26 patients (57.8%), splenomegaly in 23 (51.1%), ascites in 19 (42.2%), and jaundice in 17 patients (37.8%) [Table 2].
Table 1.
The clinical symptoms at the time of presentation in 45 children with hereditary tyrosinemia type 1 who referred for liver transplantation

Table 2.
The clinical findings at the time of referral in 45 children with hereditary tyrosinemia type 1 who referred for liver transplantation

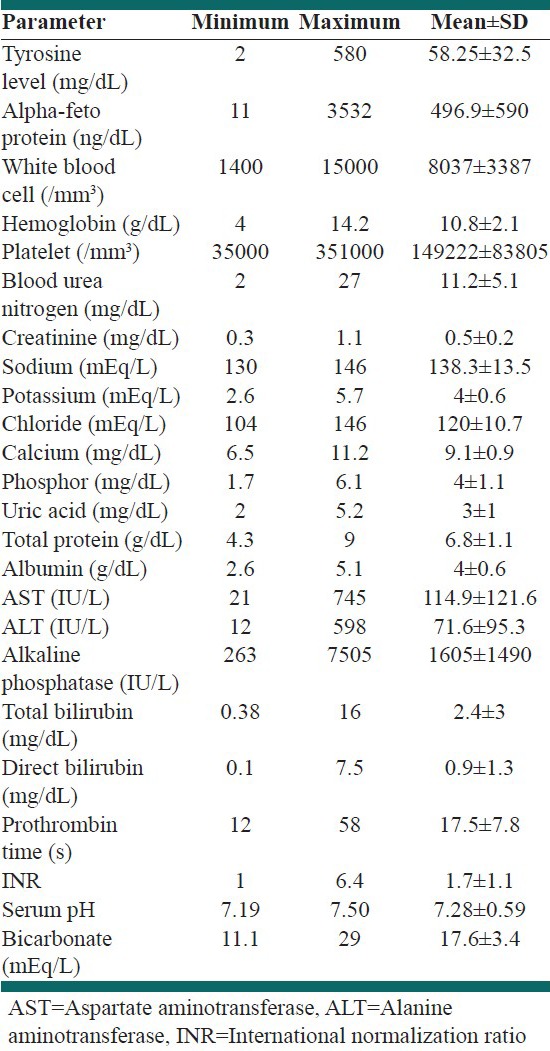
The laboratory findings are listed in Table 3. The most common paraclinical findings were positive serum SA and high serum AFP levels, which were seen in all the patients. Coagulopathy with international normalization ratio more than 2 was seen in 31 patients (68.9%). Seventeen patients (37.8%) had evidences of rickets with low serum phosphor and high AP, which in 4 of them rickets were the first presentation. The mean blood urea nitrogen and serum creatinine levels were 11.2 ± 5.1 mg/dL, and 0.5 ± 0.2 mg/dL, respectively. Furthermore, the mean values for serum sodium, potassium, chloride, calcium, phosphor, and uric acid were 138.3 ± 13.5 mEq/L, 4 ± 0.6 mEq/L, 120 ± 10.7 mEq/L, 9.1 ± 0.9 mg/dL, 4 ± 1.1 mg/dL, and 3 ± 1 mg/dL, respectively. The mean serum pH was 7.28 ± 0.59 and the mean serum bicarbonate level was 17.6 ± 3.4 mEq/L. Eight (17.8%) patients had evidence of renal tubular acidosis, which was the first presentation in two patients. Furthermore, evaluation of electrocardiograms in the 45 children with HT1 showed high voltage QRS in one child and T-inversion in another one. The echocardiographic findings included small atrial septal defect in two patients, large ventricular septal defect, mild septal hypertrophy, and small patent foramen ovale, each in one patient.
Table 3.
The laboratory data in 45 children with hereditary tyrosinemia type 1 who referred for liver transplantation
Abdominal ultrasonography of the patients showed hepatic nodules in 34 patients (75.6%), inhomogeneous hepatic parenchyma in 22 (48.9%), nephromegaly in 16 (35.6%), nephrocalcinosis in 3 (6.7%), and increased echogenicity of renal cortical parenchyma in 3 patients (6.7%). The most common findings on abdominal computed tomography scan were hyper and hypo attenuated nodules in 27 patients (60%), non-homogenous hepatic pattern in 24 (53.3%), and enlarged kidneys in 10 patients (22.2%). The histopathological findings of the liver biopsies were in favor of cirrhosis in all cases. Fourteen patients (31.1%) received Nitisinone (NTBC).
DISCUSSION
Hepatorenal tyrosinemia is characterized by early presentation with liver and kidney involvement,[21,22] and neuropathic crisis[23] in the absolute majority of patients. The mean age of our patients at the time of diagnosis was 3.7 year, which can be due to the fact that this metabolic disease is not investigated as a part of newborn screening in our country and most of the patients are diagnosed at the late stages of the disease. Paradis et al. also evaluated 16 children (7 boys and 9 girls) with HT1 with mean age of 4.3 year for possible liver transplantation, which in line with our results.[24] The current study was conducted on 29 boys and 16 girls with HT1. The disease has an autosomal recessive trait, so this result can be supposed as a random data.
The first presentations of the disease in our patients were hepatic in 80% followed by rickets in 8.9%, neurologic in 6.7%, and renal disorders in 4.4% of cases. In the study by El-Karaksy et al. on 22 children with HT1, 72.7% of the patients presented with hepatic manifestations and 22.7% with rickets.[25] El-Karaksy et al. reported clinical rickets in 63.6% of the patients; however, in our series 37.8% of the patients had rickets. Santra et al. evaluated 21 children with HT1 in the early stages of the disease with mean age at the time of starting treatment of 17 weeks. Out of these 21 patients, 9 (43%) cases presented with acute liver failure, 7 (33%) patients with chronic liver failure, and 5 (24%) cases diagnosed by screening test pre-symptomatically. In Santra et al. study all patients had evidences of renal tubular dysfunction and 4 (19%) of them had rickets that improved with Nitisinone treatment. Only one patient had nephrocalcinosis that not improved with treatment.[26]
The most common clinical features of the patients with HT1 were failure to thrive, irritability, lethargy, fever, vomiting, diarrhea, abdominal distension, hepatomegaly, edema, and coagulopathy.[27] In the present study, the most common clinical manifestations were jaundice, vomiting, irritability, weakness, and convulsion. Besides, hepatomegaly and splenomegaly were observed in 57.8% and 51.1% of the patients, respectively. Other signs were ascites in 42.2% and jaundice in 37.8% of the patients. In El-Karaksy study, hepatosplenomegaly was seen in 95.5%, jaundice in 36.4%, edema in 9.1%, and ascites in 4.5% of the patients.[25]
Renal rickets associated with hypophosphatemia and hyperphosphaturia is another important feature of HT1 that is due to reduced tubular reabsorption of phosphorus. Other signs of renal dysfunction are proteinuria, hyper aminoaciduria, and glucosuria.[27] In the present study, eight (17.8%) patients had evidence of renal tubular acidosis, which is in contrast to 100% in the study by Santra et al.[26]
In the acute stages, the patients show hypoproteinemia, hyperbilirubinemia, deterioration of liver function, and occasionally, polyneuropathic pain. Leukopenia and thrombocytopenia are usually present in the chronic phases. Serum transaminases are increased, particularly in the acute hepatic episodes, and serum AFP level is raised both in the acute and chronic stages, especially, with the development of hepatocellular carcinoma.[27] All the patients in the present study had elevated AFP as reported by El-Karaksy et al.[25] In a study on 11 Czech patients with HT1, the biochemical investigation revealed elevated liver enzymes and AFP and hypophosphatemic rickets in all cases. In addition, the metabolic investigation showed increased plasma tyrosine level, and urinary excretion of SA.[28]
Regarding findings of ultrasonography and computed tomography scan of the abdomen, hepatic nodules were seen in 75.6% and non-homogenous hepatic parenchyma in 48.9% of our patients. Paradis et al. reported non-homogenous hepatic parenchyma in 75% of the patients, hepatomegaly in 63%, hepatic nodules in 63%, and small liver in 18.7% of the patients.[24] On the other hand, El-Karaksy et al. reported 95% hepatomegaly, 72.7% non-homogenous hepatic parenchyma, and 45% focal lesions in liver.[25] Regarding kidney imaging, nephromegaly was seen in 35.6%, nephrocalcinosis in 6.7%, and increased echogenicity of the renal cortical parenchyma in 6.7% of the patients in our study, which comparable with 36.4% nephromegaly in El-Karaksy series, but they reported increased echogenicity of the renal cortical parenchyma in 54.5% of the patients, which was higher than ours.[25]
In this study, liver biopsy for all the patients showed liver cirrhosis, which can be acceptable because all the patients were candidate for liver transplantation.
The plasma tyrosine level has less diagnostic accuracy than the elevated concentration of SA in the serum or urine.[27] In neonatal screening, by measuring the blood tyrosine level, only a minority of patients with HT1 are identified. Thus, a more reliable way to identify these patients would be measuring fumarylacetoacetase using ELISA[29] or determining SA using ultra performance liquid chromatography-tandem mass spectrometry UPLC-MS/MS in dried blood spots from newborns.[30] If measuring SA and/or fumarylacetoacetase becomes a part of the newborn screening program, it might lead to an improved prognosis of the patients with HT1.
Although, the prognosis is improved dramatically under combined dietary and pharmacological treatment with Nitisinone, the patients are at risk of liver cancer with the necessity of liver transplantation.[31,32] In the present study, less than one-third of patients were treated with Nitisinone and the response to Nitisinone treatment was apparent by a steep drop in serum AFP and undetectable SA in urine within 2 months.[25]
This study has one important limitation because all the patients were referred for liver transplantation due to cirrhosis, so we evaluated the cases at the end stages of the disease.
The only possibility for decreasing the risk of complications in the patients with HT1 is early diagnosis in the extended newborn screening of hereditary metabolic diseases. HT1 diagnosis was possible only at the biochemical level without the possibility of genetic counseling and prenatal diagnosis until 2008.[33] At present, however, the molecular-genetic analysis is available.
CONCLUSIONS
We concluded that there is a delay in diagnosis and treatment of HT1 in our patients, so the newborn screening for this disease is highly suggested. It is hoped that by metabolic and molecular-genetic screening of the disease in newborns, HT1 be detected in the early stages and therefore, quality of life of these patients be improved with available treatments like Nitisinone.
ACKNOWLEDGMENTS
The present article was extracted from the thesis written by Hossein Karamnejad and was financially supported by Shiraz University of Medical Sciences grants No 89-5118.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Lindblad B, Lindstedt S, Steen G. On the enzymic defects in hereditary tyrosinemia. Proc Natl Acad Sci U S A. 1977;74:4641–5. doi: 10.1073/pnas.74.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Braekeleer M, Larochelle J. Genetic epidemiology of hereditary tyrosinemia in Quebec and in Saguenay-Lac-St-Jean. Am J Hum Genet. 1990;47:302–7. [PMC free article] [PubMed] [Google Scholar]
- 3.Kubo S, Sun M, Miyahara M, Umeyama K, Urakami K, Yamamoto T, et al. Hepatocyte injury in tyrosinemia type 1 is induced by fumarylacetoacetate and is inhibited by caspase inhibitors. Proc Natl Acad Sci U S A. 1998;95:9552–7. doi: 10.1073/pnas.95.16.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russo PA, Mitchell GA, Tanguay RM. Tyrosinemia: A review. Pediatr Dev Pathol. 2001;4:212–21. doi: 10.1007/s100240010146. [DOI] [PubMed] [Google Scholar]
- 5.Poudrier J, Lettre F, Scriver CR, Larochelle J, Tanguay RM. Different clinical forms of hereditary tyrosinemia (type I) in patients with identical genotypes. Mol Genet Metab. 1998;64:119–25. doi: 10.1006/mgme.1998.2695. [DOI] [PubMed] [Google Scholar]
- 6.Poudrier J, Lettre F, St-Louis M, Tanguay RM. Genotyping of a case of tyrosinaemia type I with normal level of succinylacetone in amniotic fluid. Prenat Diagn. 1999;19:61–3. doi: 10.1002/(sici)1097-0223(199901)19:1<61::aid-pd455>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 7.Phaneuf D, Lambert M, Laframboise R, Mitchell G, Lettre F, Tanguay RM. Type 1 hereditary tyrosinemia. Evidence for molecular heterogeneity and identification of a causal mutation in a French Canadian patient. J Clin Invest. 1992;90:1185–92. doi: 10.1172/JCI115979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kvittingen EA, Rootwelt H, Berger R, Brandtzaeg P. Self-induced correction of the genetic defect in tyrosinemia type I. J Clin Invest. 1994;94:1657–61. doi: 10.1172/JCI117509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kvittingen EA, Rootwelt H, Brandtzaeg P, Bergan A, Berger R. Hereditary tyrosinemia type I. Self-induced correction of the fumarylacetoacetase defect. J Clin Invest. 1993;91:1816–21. doi: 10.1172/JCI116393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jonkman MF. Revertant mosaicism in human genetic disorders. Am J Med Genet. 1999;85:361–4. doi: 10.1002/(sici)1096-8628(19990806)85:4<361::aid-ajmg11>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 11.Demers SI, Russo P, Lettre F, Tanguay RM. Frequent mutation reversion inversely correlates with clinical severity in a genetic liver disease, hereditary tyrosinemia. Hum Pathol. 2003;34:1313–20. doi: 10.1016/s0046-8177(03)00406-4. [DOI] [PubMed] [Google Scholar]
- 12.Noble-Jamieson G, Jamieson N, Clayton P, Bailey S, Ryalls M, Barnes ND. Neurological crisis in hereditary tyrosinaemia and complete reversal after liver transplantation. Arch Dis Child. 1994;70:544–5. doi: 10.1136/adc.70.6.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forget S, Patriquin HB, Dubois J, Lafortune M, Merouani A, Paradis K, et al. The kidney in children with tyrosinemia: Sonographic, CT and biochemical findings. Pediatr Radiol. 1999;29:104–8. doi: 10.1007/s002470050551. [DOI] [PubMed] [Google Scholar]
- 14.Mention K, Lahoche-Manucci A, Bonnevalle M, Pruvot FR, Declerck N, Foulard M, et al. Renal function outcome in pediatric liver transplant recipients. Pediatr Transplant. 2005;9:201–7. doi: 10.1111/j.1399-3046.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 15.Arora N, Stumper O, Wright J, Kelly DA, McKiernan PJ. Cardiomyopathy in tyrosinaemia type I is common but usually benign. J Inherit Metab Dis. 2006;29:54–7. doi: 10.1007/s10545-006-0203-5. [DOI] [PubMed] [Google Scholar]
- 16.Matern D, Tortorelli S, Oglesbee D, Gavrilov D, Rinaldo P. Reduction of the false-positive rate in newborn screening by implementation of MS/MS-based second-tier tests: The mayo clinic experience (2004-2007) J Inherit Metab Dis. 2007;30:585–92. doi: 10.1007/s10545-007-0691-y. [DOI] [PubMed] [Google Scholar]
- 17.la Marca G, Malvagia S, Pasquini E, Innocenti M, Fernandez MR, Donati MA, et al. The inclusion of succinylacetone as marker for tyrosinemia type I in expanded newborn screening programs. Rapid Commun Mass Spectrom. 2008;22:812–8. doi: 10.1002/rcm.3428. [DOI] [PubMed] [Google Scholar]
- 18.van Spronsen FJ, Bijleveld CM, van Maldegem BT, Wijburg FA. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifluoro-methylbenzoyl)-1, 3-cyclohexanedione treatment. J Pediatr Gastroenterol Nutr. 2005;40:90–3. doi: 10.1097/00005176-200501000-00017. [DOI] [PubMed] [Google Scholar]
- 19.Lindstedt S, Holme E, Lock EA, Hjalmarson O, Strandvik B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet. 1992;340:813–7. doi: 10.1016/0140-6736(92)92685-9. [DOI] [PubMed] [Google Scholar]
- 20.Lock EA, Ellis MK, Gaskin P, Robinson M, Auton TR, Provan WM, et al. From toxicological problem to therapeutic use: The discovery of the mode of action of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3- cyclohexanedione (NTBC), its toxicology and development as a drug. J Inherit Metab Dis. 1998;21:498–506. doi: 10.1023/a:1005458703363. [DOI] [PubMed] [Google Scholar]
- 21.Russo P, O’Regan S. Visceral pathology of hereditary tyrosinemia type I. Am J Hum Genet. 1990;47:317–24. [PMC free article] [PubMed] [Google Scholar]
- 22.van Spronsen FJ, Thomasse Y, Smit GP, Leonard JV, Clayton PT, Fidler V, et al. Hereditary tyrosinemia type I: A new clinical classification with difference in prognosis on dietary treatment. Hepatology. 1994;20:1187–91. [PubMed] [Google Scholar]
- 23.Mitchell G, Larochelle J, Lambert M, Michaud J, Grenier A, Ogier H, et al. Neurologic crises in hereditary tyrosinemia. N Engl J Med. 1990;322:432–7. doi: 10.1056/NEJM199002153220704. [DOI] [PubMed] [Google Scholar]
- 24.Paradis K, Weber A, Seidman EG, Larochelle J, Garel L, Lenaerts C, et al. Liver transplantation for hereditary tyrosinemia: The Quebec experience. Am J Hum Genet. 1990;47:338–42. [PMC free article] [PubMed] [Google Scholar]
- 25.El-Karaksy H, Fahmy M, El-Raziky M, El-Koofy N, El-Sayed R, Rashed MS, et al. Hereditary tyrosinemia type 1 from a single center in Egypt: Clinical study of 22 cases. World J Pediatr. 2011;7:224–231. doi: 10.1007/s12519-011-0287-3. [DOI] [PubMed] [Google Scholar]
- 26.Santra S, Preece MA, Hulton SA, McKiernan PJ. Renal tubular function in children with tyrosinaemia type I treated with nitisinone. J Inherit Metab Dis. 2008;31:399–402. doi: 10.1007/s10545-008-0817-x. [DOI] [PubMed] [Google Scholar]
- 27.Kitagawa T. Hepatorenal tyrosinemia. Proc Jpn Acad Ser B Phys Biol Sci. 2012;88:192–200. doi: 10.2183/pjab.88.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vondrácková A, Tesarová M, Magner M, Docekalová D, Chrastina P, Procházkova D, et al. Clinical, biochemical and molecular characteristics in 11 Czech children with tyrosinemia type I. Cas Lek Cesk. 2010;149:411–6. [PubMed] [Google Scholar]
- 29.Laberge C, Grenier A, Valet JP, Morissette J. Fumarylacetoacetase measurement as a mass-screening procedure for hereditary tyrosinemia type I. Am J Hum Genet. 1990;47:325–8. [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Dirbashi OY, Rashed MS, Jacob M, Al-Ahaideb LY, Al-Amoudi M, Rahbeeni Z, et al. Improved method to determine succinylacetone in dried blood spots for diagnosis of tyrosinemia type 1 using UPLC-MS/MS. Biomed Chromatogr. 2008;22:1181–5. doi: 10.1002/bmc.1049. [DOI] [PubMed] [Google Scholar]
- 31.Arnon R, Annunziato R, Miloh T, Wasserstein M, Sogawa H, Wilson M, et al. Liver transplantation for hereditary tyrosinemia type I: Analysis of the UNOS database. Pediatr Transplant. 2011;15:400–5. doi: 10.1111/j.1399-3046.2011.01497.x. [DOI] [PubMed] [Google Scholar]
- 32.Moini M, Mistry P, Schilsky ML. Liver transplantation for inherited metabolic disorders of the liver. Curr Opin Organ Transplant. 2010;15:269–76. doi: 10.1097/MOT.0b013e3283399dbd. [DOI] [PubMed] [Google Scholar]
- 33.Schlump JU, Mayatepek E, Spiekerkoetter U. Significant increase of succinylacetone within the first 12 h of life in hereditary tyrosinemia type 1. Eur J Pediatr. 2010;169:569–72. doi: 10.1007/s00431-009-1074-1. [DOI] [PubMed] [Google Scholar]