

Summary
Mutation is a fundamental process in tumorigenesis. However, the degree to which the rate of somatic mutation varies across the human genome and the mechanistic basis underlying this variation remain to be fully elucidated. Here, we performed a cross-cancer comparison of 402 whole genomes comprising a diverse set of childhood and adult tumors, including both solid and hematopoietic malignancies. Surprisingly, we found that the inactive X chromosome of many female cancer genomes accumulates on average twice and up to four times as many somatic mutations per megabase, as compared to the individual autosomes. Whole-genome sequencing of clonally expanded hematopoietic stem/progenitor cells (HSPCs) from healthy individuals and a premalignant myelodysplastic syndrome (MDS) sample revealed no X chromosome hypermutation. Our data suggest that hypermutation of the inactive X chromosome is an early and frequent feature of tumorigenesis resulting from DNA replication stress in aberrantly proliferating cells.
Graphical Abstract

Highlights
-
•
X chromosome has up to 4× more mutations than the autosomes in female cancer genomes
-
•
Hypermutations only affect the inactive X chromosome
-
•
X hypermutation involves somatic point mutations and indels, but not germline mutations
-
•
No X hypermutation is found in clonal expansions of normal or premalignant cells
A comparison of 402 cancer genomes identifies a surprisingly high level of somatic mutations in the inactive X chromosome of female cancer genomes. As hypermutability of the inactive X was not observed in clonal hematopoietic progenitor or preleukemic samples, it is likely that it may be a contributing factor to tumorigenesis.
Introduction
The process of somatic mutation is fundamental to cancer development. A number of causes for these mutations have been described, including intrinsic mutation processes such as damage from endogenous reactive oxygen species or incomplete fidelity of the DNA replication machinery and extrinsic factors such as environmental and lifestyle exposures. For example, UV light and tobacco exposure are both well-known factors adding to the mutational burden of somatic cells (Stratton et al., 2009).
Human germline mutation rates are not constant across the genome, varying with factors such as base composition and transcription levels (Hodgkinson and Eyre-Walker, 2011; Ellegren et al., 2003). It is also known that the X chromosome typically shows reduced variation compared with the autosomes (Malcom et al., 2003). Only recently, however, have some studies elucidated the existence of variation in genome-wide somatic mutation rates and potential causes thereof. The mutation rate varies within a cancer genome according to underlying genomic features such as GC content, CpG islands, and recombination rate (Greenman et al., 2007). Regions that are actively transcribed have mutation rates at least 25% lower than nontranscribed regions (Chapman et al., 2011) due to mechanisms of transcription-coupled repair. Chromatin organization, specifically the level of heterochromatin-associated histone modification H3K9me3, has been reported to account for more than 40% of mutation-rate variation (Schuster-Böckler and Lehner, 2012). Late-replicating regions also have a higher mutation rate than early-replicating regions in cancer as well as in the germline (Liu et al., 2013; Stamatoyannopoulos et al., 2009).
The inactive X chromosome (Xi) is one of the latest replicating regions of the human genome, being replicated distinctly later in S phase than the autosomes and its active X counterpart (Xa; Hansen et al., 1996; Morishima et al., 1962). In contrast to the autosomes, for which two active copies are present, both male and female cells carry only one active X chromosome. In mammals, dosage compensation between male and female cells is achieved by inactivating one of the two female X chromosomes (Chow and Heard, 2009; Lyon, 1961). This results in transcriptional silencing of most of the ∼1,500 genes located on the human X chromosome, although about 3%–15% of genes are known to escape X chromosome inactivation (XCI), depending on cell type (Carrel and Willard, 2005). XCI is initiated very early in embryonic stem cell differentiation and is characterized by a stochastic choice of the X chromosome subjected to inactivation (Barakat and Gribnau, 2012). The chosen inactivated copy (Xi) is then stably maintained through all subsequent cell divisions. The transcription of X-inactive-specific transcript (XIST) RNA, a 17 kb spliced and polyadenylated RNA with no coding capacity, is monoallelically upregulated at the onset of XCI and associates with the future Xi in cis (Brown et al., 1992). This XIST coating of the Xi provides the template for a series of histone modifications, including histone-H3 lysine 9 and 27 methylation and histone-H4 deacetylation and macroH2A accumulation, ultimately leading to heterochromatin formation (Plath et al., 2002). After XCI, XIST is expressed continuously and exclusively from the inactive copy of the X chromosome.
In this study, we performed a cross-cancer analysis based on 402 whole-cancer genomes, including our own published and new cancer genome data sets from six different entities (medulloblastoma [Jones et al., 2012; M.K., D.T.W.J., N.J., P.A.N., M.D.T., R.E., S.M.P., and P.L., unpublished data], pilocytic astrocytoma [Jones et al., 2013], glioblastoma [S.M.P., M.K., D.T.W.J, P.A.N., M.D.T., R.E., P.L., and A.K., unpublished data], ependymoma [S.C.M., H.W., P.A.N., D.T.W.J., N.J., S.M.P., and M.D.T., unpublished data], B cell lymphoma [Richter et al., 2012; M.S., J.R., M.H., P.L., R.E., and R.S., unpublished data], and prostate carcinoma [Weischenfeldt et al., 2013]), in addition to published mutation call sets of six different cancer types: breast cancer (Nik-Zainal et al., 2012), neuroblastoma (Molenaar et al., 2012), chronic lymphocytic leukemia (CLL, Puente et al., 2011), acute myeloid leukemia (AML, Welch et al., 2012), colorectal carcinoma (Bass et al., 2011), and retinoblastoma (Zhang et al., 2012).
In many female cancer genomes, we unexpectedly found hypermutation of the X chromosome—i.e., a clearly elevated density of mutations compared with the individual autosomes. We show that this hypermutation of the X chromosome is confined to the inactive X chromosome and involves single-nucleotide variants (SNVs) as well as small insertions and deletions (indels), which both show a marked increase in mutations along the X chromosome. Whole-genome sequencing of three independent clonal expansions of healthy hematopoietic stem/progenitor cells and one sample from myelodysplastic syndrome (MDS), which, although clonal, is considered a premalignant condition, revealed no X chromosome hypermutation. Thus, X chromosome hypermutation is a common feature of female cancer genomes occurring across a wide range of tumor types.
Results
The X Chromosome Accumulates Significantly More Mutations Than Autosomes in Medulloblastoma Genomes from Female Samples
We analyzed the genome-wide distribution of somatic SNVs in 113 primary medulloblastoma samples collected within the International Cancer Genome Consortium (ICGC) PedBrain Tumor Project. The tumors, together with matched normal DNAs, were sequenced to average 30- to 40-fold coverage (Jones et al., 2012; M.K., D.T.W.J., N.J., P.A.N., M.D.T., R.E., S.M.P., and P.L., unpublished data; Table S1 available online). To analyze the distribution of mutations in the genome, the intermutation distance (the distance between a given somatic SNV and the SNV immediately upstream) was plotted for each sample. The mutational patterns revealed by this analysis are outlined below using an exemplary female and male genome (Figures 1A and 1B), which both belong to the same tumor subgroup (Sonic Hedgehog pathway-activated medulloblastoma).
Figure 1.

Distribution of Somatic Mutations in Medulloblastoma Genomes of Female versus Male Samples
(A) Intermutation distance plot of medulloblastoma MB56 (female). Mutations are ordered on the x axis from the first variant on the short arm of chromosome 1 to the last variant on the long arm of chromosome X. The distance between each somatic SNV and the SNV immediately upstream (the intermutation distance) is plotted on the y axis on a log scale. The chromosomes are separated by thin lines; chromosome X mutations are colored in gray. See also Figure S1.
(B) Intermutation distance plot for medulloblastoma MB61 (male).
(C) To present mutational load per chromosome corrected for the size of the respective chromosome, the mutation rate per megabase was plotted for female MB56. Coloring of bars represents the ratio of the six possible nucleotide changes (C > A, C > G, C > T, T > A, T > C, and T > G) for each chromosome.
(D) Mutational load per chromosome for MB61.
A lower mean intermutation distance on the X chromosome (0.33 Mb) compared with the autosomes (1.2 Mb) was observed in the female sample (Figure 1A), corresponding to a much higher number of mutations on the X chromosome than on any of the individual autosomes. In most cases, the X chromosome harbored a higher number of SNVs than both chromosomes 1 and 2 combined (e.g., MB56 in Figure 1A), even though each of them is much larger in size than the X chromosome. Further, MB56 has a total of 2,887 somatic SNVs in its genome, 469 (16%) of which are located on the X chromosome (Figure 1A). Given the size of the X chromosome, which at ∼155 megabases is very similar in size to chromosome 7, only about 5% of SNVs would be expected to occur on the X chromosome by chance, as is the case for chromosome 7 (n = 145 SNVs, 5.0%). In the male genome, no such difference in mutation rate between the X chromosome and the autosomes was observed (Figure 1B). Both exemplary tumor genomes are diploid, with no copy number or structural variations except for partial 10q loss in the female sample (Figure S1). Therefore, copy number changes of the chromosomes do not explain the large difference of SNVs on the X chromosome.
Figure S1.
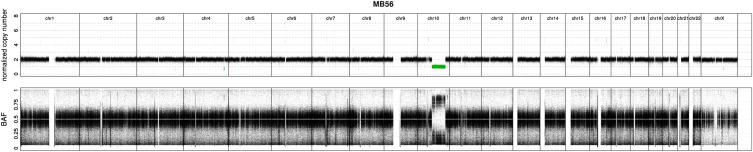
X Chromosome Hypermutation Cannot Be Explained by the Copy Number State of Chromosomes, Related to Figure 1
Genome-wide chromosomal copy number state of female diploid medulloblastoma MB56; based on whole-genome sequencing data. Lower panel: B-allele frequency.
To present mutational load per chromosome corrected for the size of the respective chromosome, the mutation rate per megabase was plotted (Figures 1C and 1D). Coloring of bars represents the proportions of the six possible nucleotide changes (C > A, C > G, C > T, T > A, T > C, and T > G) for each chromosome. This presentation of the chromosomal distribution of SNVs shows that the X chromosome accumulated 3.6-fold more somatic mutations per megabase compared to the mean of all autosomes in sample MB56 (Figure 1C). If the number of mutations per megabase on X is at least twice that of the mean mutation rate of the autosomes, we use the term “X chromosome hypermutation.” The majority of medulloblastoma cancer genomes of female patients (20/25, 80%) showed X chromosome hypermutation (Figure 2A).
Figure 2.

The X Chromosome Accumulates Significantly More Mutations Than Autosomes in Cancer Genomes of Female Samples
(A) X chromosome mutation ratio of 49 medulloblastoma genomes. The X chromosome mutation ratio (values on y axis) is calculated as mutation rate of X chromosome divided by the mean mutation rate of all autosomes. A ratio of ≥ 2 indicates X chromosome hypermutation.
(B) Age at diagnosis of the 49 medulloblastoma patients.
(C) XIST expression status of the 49 medulloblastoma genomes.
(D–G) (D) Distribution of the X chromosome mutation ratio for males (raw values and corrected for single X copy number state) and females (with both X copies) in medulloblastoma, (E) in neuroblastoma, (F) in pilocytic astrocytoma, and (G) in B cell lymphoma (n = 29, without sample 4120193). p values from t test.
See also Figure S2.
The expected number of SNVs depends on the copy number state of each chromosome. Thus, copy number changes of each chromosome have to be considered as confounding variables when comparing number of somatic SNVs per chromosome. To correct for copy number state of the autosomes, we considered only diploid medulloblastoma genomes with at most six copy number aberrant chromosomes and excluded the respective chromosomes per case; 49/113 cases fulfilled these criteria (Figure 2A). For all remaining cases, we can only infer X hypermutation to be present or not—we cannot accurately estimate the strength of X hypermutation.
In general, male medulloblastoma genomes do not show X chromosome hypermutation (Figures 1D and 2A). Male genomes have only one copy of X in the germline; therefore, the amount of mutations on the X chromosome needs to be doubled in order to correct for copy number status. Even after correcting the mutation rate of the X chromosome in males, the difference between female versus male X chromosome mutation rate is highly significant in medulloblastoma (p = 8 × 10−11, t test) and in three other tumor types (Figures 2D–2G). We note that, after this correction, a few male samples enter the range of our definition of X chromosome hypermutation (Figure 2A). This might be explained by the fact that hemizygous mutations on the single X chromosome in males can be more readily called by mutation calling algorithms than heterozygous mutations in female samples, which will typically be supported by fewer reads. However, the ratio of X chromosome versus autosome mutation rate in males and in females with X chromosome loss ranges from about 0.5 to 1, or 1- to 2-fold after correcting for the single copy of X, whereas we observe a range of ∼2- to 4-fold higher mutation rates when both Xa and Xi are present. This suggests that Xi accumulates at least twice as many mutations as Xa.
Note that the variation in strength of X chromosome hypermutation observed in the medulloblastoma female samples (Figure 2A) does not correlate with age at diagnosis (Figure 2B), which is in contrast to the finding that the overall number of somatic mutations in medulloblastoma strongly correlates with age (Jones et al., 2012).
X Hypermutation Is Confined to the Inactive X Chromosome
Most, but not all, medulloblastoma genomes from female patients display X chromosome hypermutation (Tables 1 and S1 and Figure 2A). We therefore further examined those cases not displaying this phenomenon. First, medulloblastoma genomes from females with loss of an X chromosome in the tumor (resulting in only one copy of the X chromosome) do not show X chromosome hypermutation (Figure 2A; for example, MB18). However, it is not the X copy number state in itself that determines X hypermutation. Tetraploid female sample MB6 has two copies of the X chromosome but has loss of heterozygosity and no XIST expression (Figure S2). Indeed, all medulloblastoma genomes from female samples that lost one copy of the X chromosome also show no XIST expression, indicating that Xa is kept and Xi is lost (Figure 2C). We did not observe X chromosome hypermutation in MB6 (with two copies of Xa; Figure S2B) and consistently found no X hypermutation in cases that lost Xi, regardless of the absolute copy number state of X. Further, X hypermutation is not present in male tetraploid genomes with more than one copy of the X chromosome. These analyses indicate that the copy number state of the X chromosome does not impact X hypermutation but rather indicates that it depends on the presence of Xi. Remarkably, a medulloblastoma from a male patient (MB139) with Klinefelter syndrome (47, XXY genome) also shows a trend toward X chromosome hypermutation (Figure 2A). The matching RNA data confirm XIST expression and therefore presence of Xi (Figure 2C).
Table 1.
Overview of 402 Cancer Genomes Analyzed in This Study
| Cancer Type | Cases Total | Cases Female | Cases Male | X Hyper-mutation Female | X Hyper-mutation Male | Remark | Reference |
|---|---|---|---|---|---|---|---|
| Acute myeloid leukemia | 24 | 11 | 13 | 1 | 0 | 1 female with <200 SNVs | Welch et al. (2012) |
| B cell lymphoma | 30 | 9 | 21 | 3 | 0 | 2 females with chrX loss | Richter et al. (2012); M.S., J.R., M.H., P.L., R.E., and R.S., unpublished data |
| Breast cancer | 21 | 21 | NA | 2 | NA | 15 cases have partial or complete chrX loss | Nik-Zainal et al. (2012) |
| Chronic lymphocytic leukemia | 4 | 2 | 2 | 0 | 0 | Puente et al. (2011) | |
| Colorectal adenocarcinoma | 9 | 6 | 3 | 0 | 0 | Bass et al. (2011) | |
| Ependymoma | 5 | 1 | 4 | 1 | 0 | S.C.M., H.W., P.A.N., D.T.W.J., N.J., S.M.P., and M.D.T., unpublished data | |
| Glioblastoma | 1 | 1 | NA | 1 | NA | S.M.P., M.K., D.T.W.J, P.A.N., M.D.T., R.E., P.L., and A.K., unpublished data | |
| Medulloblastoma | 113 | 48 | 65 | 29 | 0 | 14 females with chrX loss; 1 female with <200 SNVs | Jones et al. (2012); M.K., D.T.W.J., N.J., P.A.N., M.D.T., R.E., S.M.P., and P.L., unpublished data; P.A.N., M.K., D.T.W.J., N.J., M.D.T., R.E., S.M.P., P.L., unpublished data |
| Neuroblastoma | 84 | 36 | 48 | 8 | 0 | 6 females with chrX loss; 8 females with <200 SNVs | Molenaar et al. (2012) |
| Pilocytic astrocytoma | 96 | 54 | 42 | 10 | 0 | 36 females with <200 SNVs | Jones et al. (2013) |
| Prostate carcinoma | 11 | NA | 11 | NA | 0 | Weischenfeldt et al. (2013) | |
| Retinoblastoma | 4 | 2 | 2 | 1 | 0 | 1 female with <200 SNVs | Zhang et al. (2012) |
| Summary | 402 | 191 | 211 | 56 | 0 | additional 27 females with increased X mutation rate; see Table S1 |
See also Table S1.
Figure S2.

Lack of X Chromosome Hypermutation in Cancer Genomes of Female Samples with X Chromosome Loss, Related to Figure 2
(A) Genome-wide chromosomal copy number state of tetraploid female medulloblastoma MB6; based on whole-genome sequencing data.
(B) Mutations per megabase per chromosome for MB6.
(C) XIST expression versus copy number state of the X chromosome in medulloblastoma.
To further study the confinement of hypermutation to Xi, we performed two different approaches. First, for two samples (MB101 and lymphoma 4120193) with high mutational load and imbalanced copy number states of Xi versus Xa, we assigned individual SNVs to the active/inactive X chromosome by in silico haplotype phasing of mutations. Second, we performed chromatin immunoprecipitation sequencing (ChIP-seq) for histone marks H3K36me3 and histone variant macroH2A1 in order to haplotype the X chromosome of two additional samples (MB59 and GBM103).
For haplotype phasing, we attempted to phase mutations that are sufficiently close together to be spanned by single read pairs. The first 52 megabases of chromosome arm Xp in female sample MB101 are present at three copies in the tumor. RNA sequencing (RNA-seq)-based allele frequencies of germline variants clearly identify Xi being present with two copies and Xa with a single copy. In total, 222 somatic SNVs were called in this region. Many somatic mutations were sufficiently close to heterozygous germline SNPs that individual sequence read pairs spanned both, thus allowing the mutation to be phased with the SNP. Here, a germline SNP with an allele frequency of about 1/3 indicates that this mutation is on Xa, whereas an allele frequency of 2/3 indicates that this mutation is located on Xi. In total, 58 somatic SNVs were haplotyped by this approach, of which 46 unambiguously mapped to Xi and 12 to Xa (p < 0.0001, permutation test). Even when doubling the amount of Xa mutations in order to correct for the single copy state of Xa in MB101, the difference is still significant (24 Xa versus 46 Xi mutations; p = 0.006).
The same haplotyping approach was applied to X chromosome hypermutated lymphoma sample 4120193 (female), for which most of the Xq arm is present at three copies in the tumor (Figure 3). Here, RNA-seq-based allele frequencies of germline variants indicate that Xi has two copies and Xa has a single copy. The sample has a high genome-wide mutational load with 3,756 somatic SNVs on the X chromosome alone. Of those SNVs, 444 were haplotyped, of which 389/444 SNVs unambiguously mapped to Xi and 55/444 mapped to Xa (p < 0.0001, permutation test). Doubling the amount of Xa mutations (n = 110) in order to correct for the single copy state of Xa still results in 3.5× more mutations on Xi. Interestingly, this sample also shows focal regions of higher intermutation distance on the X chromosome (Figure 3A), corresponding to lower mutation rates. These regions with fewer mutations harbor genes escaping X inactivation, as well as the pseudoautosomal regions PAR1 and PAR2 (Figure 3B). In addition, we compared medulloblastoma female samples with X hypermutation (n = 10) with male samples (n = 10) of the same tumor subtype and ploidy and found that regions escaping X chromosome inactivation have a mean mutation rate of 1/Mb in both male and female samples. Thus, regions escaping X chromosome inactivation are not X hypermutated, further supporting our finding that X hypermutation is confined to heterochromatic regions of the inactive X chromosome.
Figure 3.

Fewer Mutations in Regions Escaping X Chromosome Inactivation
(A) Genome-wide intermutation distance plot of lymphoma 4120193 (DLBCL subtype, female).
(B) X chromosome-wide intermutation distance plot of somatic mutations along the X chromosome coordinates. Genes that escape X inactivation and are expressed in this sample as determined by RNA-seq data show fewer mutations (marked in gray).
(C) X chromosome copy number plot of lymphoma 4120193; 81 Mbs of the Xq arm are present at three copies in the tumor.
The asterisk in all three panels marks the same mutation (the first mutation after the centromere).
To haplotype the X chromosome of two additional samples (MB59 and GBM103), we performed ChIP-seq for histone marks H3K36me3 and macroH2A1. Histone variant macroH2A1 is known to show an ∼1.5-fold uniform enrichment along the inactive X chromosome (Mietton et al., 2009). In contrast, H3K36me3 is enriched in actively transcribed regions and therefore on Xa. Thus, by sequencing both histone marks to sufficiently high depth to infer allele frequencies of mutations, a mutation on Xi should have a high allele frequency in the macroH2A1 reads and a low allele frequency in H3K36me3, which we clearly observed for the germline mutations (data not shown). Integrating matching RNA-seq data with mutations showing particularly high/low macroH2A1/H3K36me3 allele frequencies (Extended Experimental Procedures), 31 SNVs were haplotyped in glioblastoma GBM103, with only 3 SNVs locating to Xa (p < 0.0001, permutation test). In medulloblastoma MB59, eight SNVs were haplotyped, of which only two mapped to Xa.
Extended Experimental Procedures.
All main text and supplemental figures were created with R (http://www.R-project.org/).
Samples
Informed consent and an ethical vote (Institutional Review Board) for our new and published cancer samples and one MDS sample were obtained according to ICGC guidelines (www.icgc.org). No patient underwent chemotherapy or radiotherapy prior to the surgical removal of the primary tumor.
Bone marrow mononuclear cells from a 73 year old healthy female were taken from a biobank of human bone marrow samples collected from the proximal femur during hip replacement surgery. Biobanking was performed under precepts established by the Helsinki Declaration and approved by the ethics committee of the Medical Faculty of Heidelberg University and the ethics committee of the Landesärztekammer Rheinland-Pfalz. Approval from local ethics committee and informed consent of the donor was obtained specifically for whole genome sequencing.
A progenitor cell clone was raised from a peripheral blood sample of a 39 year old healthy female participating in a prospective mother-child study (Weisse et al., 2012), which had informed consent regarding genetic analyses. The study was approved by the Ethics Committee of the University of Leipzig (file ref # 046-2006, 160-2008, 160b/2008).
Sequencing Library Preparation by Tagmentation Used for the Single-Cell Expansions of Healthy Somatic Tissue
Tagmentation-based sequencing library preparation using 25 ng of genomic DNA was performed as described for whole genome bisulfite sequencing (Adey and Shendure, 2012; Wang et al., 2013) with some modifications. The adaptor for tagmentation was assembled from oligonucleotides Tn5mC-Apt1 and Tn5mC1.1-A1block, and for the oligo replacement/gap repair step Tn5mC-ReplO1 was used (see table below). The transposome was generated from the tagmentation adaptor and Tn5 transposase (Epicenter via Biozym, Hessisch Oldendorf, Germany). After oligo replacement/gap repair, the purified DNA was split into two halves, and from each half a library was prepared using primers Tn5mCP1 and Tn5mCBar with 12 PCR cycles on a Lightcycler 480 (Roche, Mannheim, Germany). Purified libraries were size-selected for a range of 300 bp and sequenced on an Illumina HiSeq 2500 in the 101-bases paired-end rapid mode.
Oligonucleotides for tagmentation-based sequencing library preparation:
Tn5mC-Apt1: T[5mC]GT[5mC]GG[5mC]AG[5mC]GT[5mC]AGATGTGTATAAGAGA[5mC]AG
Tn5mC1.1-A1block: [Phos]-CTGTCTCTTATACA[ddC]
Tn5mC-ReplO1: [Phos]-[5mC]TGT[5mC]T[5mC]TTATA[5mC]A[5mC]AT[5mC]T[5mC]- [5mC]GAG[5mC][5mC][5mC]A[5mC]GAGA[5mC] [inv dT]
Tn5mCP1: AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTC
Tn5mCBar∗: CAAGCAGAAGACGGCATACGAGATNNNNNNNNNGTCTCGTGGGCTCGG
∗Barcodes for PCR primer Tn5mCBar: Bar1, GGATGTTCT; Bar2, CTTATCCAG; Bar3, GTAAGTCAC; Bar4, TTCAGTGAG; Bar5, CTCGTAATG; Bar6, CATGTCTCA; Bar7, AATCGTGGA; Bar8, GTATCAGTC.
Isolation of Analytes for ICGC PedBrain Samples
Analyzed DNA and total RNA for whole-genome sequenced samples were isolated using a QIAGEN Allprep DNA/RNA/Protein Mini Kit. For simultaneous DNA and RNA isolation by Allprep DNA/RNA/Protein Mini Kit, on average 125 mg of homogenized (TissueLyser, QIAGEN) tumor tissue was used for isolation of analytes. The manufacturer’s protocol was adapted to allow for DNA and total RNA isolation. DNA from patient-matched blood samples was extracted using QIAGEN Blood and Cell Culture Midi Kit according to the manufacturer’s protocol.
DNA and RNA Library Preparation and Sequencing of ICGC PedBrain Samples
Paired-end (PE) DNA library preparation was carried out using Illumina protocols. In brief, 50ng - 5 μg of genomic DNA were fragmented to ∼300 bp (PE) insert-size with a Covaris device, followed by size selection through agarose gel excision. Deep sequencing was carried out with Illumina HiSeq 2000 and 2500 instruments. RNA-seq libraries for medulloblastoma were prepared and analyzed as described in Jones et al., 2012 (for medulloblastoma) and Jones et al., 2013 (for astrocytoma).
Mapping and Analysis of Paired-End DNA Sequences
As previously described (Jones et al., 2013), paired-end DNA sequencing reads were mapped to the hg19 (NCBI build 37.1, downloaded from the UCSC genome browser at http://genome.ucsc.edu/) assembly of the human reference genome using BWA version 0.5.9-r16 (Li and Durbin, 2009) and processed with samtools (version 0.1.17) and Picard tools (version 1.61) (Li et al., 2009).
For detection of single nucleotide variants (SNVs) and small insertions or deletions (indels) we applied our in-house analysis pipeline based on samtools mpileup and bcftools (Li et al., 2009) with parameter adjustments to allow calling of somatic variants. For indels, the matched control sample was also analyzed by samtools mpileup at tumor indel positions ± 20 bp. Indels were classified as “somatic” if samtools mpileup did not call an indel or multibase variant in this extended neighborhood region of the tumor indel in the control sample.
Integration of SNVs with RNA Sequencing Data
As previously described (Jones et al., 2013), paired-end RNA sequencing reads were mapped to the hg19 assembly of the human reference genome using BWA. Gene expression levels were calculated per exon according to reads per kilobase of exon model per million mapped reads (RPKM) using BEDTools (Quinlan and Hall, 2010) and custom Perl scripts. Where available, candidate DNA variant positions were annotated with RNA information by generating a pileup of the DNA variant position in the RNA BAM alignment file.
The following 16 medulloblastoma female samples had somatic SNVs found in the DNA sequencing data sufficiently covered in the patient-matched RNA sequencing data:
MB20, MB23, MB31, MB46, MB50, MB56, MB57, MB59, MB68, MB83, MB101, MB113, MB117, MB124, MB130, MB132
Expression Array Analysis for XIST Expression Status
Affymetrix U133 Plus 2.0 expression array data for XIST was extracted via the R2 software tool for analysis and visualization of genomics data (http://r2.amc.nl). Sample library preparation, hybridization, and quality control were performed according to protocols recommended by the manufacturer. The MAS5.0 algorithm of the GCOS program (Affymetrix Inc) was used for normalization. Detection p-values were assigned to each probe set using the MAS5.0 algorithm.
In Figure 2B, XIST expression values were plotted for each sample. For cases where no Expression Array data were available, but RNA-seq data, the XIST RPKM value was plotted (after normalization). For MB69, neither Expression Array nor RNA-seq data were available.
RNA-Seq-Based Analysis of X Inactivation Escape Regions
We determined regions of X inactivation escape in a specific tumor sample by integrating patient-matched RNA-seq data, since a subset of genes exhibit tissue-specific differences in escape from X inactivation. If a heterozygous mutation of the DNA was present in the RNA-seq data with a homozygous allele frequency, we concluded mono-allelic expression and hence X inactivation of the underlying gene. If the heterozygous mutation of the DNA was also heterozygous in the RNA, we classified the site as X inactivation escape region. As a positive control, this approach was able to correctly identify known escape genes like ZFX, DDX3X or KDM6A. Note that this approach can only identify escape genes that harbor a heterozygous mutation within transcribed regions. Therefore, we also used the list of escape genes from Carrel and Willard (2005) and required an RPKM > 5 of the respective gene to define an escape region (Figure 3, escape regions marked in gray).
H3K36me3 and macroH2A1 ChIP-Seq of Frozen Tissue for Samples GBM103 and MB59
Frozen tissue was crushed with a pre-cooled douncer on dry ice. After crushing the tissue was fixed with freshly prepared 1% formaldehyde in PBS for 12 min. The reaction was stopped with 125 mM glycine for 5 min. To gain nuclei the fixed tissue was dounced again and washed 3 times with PBS. The tissue was then resuspended in MNase buffer (25 mM KCl, 4 mM MgCl2, 1 mM CaCl2, 50 mM Tris/HCl pH 7.4) and 10 U MNase per 15 mg tissue was added. After 15 min, incubation at 37°C MNase was stopped by adding 10x covaris buffer (100 mM Tris pH 8.0, 2 M NaCl, 10 mM EDTA, 5% N-lauroylsarcosine, 1% Na-deoxycholate, supplemented with protease inhibitors). The samples were sonicated for 25 min with the following parameters with a Covaris S2 system: burst 200, cycle 20%, and intensity 8. After centrifugation the supernatant was collected and directly used for IP.
After IgG preclearance the sheared chromatin was incubated with protein G magnetic beads (Cell signaling, 9006) and 4 μg of either H3K36me3 (Abcam, ab9050) or macroH2A (Abcam, ab37264) antibody overnight. After washes with 1x covaris buffer (10 mM Tris–HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5% N-lauroylsarcosine, 0.1% Na–deoxycholate), high-salt-buffer (50 mM HEPES pH 7.9, 500 mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% Na–deoxycholate, 0.1% SDS), lithium buffer (20 mM Tris–HCl pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% Na–deoxycholate) and 10 mM Tris–HCl, chromatin was eluted from the magnetic beads (elution buffer: 50 mM Tris pH 8.0, 1 mM EDTA, 1% SDS, 50 mM NaHCO3) and the crosslink was reversed overnight. After RNase A and proteinase K digestion, DNA was purified and cloned in a barcoded sequencing library for the Illumina sequencing platform. In brief, after DNA repair and A-addition NEBNext adapters (NEB, E7335) were ligated and digested with the USER enzyme. Barcodes (NEB, E7335) were introduced via PCR with a maximum of 14 cycles by the NEBNext polymerase (NEB, M0541). Size selection for mononucleosomal insert fragments was done with Ampure XP beads (Agencourt, A63880).
Each ChIP-seq library was sequenced with two complete lanes on the Illumina HiSeq 2500 in the 101-bases paired-end rapid mode and aligned to hg19 using bwa. This resulted in the following coverage values (genome-wide, after deduplication, including all uniquely mapping reads):
GBM103 macroH2A1: 17x H3K36me3: 20x
MB59 macroH2A1: 11x H3K36me3: 11x
Analysis of ChIP-Seq Data
For peak calling, MACS version 1.4 (Zhang et al., 2008) was used without control and without local lambda calculation. On the X chromosome, a total of 8.8 Mbs of H3K36me3 peak regions were called in GBM103, and 4.5 Mbs in MB59. Peak calling for macroH2A1 was not possible, due to the uniform enrichment along the X chromosome (Mietton et al., 2009). Therefore, we restricted haplotype assignment (to Xi or Xa) of mutations to the H3K36me3 peak regions. MacroH2A1 is enriched on Xi (Mietton et al., 2009), while H3K36me3 is enriched in actively transcribed regions and therefore on Xa. Thus, a mutation located on Xi is expected to have a high allele frequency in the macroH2A1 data and a low allele frequency in H3K36me3, which we clearly observed for the heterozygous germline mutations on the X chromosome (data not shown). In addition, patient-matched RNA-seq data further supported this in silico haplotying approach of the X chromosome: heterozygous DNA mutations that showed ∼100% allele frequency in the RNA-seq data had high allele frequencies in H3K36me3 data (or ∼0% allele frequency in the RNA-seq correlating with low allele frequency in H3K36me3 data).
PCA
In order to compare the mutation spectrum of the combined autosomes and the hypermutated X chromosome of individual samples (as shown in Figure S4A), PCA was performed on a matrix of 68 rows representing 34 samples (separated each for the autosomes and the X chromosome) and six columns, corresponding to the ratio of the six possible nucleotide changes (C > A, C > G, C > T, T > A, T > C, and T > G). Calculations were performed in R using the prcomp function. All feature vectors were scaled to be zero-centered and to have unit variance. The first three principal components accounted for 84% variance of the data.
RepliSeq-Based Replication Timing Analysis for the Autosomes
We correlated the number of somatic mutations binned into windows of sizes 100Kb, 1Mb, and 5Mb with genome-wide replication timing data (Repli-Seq; Hansen et al., 2010). The Repli-Seq data used in this analysis are a wavelet-smoothed, weighted average signal where high (and low) values indicate early (and late) replication during S-phase. Values < 38 indicate late replication timing. RepliSeq replication timing data were downloaded from http://genome.ucsc.edu/ENCODE for 10 different cell lines:
Gm06990, Gm12801, Gm12812, Gm12813, Gm12878, Hepg2, Huvec, K562, Mcf7, Nhek.
We used the mean value of genomic regions that maintain similar replication timing between these different cell types, determined by low standard deviation per window.
For B cell lymphoma (in Figures 6B and S5), we only used the Repli-Seq data for lymphoblastoid cell lines (n = 5) to match the tumor cell of origin:
Gm06990, Gm12801, Gm12812, Gm12813, Gm12878.
Most X hypermutated samples have no RNA-seq read support at mutated positions because these mutations localize mainly to intergenic regions. However, a pool of 16 medulloblastoma samples had in total 22 somatic SNVs sufficiently covered in the patient-matched RNA sequencing data (excluding genes that escape X inactivation). Of these 22 mutations, 21/22 mutations were not present in the RNA despite sufficient RNA coverage at the mutated position and are therefore likely to be present on the inactive X (p < 0.0001, permutation test).
X chromosome hypermutation was observed in all medulloblastoma samples of female patients in which the inactive X chromosome was present, and haplotyping of four individual samples clearly indicates that X hypermutation is confined to the inactive X chromosome.
X Chromosome Hypermutation Is a Feature of Many Different Cancer Types
To assess whether X chromosome hypermutation is a general feature of multiple tumor types, we analyzed the somatic mutation rate of the X chromosome in an additional ∼300 whole-cancer genomes, including our own published and unpublished data from five different cancer entities (pilocytic astrocytoma, glioblastoma, ependymoma, B cell lymphoma, and prostate carcinoma) complemented by published mutation call sets of six different cancer types: breast cancer (Nik-Zainal et al., 2012), neuroblastoma (Molenaar et al., 2012), chronic lymphocytic leukemia (Puente et al., 2011), acute myeloid leukemia (Welch et al., 2012), colorectal carcinoma (Bass et al., 2011), and retinoblastoma (Zhang et al., 2012). We observed X chromosome hypermutation in a significant fraction of cancer genomes from female samples (56/191, 29%) comprising nine different cancer entities across a diverse set of childhood tumors as well as adult solid and hematopoietic malignancies (Tables 1 and S1 and Figure 4).
Figure 4.

X Chromosome Hypermutation Is a Feature of Many Different Cancer Types
Mutations per chromosome plot for exemplary genomes from female samples of (A) breast cancer, (B) retinoblastoma, (C) B cell lymphoma, (D) neuroblastoma, (E) glioblastoma, and (F) AML. See also Figure S3.
In contrast to medulloblastoma, not every female cancer genome containing Xi in the other cancer types displays X hypermutation, as far as information about Xi status was available. In the two female cases of CLL, no X hypermutation was detected. This was also the case for the six female samples of colorectal carcinoma by the strict definition given above, but in two genomes of female samples, the X chromosome showed the highest mutation rate of all chromosomes. If we include all female cases of the different cancer types with an increased mutation rate, but not X chromosome hypermutation by our strict definition, then 83/191 (43%) females are affected. Some of the cancer genomes had extremely low amounts of somatic mutations (<200 SNVs), specifically in the pediatric tumors pilocytic astrocytoma and neuroblastoma (Table S1), which makes detection of X chromosome hypermutation impossible for a single sample.
Interestingly, a breast cancer genome with about 10,000 mutations showed clear X chromosome hypermutation, as did a retinoblastoma genome of a 21-month-old patient with only 258 mutations (Figures 4A and 4B). Thus, the occurrence of X chromosome hypermutation cannot simply be explained by the total number of somatic mutations or by age at diagnosis. Some cancer genomes from females with a very high number of somatic mutations, like some of the breast cancer (Nik-Zainal et al., 2012) and colorectal adenocarcinoma genomes (Bass et al., 2011), do not show X hypermutation. It may be that, in some tumors, local hypermutation due to processes such as kataegis (Nik-Zainal et al., 2012) might mask the general accumulation of mutations on Xi. We also observed X chromosome hypermutation in a recurrence of medulloblastoma MB34 (Jones et al., 2012), with the same degree of hypermutation as in the primary tumor despite a higher number of mutations genome wide (data not shown).
Our analyses thus indicate that X chromosome hypermutation is present in many diverse types of cancer genomes from female patients but to varying degrees within each cancer type in terms of strength of X hypermutation and number of affected samples.
Characteristics of X Chromosome Hypermutation in Medulloblastoma
Next, we examined whether X chromosome hypermutation is an early or late event in tumorigenesis. Because tetraploidy is known to be a frequent early event in medulloblastoma tumorigenesis (Jones et al., 2012), we assessed whether X hypermutation is already present before tetraploidy occurs. For this analysis, we considered only mutations with a clear ∼50% allele frequency because allele frequencies of ∼25% indicate a mutation on only one of the four alleles present after genome duplication. In the tetraploid genome of MB101 (Figure 5A), only 7% of genome-wide mutations are ∼50% heterozygous—i.e., present before tetraploidy occurred—indicating that most mutations arise after tetraploidy. However, X hypermutation is already clearly observable in the fraction of SNVs present before genome duplication (Figure 5B), demonstrating that this phenomenon develops even earlier during tumor evolution.
Figure 5.

Characteristics of X Chromosome Hypermutation in Medulloblastoma
(A and B) Mutational load per chromosome for female tetraploid medulloblastoma MB101 (A) for all somatic SNVs and (B) only for somatic SNVs with a clear ∼50% allele frequency, demonstrating X hypermutation to occur before tetraploidy.
(C) Intermutation distance plot for genome-wide somatic indels of MB101.
(D) X hypermutation is observed for indels (y axis) in those genomes that display X hypermutation based on SNVs (x axis). Values on axes calculated as mutation rate of X chromosome divided by the mean mutation rate of all autosomes.
(E) Mutational load per chromosome for MB101 using all germline single nucleotide substitutions shows that the X chromosome has less germline substitutions than the autosomes.
(F) Distribution of somatic SNVs per chromosome into different genomic regions.
See also Figure S4.
Interestingly, X chromosome hypermutation is not confined to SNVs but is also observed for somatic indels in those genomes that display X hypermutation based on SNVs (Figures 5C and 5D).
X chromosome hypermutation is also clearly a somatic phenomenon, as notably fewer germline polymorphisms occur on the X chromosome compared to the autosomes in our medulloblastoma samples (Figure 5E). Indeed, it is a well-known phenomenon that germline variation is found to be lowest on the X chromosome, with an ∼ 30% lower rate than for the autosomes (Hodgkinson and Eyre-Walker, 2011).
The distribution of mutations between exons, introns, and intergenic DNA is not different on the hypermutated X chromosome compared to the autosomes (Figure 5F). Lack of transcription-coupled repair on the largely transcriptionally silent Xi is unlikely a cause for X hypermutation given the predominantly intergenic location. Further, X chromosome hypermutation cannot be linked to hypermethylation of Xi at gene promoters, leading to spontaneous deamination of cytosine because no increase in the fraction of C > T mutations was found (Figures 1C and 4).
Similar Mutation Spectrum on Autosomes and the Hypermutated X
Within an individual cancer genome, there is no substantial variation between the autosomes and X chromosome in the relative contributions of each of the six classes of base substitution (C > A, C > G, C > T, T > A, T > C, and T > G) (Figure 4). Thus, the hypermutated X chromosome has the same mutation spectrum as the autosomes. This holds true even in those cases of lymphoma or neuroblastoma that have a very unique mutation spectrum (Figures 4C and 4D). To provide further insight into the underlying mutational processes, we incorporated the sequence context in which mutations occurred by considering the bases immediately 5′ and 3′ to each mutated base, giving 96 possible trinucleotide contexts for a mutation. The resulting heatmap reveals that the mutational signatures on the autosomes and the X chromosome are very similar (Figure S3).
Figure S3.

Genomic Heatmap for Medulloblastoma Female Samples with X Chromosome Hypermutation, Related to Figure 4
Presented is the fraction of mutations at each of the 96 mutated trinucleotides as a heatmap for each chromosome (1-22, X), normalized according to the prevalence of each trinucleotide on the respective chromosome. Log-transformed values of these ratios were plotted in the heatmap, where red indicates a high mutations fraction, yellow low and white no mutations. The 5′ base to each mutated base is shown on the vertical axis and 3′ base on the horizontal axis. Somatic SNVs pooled from 25 medulloblastoma females.
Further, principal component analysis (PCA) shows that the mutation spectrum of the autosomes and the hypermutated X chromosome of individual samples within one tumor type cluster closer together than the hypermutated X chromosomes of different tumors (Figure S4A). In addition, the distribution of mutations along the hypermutated X chromosome is very similar to the distribution observed on X in males of the same tumor type (e.g., for B cell lymphoma r2 = 0.64, p < 2 × 10−16, Pearson’s product-moment correlation; Figures S4B and S4C).
Figure S4.

Mutations on the Hypermutated X Chromosome Do Not Show a Unique Mutation Spectrum or Distribution Pattern, Related to Figure 5
(A) Principal component analysis (PCA) comparing the mutation spectrum of the combined autosomes and the hypermutated X of individual samples (n = 34) of four different tumor types. The first three principal components (PC1, PC2, and PC3) separate the different tumor types, but not the autosomes from the hypermutated X chromosomes.
(B) The distribution of somatic mutations per 1 Mb window along the X chromosome in B cell lymphoma for a set of females (red) and males (blue).
(C) The distribution of somatic mutations per 1 Mb window shows a higher correlation between males and females of the same tumor type as compared to females with X hypermutation of different tumor types.
We conclude that Xi is hypermutated during tumorigenesis by the same processes that cause mutations on the autosomes and Xa as opposed to Xi-specific mutational processes.
Autosomal Regions as Highly Mutated as the X Chromosome Are Late Replicating
Late-replication timing is known to increase the mutation rate (Liu et al., 2013). Because regions subject to X inactivation are known to be late replicating (Hansen et al., 1996; Morishima et al., 1962), we analyzed whether the autosomes have late-replicating regions as strongly mutated as the hypermutated X chromosome. We used the RepliSeq replication timing data (http://genome.ucsc.edu/ENCODE) for 10 different cell lines and focused on genomic regions that maintain similar replication timing between these different cell types. Similar to the definition of X chromosome hypermutation, we defined an autosomal region to be hypermutated if this region had at least twice the number of mutations compared to the mean mutation rate of the autosomes. At the 1 Mb scale, the highest mutated regions are almost all late replicating, with very few exceptions (Figure 6). These conclusions were upheld when using alternative genomic window sizes of 100 kb or 5 Mb (Figure S5). Not every late replicating autosomal region, however, displayed hypermutation.
Figure 6.

Autosomal Regions as Highly Mutated as the X Chromosome Are Late Replicating
(A) RepliSeq replication timing data (y axis) versus somatic mutations per 1 Mb window (x axis) of the merged mutation set of medulloblastoma genomes (n = 113) including only the autosomes. An autosomal region in this analysis is defined as hypermutated if ≥2-fold mutation rate compared to the mean mutation rate of the autosomes (here, >118 SNVs per 1 Mb). The RepliSeq data are a wavelet-smoothed, weighted average signal where high and low values indicate early and late replication during the S phase, respectively (y axis).
(B) Replication timing correlation for B cell lymphoma (n = 29, without sample 4120193). Autosomal regions with >125 SNVs per 1 Mb are defined here as hypermutated.
See also Figure S5 for alternative genomic window sizes of 100 Kb and 5 Mb.
Figure S5.

Autosomal Regions as Highly Mutated as the X Chromosome Are Late Replicating, Related to Figure 6
Replication timing correlation for medulloblastoma (n = 113) for (A) 5 Mb and (C) 100 Kb window size, and for B cell lymphoma (n = 29) for (B) 5 Mb and (D) 100 Kb window size.
For B cell lymphoma (Figure 6B), we used only the RepliSeq data for lymphoblastoid cell lines (n = 5) to match the tumor cell of origin. The immunoglobulin loci IGH, IGK, and IGL, which are known to undergo AID-mediated somatic hypermutation in germinal center B cells, were excluded (and showed early replication timing). However, additional regions affected by AID-mediated somatic hypermutation in germinal center B-cell-derived lymphomas, such as the MYC gene region in Burkitt lymphomas and the BCL2 gene region in follicular lymphomas, were not excluded and appear as early-replicating, hypermutated regions in this analysis (Figure 6B).
Notably, very few regions on the autosomes are mutated at a high frequency as the hypermutated X chromosome. Thus, autosomes are also affected by hypermutation in late-replicating regions but to a much lesser extent than Xi.
Whole-Genome Sequencing of Nonmalignant Somatic Cells Reveals No X Chromosome Hypermutation
Because X chromosome hypermutation is an early event in tumorigenesis of medulloblastoma and is observed across a variety of very diverse cancer types, we sought to elucidate whether X hypermutation is a general feature of normal somatic cells from females, arising independent of tumorigenesis. We therefore performed whole-genome sequencing of two clonally expanded, single CD34+ CD38− hematopoietic stem/progenitor cells (HSPCs) derived from bone marrow mononuclear cells of a healthy 73-year-old female. The two genomes harbored about 1,300 and 1,500 somatic point mutations, respectively, which we were able to call as “somatic” by comparing against whole-genome sequence data derived from the matching bulk bone marrow cells. Despite the relatively high number of somatic SNVs in these two healthy cells, we did not observe X chromosome hypermutation (Figures 7A and 7B). In addition, one HSPC clone derived from peripheral blood of a healthy 39-year old female was sequenced and compared against the matching peripheral blood, but X hypermutation was not observed in the 442 somatic mutations (Figure 7C).
Figure 7.

Whole-Genome Sequencing of Nonmalignant Somatic Cells Reveals No X Chromosome Hypermutation
Mutational load per chromosome plot for genomes from female samples of (A) HSPC clone B6 and (B) HSPC clone G2 of a healthy 73-year-old female, (C) HSPC clone of a 39-year-old healthy female, (D) MDS of Li-Fraumeni syndrome case LFS-MB1, and (E) medulloblastoma of Li-Fraumeni syndrome case LFS-MB1.
Next, we studied one sample derived from secondary MDS arising after treatment for medulloblastoma in an 11-year-old female Li-Fraumeni syndrome case (LFS-MB1). Although clonal, MDS is considered a premalignant condition with a propensity to progress to AML when additional genetic abnormalities are acquired. We did not observe X hypermutation in this MDS genome, despite a total of 825 somatic point mutations (Figure 7D). In contrast, the medulloblastoma genome of this Li-Fraumeni syndrome case is one of the strongest X hypermutated samples in our data set (Figure 7E).
Thus, from the four samples studied, we did not find evidence for X chromosome hypermutation in noncancerous, clonally expanded cell populations.
Discussion
Hypermutation of the Inactive X Chromosome in Cancer Genomes
Our analysis of more than 400 cancer genomes from 12 different cancer entities revealed the finding that the X chromosome of female patients frequently accumulates twice as many somatic mutations per megabase compared to the autosomes, whereas some tumors even accumulate up to four times more mutations on the X chromosome. We provide strong evidence that this X chromosome hypermutation is confined to the inactive X chromosome (Xi). Further, X hypermutation has no similarities with the focal somatic hypermutation present in normal immune cells or with the recently described kataegis phenomenon observed in some tumors, which is thought to be linked to APOBEC family cytidine deaminase activity (Nik-Zainal et al., 2012).
We used data from different sequencing technologies (Illumina and Complete Genomics) sequenced at different centers and analyzed with different alignment algorithms and mutation calling pipelines. In addition, all genomic somatic mutations from the University of Washington (AML and retinoblastoma; Welch et al., 2012; Zhang et al., 2012) were validated by orthogonal sequencing technologies, which preclude X chromosome hypermutation being an artifact of next-generation sequencing technology and/or analysis methodology.
We sequenced four nonmalignant genomes (three female samples of HSPCs and one female MDS case) and found no X hypermutation, despite a sufficiently high number of somatic mutations, which would have allowed for the identification of X hypermutation if it were present. Although we cannot conclusively exclude, based on the analysis of these four genomes, that the higher accumulation of mutations on Xi is due to random background mutations in healthy somatic cells from females, we hypothesize that X chromosome hypermutation is a cancer-specific feature.
We propose a model in which Xi accumulates somatic mutations during tumorigenesis not by any specific process acting exclusively on Xi but by a higher burden of the same processes mutating the autosomes, thereby resulting in the same mutational spectrum as observed on the autosomes. If Xi hypermutation would be attributed solely to the unique heterochromatin structure of Xi, one would expect Xi hypermutation to occur also in normal cells and cancer genomes from female samples in general, which we did not observe. The mutations on Xi seem not to be detected and/or repaired as efficiently as on the autosomes and Xa. Alternatively, it has been reported that a shortage of nucleotides during late S phase could lead to erroneous incorporation of nucleotides, thus suggesting rather elevated mutagenesis than impaired repair causing X hypermutation (Burrell et al., 2013). Xi is known to be extremely late replicating in S phase (Hansen et al., 1996; Morishima et al., 1962), and other late-replicating regions on the autosomes also displayed markedly higher mutation rates. Thus, Xi might not be properly repaired in aberrantly proliferating cells as a result of a shorter and/or compromised late S phase caused by DNA replication stress.
This hypothesis is supported by our analysis of retinoblastoma, an aggressive childhood cancer initiated by the biallelic loss of RB1. Tumors progress very quickly following RB1 inactivation and thereby experience DNA replication stress (Tort et al., 2006). The retinoblastoma genome of a 21-month-old patient with only 258 mutations genome wide (Zhang et al., 2012) already shows strong X chromosome hypermutation, with a three times higher mutation rate on chromosome X than on the autosomes (Figure 4B).
A second rapidly growing, malignant childhood tumor, medulloblastoma (Northcott et al., 2012), is also one of the entities most commonly displaying X chromosome hypermutation. All medulloblastoma genomes from females with two different copies of the X chromosome (i.e., a maternal and a paternal copy) show X hypermutation or an increased rate of X chromosomal mutations. Further, X hypermutation is already present before tetraploidy, a known early event in this tumor (Jones et al., 2012). This would be in keeping with our hypothesis that replication stress may be one of the prerequisites for X chromosome hypermutation because replication stress is one of the earliest events in tumorigenesis (Bester et al., 2011) and is also known to cause tetraploidy.
Remarkably, the medulloblastoma genome of female Li-Fraumeni syndrome case LFS-MB1 is one of the strongest X hypermutated samples, although the patient’s secondary MDS genome shows no X hypermutation (Figure 7D and 7E), supporting our suggestion that X hypermutation is a cancer-specific feature.
Given the data presented here, we hypothesize that hypermutation of Xi is one of the earliest events in tumorigenesis occurring in response to replication stress in cancer cells. Future investigation into the detailed mechanistic basis and extent of hypermutation on the inactive X chromosome may therefore provide novel insights into how tumor cells, and in particular the DNA repair machinery, respond to early oncogenic stresses. Our study makes an important contribution toward ongoing efforts to solve the fundamental question of where and how cancer genomes acquire mutations.
Experimental Procedures
Sequence Variant Discovery and Analysis
For our own data sets (medulloblastoma, astrocytoma, B cell lymphoma, ependymoma, glioblastoma, and prostate carcinoma), Illumina sequence data were aligned to the hg19 human reference genome assembly using BWA (Li and Durbin, 2009); duplicate and nonuniquely mapping reads were excluded. We subsequently detected SNVs and InDels with complementary computational approaches (see Extended Experimental Procedures). Somatic genome-wide single-nucleotide variants of published cancer data sets from other institutes were obtained from the supplemental data files of the respective publications; no additional filtering of these mutation call sets was performed.
Clonal Expansion of Single Healthy Somatic Cells
Bone marrow mononuclear cells from a healthy 73-year-old female were thawed and labeled with Alexa-Fluor 488-conjugated anti-CD34 (581, Biolegend), Alexa-Fluor 700-conjugated anti-CD38 (HIT2, eBioscience), a cocktail of APC-conjugated lineage antibodies consisting of anti-CD4 (RPA-T4), anti-CD8 (RPA-T8), anti-CD11b (ICRF44), anti-CD20 (2H7), anti-CD56 (B159, all BD Biosciences), anti-CD14 (61D3), anti-CD19 (HIB19), and anti-CD235a (HIR2, all eBiocience), and 1 μg/ml propidium iodide (Sigma). Using a BD FACSAria cell sorter, single Lin−CD34+CD38−PI− cells were individually sorted into low-adhesion 96-well tissue culture plates (Corning) containing 100 μl of StemSpan Serum-Free Expansion Medium (Stemcell technologies) supplemented with 100 ng/ml of human SCF and FLT-3L, 50 ng/ml of human TPO, 20 ng/ml of human IL-3, IL-6, and G-CSF (all cytokines from Peprotech), and 50 U/ml of penicillin and 50 μg/ml of streptomycin (Sigma). Cells were incubated at 37°C in a humidified atmosphere with 5% CO2 in air. After 5 days in culture, another 100 μl of cytokine-containing medium were added. 13 days after seeding, clones B6 and G2 had expanded to ∼105 cells and were selected for whole-genome sequencing (2 × 101 bp, paired-end, Illumina HiSeq2500) after tagmentation-based library preparation (see Extended Experimental Procedures) for clone B6 and standard library preparation for clone G2. For germline-control, ∼106 unsorted bone marrow mononuclear cells from the same donor were used for sequencing. An average of 30-fold sequence coverage for each the clones and the matching control was obtained.
A progenitor cell clone was raised from a peripheral blood sample of a 39-year-old healthy female. Frozen peripheral blood mononuclear cells (PBMCs) were isolated from 2 ml heparinised peripheral blood via Ficoll Paque density centrifugation. A methylcellulose assay was performed as described earlier (Weisse et al., 2012). In brief, nonadherent mononuclear cells were incubated in the presence of the recombinant human cytokines IL-3, IL-5, and GM-CSF (R&D systems) over 14 days to induce colony formation. Colonies were detected under an inverted light microscope and were plucked by a pipette when colonies had ∼10,000 cells/CFU. Each colony was washed three times in PBS and finally frozen as a cell pellet in −80°C. Genomic DNA was isolated using the QIAamp DNA micro kit according to the instructions of the manufacturer (QIAGEN, Hilden). Whole-genome sequencing (2 × 101 bp, paired-end, Illumina HiSeq2500) was performed for colony 4 after tagmentation-based library preparation and resulted in 15-fold sequence coverage for each the colony and the matching whole blood.
Acknowledgments
We would like to acknowledge the members of the PedBrain Tumor and MMML-Seq Project contributing to the ICGC. For technical support and expertise, we thank the DKFZ Genomics and Proteomics Core Facility, specifically Sabine Schmidt and Sasithorn Chotewutmontri. We acknowledge assistance provided by Hans-Jörg Warnatz for RNA sequencing and analysis at the Max Planck Institute for Molecular Genetics. We especially express our gratitude to Edith Heard for very helpful comments on our manuscript. This work was principally supported by the PedBrain Tumor Project and the MMML-Seq Project contributing to the ICGC, funded by German Cancer Aid (109252) and by the German Federal Ministry of Education and Research (BMBF, grants 01KU1201A, MedSys 0315416C and 01KU1002A to 01KU1002J). Additional support came from the German Cancer Research Center Heidelberg Center for Personalized Oncology (DKFZ-HIPO), the Max Planck Society, Genome Canada, and the Canadian Institute for Health Research (CIHR) with cofunding from Genome BC, Genome Quebec, CIHR-ICR (Institute for Cancer Research), and the Dietmar Hopp Foundation.
Published: October 17, 2013
Footnotes
Supplemental Information includes Extended Experimental Procedures, five figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2013.09.042.
Accession Numbers
Next-generation sequencing data have been deposited at the European Genome-phenome Archive (EGA, https://www.ebi.ac.uk/ega/) hosted by the EBI under accession numbers EGAS00001000394 and EGAS00001000565.
Supplemental Information
List of identifiers of each individual cancer sample analyzed, including clinical data, number of somatic mutations, and status of X chromosome hypermutation.
References
- Barakat T.S., Gribnau J. X chromosome inactivation in the cycle of life. Development. 2012;139:2085–2089. doi: 10.1242/dev.069328. [DOI] [PubMed] [Google Scholar]
- Bass A.J., Lawrence M.S., Brace L.E., Ramos A.H., Drier Y., Cibulskis K., Sougnez C., Voet D., Saksena G., Sivachenko A. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat. Genet. 2011;43:964–968. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bester A.C., Roniger M., Oren Y.S., Im M.M., Sarni D., Chaoat M., Bensimon A., Zamir G., Shewach D.S., Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C.J., Hendrich B.D., Rupert J.L., Lafrenière R.G., Xing Y., Lawrence J., Willard H.F. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71:527–542. doi: 10.1016/0092-8674(92)90520-m. [DOI] [PubMed] [Google Scholar]
- Burrell R.A., McClelland S.E., Endesfelder D., Groth P., Weller M.-C., Shaikh N., Domingo E., Kanu N., Dewhurst S.M., Gronroos E. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–496. doi: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel L., Willard H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Chapman M.A., Lawrence M.S., Keats J.J., Cibulskis K., Sougnez C., Schinzel A.C., Harview C.L., Brunet J.-P., Ahmann G.J., Adli M. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow J., Heard E. X inactivation and the complexities of silencing a sex chromosome. Curr. Opin. Cell Biol. 2009;21:359–366. doi: 10.1016/j.ceb.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Ellegren H., Smith N.G.C., Webster M.T. Mutation rate variation in the mammalian genome. Curr. Opin. Genet. Dev. 2003;13:562–568. doi: 10.1016/j.gde.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Greenman C., Stephens P., Smith R., Dalgliesh G.L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen R.S., Canfield T.K., Fjeld A.D., Gartler S.M. Role of late replication timing in the silencing of X-linked genes. Hum. Mol. Genet. 1996;5:1345–1353. doi: 10.1093/hmg/5.9.1345. [DOI] [PubMed] [Google Scholar]
- Hodgkinson A., Eyre-Walker A. Variation in the mutation rate across mammalian genomes. Nat. Rev. Genet. 2011;12:756–766. doi: 10.1038/nrg3098. [DOI] [PubMed] [Google Scholar]
- Jones D.T.W., Jäger N., Kool M., Zichner T., Hutter B., Sultan M., Cho Y.-J., Pugh T.J., Hovestadt V., Stütz A.M. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T.W., Hutter B., Jäger N., Korshunov A., Kool M., Warnatz H.-J., Zichner T., Lambert S.R., Ryzhova M., Quang D.A.K., International Cancer Genome Consortium PedBrain Tumor Project Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 2013;45:927–932. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., De S., Michor F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun. 2013;4:1502. doi: 10.1038/ncomms2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- Malcom C.M., Wyckoff G.J., Lahn B.T. Genic mutation rates in mammals: local similarity, chromosomal heterogeneity, and X-versus-autosome disparity. Mol. Biol. Evol. 2003;20:1633–1641. doi: 10.1093/molbev/msg178. [DOI] [PubMed] [Google Scholar]
- Mietton F., Sengupta A.K., Molla A., Picchi G., Barral S., Heliot L., Grange T., Wutz A., Dimitrov S. Weak but uniform enrichment of the histone variant macroH2A1 along the inactive X chromosome. Mol. Cell. Biol. 2009;29:150–156. doi: 10.1128/MCB.00997-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar J.J., Koster J., Zwijnenburg D.A., van Sluis P., Valentijn L.J., van der Ploeg I., Hamdi M., van Nes J., Westerman B.A., van Arkel J. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483:589–593. doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- Morishima A., Grumbach M.M., Taylor J.H. Asynchronous duplication of human chromosomes and the origin of sex chromatin. Proc. Natl. Acad. Sci. USA. 1962;48:756–763. doi: 10.1073/pnas.48.5.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A., Breast Cancer Working Group of the International Cancer Genome Consortium Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott P.A., Jones D.T.W., Kool M., Robinson G.W., Gilbertson R.J., Cho Y.-J., Pomeroy S.L., Korshunov A., Lichter P., Taylor M.D., Pfister S.M. Medulloblastomics: the end of the beginning. Nat. Rev. Cancer. 2012;12:818–834. doi: 10.1038/nrc3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K., Mlynarczyk-Evans S., Nusinow D.A., Panning B. Xist RNA and the mechanism of X chromosome inactivation. Annu. Rev. Genet. 2002;36:233–278. doi: 10.1146/annurev.genet.36.042902.092433. [DOI] [PubMed] [Google Scholar]
- Puente X.S., Pinyol M., Quesada V., Conde L., Ordóñez G.R., Villamor N., Escaramis G., Jares P., Beà S., González-Díaz M. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J., Schlesner M., Hoffmann S., Kreuz M., Leich E., Burkhardt B., Rosolowski M., Ammerpohl O., Wagener R., Bernhart S.H., ICGC MMML-Seq Project Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012;44:1316–1320. doi: 10.1038/ng.2469. [DOI] [PubMed] [Google Scholar]
- Schuster-Böckler B., Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–507. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- Stamatoyannopoulos J.A., Adzhubei I., Thurman R.E., Kryukov G.V., Mirkin S.M., Sunyaev S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009;41:393–395. doi: 10.1038/ng.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton M.R., Campbell P.J., Futreal P.A. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tort F., Bartkova J., Sehested M., Orntoft T., Lukas J., Bartek J. Retinoblastoma pathway defects show differential ability to activate the constitutive DNA damage response in human tumorigenesis. Cancer Res. 2006;66:10258–10263. doi: 10.1158/0008-5472.CAN-06-2178. [DOI] [PubMed] [Google Scholar]
- Weischenfeldt J., Simon R., Feuerbach L., Schlangen K., Weichenhan D., Minner S., Wuttig D., Warnatz H.-J., Stehr H., Rausch T. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell. 2013;23:159–170. doi: 10.1016/j.ccr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Weisse K., Lehmann I., Heroux D., Kohajda T., Herberth G., Röder S., von Bergen M., Borte M., Denburg J. The LINA cohort: indoor chemical exposure, circulating eosinophil/basophil (Eo/B) progenitors and early life skin manifestations. Clin. Exp. Allergy. 2012;42:1337–1346. doi: 10.1111/j.1365-2222.2012.04024.x. [DOI] [PubMed] [Google Scholar]
- Welch J.S., Ley T.J., Link D.C., Miller C.A., Larson D.E., Koboldt D.C., Wartman L.D., Lamprecht T.L., Liu F., Xia J. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Benavente C.A., McEvoy J., Flores-Otero J., Ding L., Chen X., Ulyanov A., Wu G., Wilson M., Wang J. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–334. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Supplemental References
- Adey A., Shendure J. Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res. 2012;22:1139–1143. doi: 10.1101/gr.136242.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen R.S., Thomas S., Sandstrom R., Canfield T.K., Thurman R.E., Weaver M., Dorschner M.O., Gartler S.M., Stamatoyannopoulos J.A. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Gu L., Adey A., Radlwimmer B., Wang W., Hovestadt V., Bähr M., Wolf S., Shendure J., Eils R. Tagmentation-based whole-genome bisulfite sequencing. Nat. Protoc. 2013;8:2022–2032. doi: 10.1038/nprot.2013.118. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of identifiers of each individual cancer sample analyzed, including clinical data, number of somatic mutations, and status of X chromosome hypermutation.