

Abstract
In various autoimmune diseases, anti-tumour necrosis factor (TNF)-α treatment has been shown to reduce both clinical disease severity and T helper type 1 (Th1)1/Th17 responses. In experimental autoimmune encephalomyelitis (EAE), however, the role of TNF-α has remained unclear. Here, C57BL/6 mice were immunized with myelin oligodendrocyte glycoprotein (MOG) peptide 35–55 and treated with anti-TNF-α, control antibody or vehicle. The clinical disease course, incidence and severity were assessed. On day 20 after immunization the antigen-specific Th1/Th17 response was evaluated by enzyme-linked immunospot (ELISPOT) in spleen and central nervous system (CNS). Also, the extent of spinal cord histopathology was analysed on semi- and ultrathin sections. Our results demonstrate that anti-TNF-α treatment reduced the incidence and delayed the onset of EAE, but had no effect on disease severity once EAE had been established. Whereas anti-TNF-α treatment induced an increase in splenic Th1/Th17 responses, there was no effect on the number of antigen-specific Th1/Th17 cells in the spinal cord. Accordingly, the degree of CNS histopathology was comparable in control and anti-TNF-α-treated mice. In conclusion, while the anti-TNF-α treatment had neither immunosuppressive effects on the Th1/Th17 response in the CNS nor histoprotective properties in EAE, it enhanced the myelin-specific T cell response in the immune periphery.
Keywords: EAE/MS, inflammation, monocytes/macrophages, neuroimmunology, T cells
Introduction
Tumour necrosis factor (TNF)-α is a cytokine with pleiotropic functions and has been implicated in various autoimmune diseases. The main sources of TNF-α are activated mononuclear phagocytes and, to a lesser degree, T cells. TNF-α is synthesized as a transmembrane protein, and cleavage of the extracellular domain results in the release of soluble TNF-α. TNF-α can bind to two receptors, TNF receptor (TNFR) p55 (TNFR I) and TNFR p75 (TNFR II). TNFR p55 is expressed on almost every cell type; TNFR p75, in contrast, is restricted mainly to immune cells, but is also found on oligodendrocytes 1 and neurones 2. While the extracellular domains of the two receptors are structurally similar, the intracellular domains have little homology. TNFR p55 contains an intracellular death domain, which ultimately activates caspases and triggers apoptosis. In contrast, TNFR p75 lacks a death domain, but activates mitogen-activated protein kinase (MAPK), nuclear factor (NF)-κB and activator protein (AP)-1 pathways, leading to transcription of genes involved in inflammation or cell survival 3. However, there is thought to be a degree of cross-talk between the two TNF receptors, so that both contribute to gene transcription and apoptosis 3.
Aberrant TNF production 4 and defects in TNF receptor signalling 3 have been detected in various autoimmune diseases. To date, TNF-α inhibitors are licensed for rheumatoid arthritis, psoriasis, psoriatic arthritis, ankylosing spondylitis and Crohn's disease 5–9. However, in a clinical trial with multiple sclerosis (MS) patients, an anti-TNF agent led to aggravation of the relapse rate 10. In other patients undergoing anti-TNF treatment, demyelinating side effects and autoantibody production (e.g. against DNA or nuclear proteins) have been observed. Even new onset of MS or of other autoimmune disorders has been reported 11, highlighting the complex role played by TNF-α in autoimmunity.
In-depth analyses of treatment effects in patients are hampered by the difficulty in obtaining cells from lymphoid organs and inflammatory sites. Therefore, research relies largely upon suitable animal models. The most widely used animal model for multiple sclerosis is experimental autoimmune encephalomyelitis (EAE), in which immunization of susceptible animal strains with myelin antigens emulsified in complete Freund's adjuvant induces an autoreactive immune response, spinal cord histopathology and finally clinical symptoms 12.
Collagen-induced arthritis (CIA) is a well-validated model of rheumatoid arthritis that has been used widely to study the effects of anti-TNF-α treatment. Anti-TNF-α treatment reduced paw swelling and the severity of joint histopathology 13. However, more recent studies showed that TNF-α antibody had a dual effect on the T cell response: while treatment reduced T helper type 1 (Th1)/Th17 infiltration of the joint, it led to increased numbers of Th1/Th17 cells in the lymph nodes 14. The literature on TNF-α abrogation in EAE presents a controversial picture. While anti-TNF-α antibody treatment in general conveys some clinical disease amelioration, not all studies found the same features to be affected in the same way 15–18. Data on the antigen-specific Th1/Th17 response in lymphoid organs and the central nervous system (CNS) are heterogeneous 11–12, as is the effect of TNF-α neutralization on axonal damage 11.
In this study we characterized simultaneously the effects of anti-TNF-α treatment on clinical, immunological and histopathological disease development in the traditional myelin oligodendrocyte glycoprotein peptide 35–55 (MOG:35–55)-induced EAE model. We analysed whether or not the treatment provoked a dual effect on the T cell response similar to the one observed in collagen-induced arthritis, which would predict increased Th1/Th17 frequencies in the spleen and reduced Th1/Th17 frequencies in the CNS. Additionally, we investigated the effect of anti-TNF-α treatment on axon and myelin integrity. The assessment of histopathology is of particular importance, as neurodegeneration is the morphological correlate of irreversible clinical symptoms in MS patients 19. Currently, TNF-α blocking agents are not considered as candidate drugs for MS because of the dramatic failure of a clinical trial in 1999 10. It is therefore important to elucidate the general effects of anti-TNF-α treatment on immune responses in EAE versus its effect on inflammation in the CNS. In this way we may ultimately gain invaluable insights towards successful CNS immune modulation.
Material and methods
Animals
C57BL/6 mice (6–8 weeks old) were purchased from the Harlan Laboratories (Sulzfeld, Germany) and Janvier (Saint Berthevin Cedex, France) and maintained in individually ventilated cages at the animal facilities of the Department of Anatomy of Cologne University. Incomplete Freund's adjuvant (IFA) was prepared as a mixture of paraffin oil (EMScience, Gibbstown, NJ, USA) and mannide monooleate (Sigma, Schnelldorf, Germany). Complete Freund's adjuvant (CFA) was obtained by adding Mycobacterium tuberculosis H37RA (Difco Laboratories, Franklin Lakes, NJ, USA) at 5 mg/ml to IFA. Animals were immunized subcutaneously in both sides of the flank with a total dose of 100 μg MOG:35–55 (EZBiolab, Carmel, IN, USA) emulsified in CFA (injection volume = 200 μl). Each mouse received 200 ng pertussis toxin (List Biological Laboratories, Hornby, Ontario, Canada) in 500 μl sterile phosphate-buffered saline (PBS) on the day of immunization and 48 h later. Clinical symptoms were evaluated daily according to the standard EAE scale: 0, no symptoms; 1, floppy tail; 2, hind limb weakness; 3, hind limb paralysis; 4, quadriplegia; and 5, death. Mice were euthanized with CO2 on day 20 post-immunization. For treatment, mice were injected intraperitoneally every other day, starting from day 3 post-immunization, either with 100 μg Enbrel®, 100 μg Humira® or PBS (injection volume of 500 μl). Enbrel® is a fusion protein between the extracellular domain of the TNFR2/p75 and the Fc fragment of human immunoglobulin (Ig)G1. Humira® is of the same isotype as Enbrel, but does not neutralize murine TNF. All experiments were approved by the German Animal Welfare Act.
Cell preparation
The spleen and spinal column were removed, and the spinal cord was flushed out with Dulbecco's modified Eagle's medium (DMEM) (PAA, Pasching, Austria). Specimens were disintegrated mechanically and filtered through a 70-μm nylon cell strainer (BD Falcon, Heidelberg, Germany). After washing the cells with RPMI-1640 (Biochrom AG, Berlin, Germany) and counting them with acridine orange (0·1%, Sigma)/ethidium bromide (0·1%, Serva, Heidelberg, Germany), cells were resuspended in HL-1 (Lonza, Cologne, Germany) supplemented with 1% glutamine (Sigma) and 1% penicillin/streptomycin (Sigma).
ELISPOT assays
Low-volume Unifilter Whatman plates (Whatman Inc., Florham Park, NJ, USA) were coated overnight with the capture antibodies rat anti-mouse interferon (IFN)-γ (final concentration 3 μg/ml, clone AN-18; eBioscience, San Diego, CA, USA) and rat anti-mouse interleukin (IL)-17 (final concentration 4 μg/ml, clone TC-11-18H10; BD Biosciences, San Diego, CA, USA) in PBS as follows: first, plates were precoated with anti-IFN-γ, after 10 min anti-IL-17 was added. Plates were washed with PBS, and blocked with 1% bovine serum albumin (BSA) in PBS for 2 h at room temperature. Spleen cells were plated at 5 × 105 cells/well and spinal cord cells at 1 × 105 cells/well. Antigen-presenting cells were obtained by irradiating spleen cells from naive C57BL/6 mice with 26 Gy and added to the spinal cord cells at a concentration of 2·5 × 105 cells/well. Cells were incubated with either medium or MOG:35–55 (final concentration: 15 μg/ml) at 7% CO2 and 37°C for 24 h. Plates were washed and incubated with fluorescein isothiocyanate (FITC)-conjugated anti-IFN-γ (0·5 μg/ml; gift from M. Tary-Lehmann, clone R4-6A2) and biotin-conjugated anti-IL-17 (0·5 μg/ml; Pharmingen, San Diego, CA, USA; clone TC-11-8H4·1) overnight at 4°C. After washing, plates were incubated for 2 h with anti-FITC-labelled alkaline phosphatase (1/500; Dako, Glostrup, Denmark) and streptavidin-conjugated horseradish peroxidase (1/1000; Dako). Plates were developed with Vector Blue (Vector Laboratories, Burlingame, CA, USA) and AEC (Vector Laboratories) solution according to the vendor's instructions. Plates were air-dried overnight and spots were counted with an ImmunoSpot Series 5 UV Analyzer (Cellular Technology Limited, Shaker Heights, OH, USA). All results were medium-subtracted and normalized to 106 cells per well.
Histology
In order to investigate the effects of anti-TNF-α treatment on spinal cord histopathology, semi- and ultrathin sections of the lumbar spinal cord of PBS-, Enbrel®- and Humira®-treated mice were obtained as described previously 20. Briefly, mice were perfused intracardially on day 20 after EAE induction with a 4% paraformaldehyde/4% glutaraldeyhde/PBS solution. The lumbar spinal cord was post-fixed, rinsed in cacodylate buffer and treated with 1% osmium tetroxide (Chempur, Karlsruhe, Germany). Tissues were treated with 1% uranyl acetate (Plano GmbH, Wetzlar, Germany) in 70% ethanol for contrast enhancement overnight. Subsequently, specimens were embedded in epon (Fluka, St Louis, MO, USA) and polymerized at 60°C for at least 72 h. Eighty nm-thick ultrathin and 500 nm-thick semithin sections of each plastic-embedded spinal cord sample were cut on a Leica Ultracut UCT ultramicrotome (Leica Microsystems, Wetzlar, Germany). For light-microscopic analysis, sections were stained with methylene blue and for electron microscopy with 1% aqueous uranyl acetate solution for 20 min and Reynold's lead citrate solution (Merck KGaA, Darmstadt, Germany) for 7 min. Specimens were examined with a Zeiss EM 902 A transmission electron microscope at 80 kV acceleration voltage and images were taken with an EM digital camera system (MegaView III; Olympus Soft Imaging Systems GmbH, Münster, Germany) at ×7000 magnification. Images of score-matched Enbrel®- (n = 4), Humira® (n = 3)- and non-immunized control mice (n = 4) (10 images per mouse) from the ventrolateral tract were analysed for the extent of myelin and axonal pathology. The numbers of physiological, demyelinated and axolytic nerve fibres were counted with ImagePro Plus software (Media Cybernetics, Inc., Rockville, MD, USA). Methylene blue-stained sections were observed using a Leica DM LB2 microscope and digital images were acquired at ×20 magnification with an AxioCam camera (Zeiss, Oberkochen, Germany) and Zeiss software (AxioVision 40 4·7). The degree of inflammation was determined semiquantitatively according to the following scoring system: 0, no infiltrates (resembling naive mice); 1, partial meningeal and perivascular infiltration; 2, pronounced meningeal and perivascular infiltration; 3, pronounced meningeal and perivascular infiltration and some parenchymal infiltration; and 4, pronounced meningeal and perivascular and widespread parenchymal infiltration.
Statistical analyses
SigmaPlot Software version 12·0 (Chicago, IL, USA) was used for all statistical analyses. For evaluation of differences in the clinical score, disease onset, antigen-specific IFN-γ and IL-17 production by enzyme-linked immunospot (ELISPOT) and CNS histopathology the Wilcoxon–Mann–Whitney test was used. Fisher's exact test was used for evaluating differences in disease incidence. Spearman's rank-order correlation was used for correlating antigen-specific cytokine production and clinical score. The limit of statistical significance was P ≤ 0·05.
Results
TNF-α blockade reduces the incidence and delays the onset of EAE
For the induction of EAE, C57BL/6 mice were immunized with 100 μg MOG:35–55 in CFA. Pertussis toxin was administered at 200 ng per mouse on days 0 and 2 post-immunization. Mice were scored daily for the occurrence of clinical symptoms. Starting on day 3 post-immunization, n = 15 mice were treated with Enbrel® every other day, while n = 19 mice received sham injections (PBS). Ten mice were treated with Humira® as control antibody. Figure 1a shows the clinical course of a representative cohort of Enbrel®-, Humira®- or PBS-treated mice. In comparison to PBS- or Humira®-treated mice, mice that received Enbrel® showed reduced EAE incidence (Fig. 1b). While 95% of the PBS- and 100% of the Humira®-treated mice exhibited symptoms, only 53% of the Enbrel®-treated mice developed EAE (P < 0·05, Fisher's exact t-test). Enbrel® also caused delayed EAE onset (Fig. 1c). In PBS-treated mice, EAE developed on day 10 (± 1 day) and mice that received Humira® showed an EAE onset on day 12 (± 0·6 days), whereas in Enbrel®-treated mice EAE onset was on day 17 (± 3 days) (P < 0·001 and P < 0·01, respectively, rank sum t-test). However, once clinical EAE had been established, there was no significant difference in the mean clinical scores between the three groups (Fig. 1d).
Figure 1.
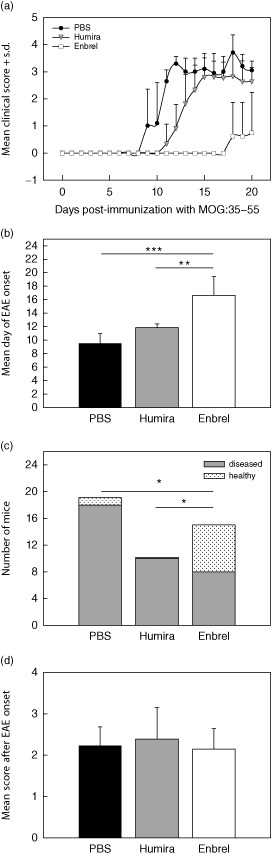
Tumour necrosis factor (TNF)-α blockade reduces the incidence and delays the onset of experimental autoimmune encephalomyelitis (EAE). (a) Clinical course of a representative cohort of Enbrel®-treated mice (white squares) and mice that received phosphate-buffered saline (PBS) (black circles) or Humira® injections (grey triangles). The Enbrel® and PBS cohort included mice that did not develop EAE. (b) Mean day of EAE onset (***P < 0·001, rank sum test). (c) EAE incidence with solid bar segments referring to diseased mice and dotted segments referring to healthy mice 20 days post-immunization (*P < 0·05, Fisher's exact test). (d) Mean score after EAE onset of all diseased mice. The data refer to n = 15 Enbrel®-, n = 10 Humira®- and n = 19 PBS-treated mice.
TNF-α blockade increases the antigen-specific Th1/Th17 response in the spleen, but has no effect on the antigen-specific Th1/Th17 response in the spinal cord
In order to determine if the clinical amelioration observed with anti-TNF-α treatment was associated with changes in the antigen-specific T cell response, mice were killed on day 20 post-immunization, after the onset of full-blown EAE. ELISPOT assays were performed with spleen and spinal cord cells. Figure 2 shows the number of antigen-specific IFN-γ- and IL-17-producing cells of PBS-, Humira®- and Enbrel®-treated mice. With Enbrel® treatment, there was a significant increase in the splenic antigen-specific Th1 (P < 0·01, Wilcoxon's rank-sum test) and Th17 (P < 0·05, Wilcoxon's rank-sum test) response in comparison to PBS or Humira® treatment (Fig. 2a). However, we observed no difference in the antigen-specific Th1/Th17 response with Enbrel® treatment in the spinal cord (Fig. 2b).
Figure 2.
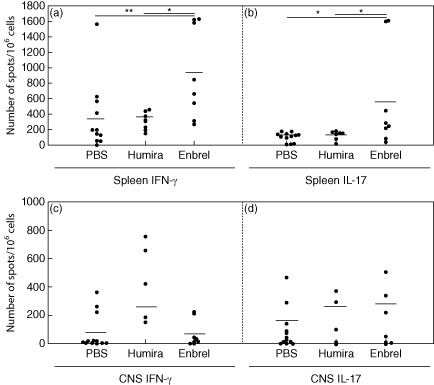
Effects of tumour necrosis factor (TNF)-α blockade on the frequencies of antigen-specific Th1/Th17 cells in spleen and central nervous system (CNS). (a) Results of enzyme-linked immunospot (elispot) assays in phosphate-buffered saline (PBS) (n = 12)-, Humira® (n = 8)- and Enbrel®-treated mice (n = 8) for interferon (IFN)-γ (**P < 0·01, rank sum test) and interleukin (IL-17 (*P < 0·05, rank sum test) in the spleen. (b) Results of IFN-γ/IL-17 elispot assays in PBS (n = 13)-, Humira® (n = 5)- and Enbrel®-treated mice (n = 8 IFN-γ; n = 7 IL-17) in the spinal cord.
In a next step, we analysed the extent to which the antigen-specific Th1/Th17 response correlated with disease severity. While there was a significant correlation between the antigen-specific Th1/Th17 response in the spinal cord and the clinical score (rs = 0·668, P < 0·05 for IFN-γ; rs = 0·6, P < 0·05 for IL-17; Spearman's rank-order correlation), no such correlation could be found in the spleen (rs = 0·567, P = 0·051 for IFN-γ; rs = 0·251, P = 0·415 for IL-17; Spearman's rank-order correlation).
These findings indicate that inhibition of TNF-α increased the numbers of antigen-specific Th1/Th17 cells in the spleen, although these cells appeared not to be associated with the disease process. In the CNS, however, no protective effects of Enbrel® treatment could be observed, which was mirrored by the similarity of disease severity in the various treatment groups. To validate the lack of a neuroprotective effect of TNF-α blockade, we performed in-depth histopathological analysis of the spinal cord.
TNF-α blockade has no protective effect on inflammatory activity, myelin pathology and axonal damage in MOG:35–55-induced EAE
Twenty days after EAE induction, n = 4 PBS-, n = 3 Humira®- and n = 4 Enbrel®-treated mice were killed. The spinal cord was epon-embedded and semi- and ultrathin sections were obtained. Representative images of semithin sections are shown in Fig. 3a–d. In these sections, we quantified the mean lesion size per total white matter. Table 1 shows that there was no difference in the overall lesion size between PBS-, Humira®- and Enbrel®-treated mice. In addition, the extent of inflammation was assessed semiquantitatively. Again, no difference in the two inflammatory parameters was detected between the treatment groups (Table 1).
Figure 3.
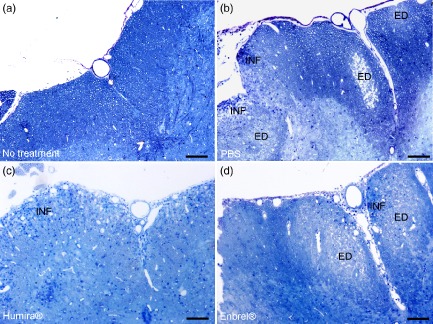
Light-microscopic analysis of spinal cord histopathology in anti-tumour necrosis factor (TNF)-α-treated mice. C57BL/6 mice were immunized with MOG:35–55, treated with phosphate-buffered saline (PBS) (n = 4), Humira® (n = 3) or Enbrel® (n = 4) and killed on day 20 post-immunization. Spinal cords were removed carefully from the vertebral canal, epon-embedded and semi-thin sections of the lumbar part were obtained. (a–d) Representative images of non-immunized control mice (a) and PBS- (b), Humira®-(c) or Enbrel®-treated MOG:35–55-immunized mice (d). The scale bar denotes 100 μm. ED = oedema; INF = blue dots represent immune cells infiltrating the central nervous system (CNS).
Table 1.
Central nervous system (CNS) histopathology in phosphate-buffered saline (PBS)-, Humira®- and Enbrel®-treated mice
| Treatment | No. of mice | Clinical score | Degree of inflammation | Lesion size [%] | No. of intact axons/mm2 | No. of axolytic axons/mm2 | No. of demyelinated axons/mm2 |
|---|---|---|---|---|---|---|---|
| PBS | 4 | 2·50 ± 0·41 | 2·00 ± 1·08 | 31·92 ± 20·37 | 2·17 × 105 ± 2·75 × 104 | 3·39 × 104 ± 3·51 × 103 | 6·17 × 103 ± 6·64 × 103 |
| Humira® | 3 | 2·92 ± 0·14 | 2·33 ± 0·94 | 29·73 ± 10·36 | 1·40 × 105 ± 1·40 × 105 | 5·21 × 104 ± 2·83 × 103 | 5·58 × 105 ± 9·33 × 105 |
| Enbrel® | 4 | 2·69 ± 0·13 | 2·88 ± 0·85 | 55·76 ± 25·47 | 1·60 × 105 ± 1·11 × 105 | 4·06 × 104 ± 3·72 × 104 | 4·96 × 103 ± 5·02 × 103 |
Light-microscopic analysis can give a general overview of the pathological traits, while falling short of providing details about the fine characteristics of nerve fibre damage. Previous studies have reported changes in the extent of axonal damage and demyelination after anti-TNF-α treatment 16–18. However, these studies relied upon light-microscopic and immunohistochemical techniques. In order to provide more profound information about axon and myelin pathologies, we performed ultrastructural analysis of PBS-, Humira®- and Enbrel®-treated mice. Representative images of non-immunized control mice and of PBS-, Humira®- and Enbrel®-treated mice are depicted in Fig. 4a–d. To begin with, we determined the number of demyelinated nerve fibres per mm2 lesion area. There was no statistically significant difference in the number of demyelinated nerve fibres between PBS-, Humira®- and Enbrel®-treated mice (Table 1). We next determined the extent of axonal loss by counting the numbers of axons per mm2 lesion area. There was no statistically significant difference in axonal density between the treatment groups (Table 1). The same applied to gross axonal damage as characterized by axolysis (Table 1).
Figure 4.
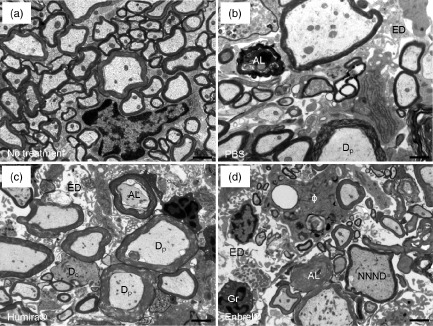
Ultrastructural analysis of spinal cord histopathology in anti-tumour necrosis factor (TNF)-α-treated mice. Mice were immunized and treated as described in Fig. 3. After epon-embedding, ultrathin sections of the lumbar spinal cord of non-immunized control, phosphate-buffered saline (PBS)-, Humira®- and Enbrel®-treated mice were obtained and assessed by electronmicroscopy. Representative images are shown in (a–d). The scale bar depicts 1 μm. AL = axolysis; ED = oedema; Dc = demyelinated nerve fibre; Dp = nerve fibre in the process of demyelination; Gr = granulocyte; Φ = macrophage with myelin debris in the cytoplasm; NNND = nerve fibre with decreased nearest-neighbour neurofilament distance indicating axonal pathology.
Discussion
Here we explored the clinical, immunological and histopathological consequences of treatment with a TNF-α inhibitor in EAE. The results of this study suggest that anti-TNF-α treatment may be helpful in delaying or even preventing EAE outbreak, but once the disease has been established, TNF-α blockade provides no further clinical benefit. More importantly, anti-TNF-α treatment led to the accumulation of antigen-specific Th1/Th17 cells in the spleen, which could have deleterious consequences when the anti-TNF-α antibody is withdrawn.
TNF-α is known to be required for the functioning of regulatory T cells (Treg), which suppress Th1/Th17 cells via IL-10 secretion 21–24. Anti-TNF-α treatment may therefore have impaired Treg function, thereby reducing their suppressive capacity towards Th1/Th17 cells. This may explain the increased splenic antigen-specific Th1/Th17 response in our study. Along these lines, it has been reported that TNF-signalling is particularly important for natural, but not inducible, Tregs 25. This offers a possible explanation as to why anti-TNF-α treatment has detrimental effects in some human diseases, but not in others: conceivably, the diseases differ in the relevance of natural and/or inducible Tregs. However, it has also been shown that TNF-α inhibits the expression of IL-12/IL-23 p40 14, and this provides an equally plausible explanation for the increased numbers of Th1/Th17 cells observed. It is interesting to note that increased numbers of Th1/Th17 cells were observed in TNFR1−/− mice with reactive arthritis, which was associated with enhanced p40 expression 26. In experimental colitis, IL-17 has been shown to suppress T-bet and limit TH1 activity 27. While an increase in Th17 immunity may therefore have beneficial effects, in the present study the increased splenic Th1/Th17 response appeared to have no impact on the clinical EAE outcome. Our results show that, in MOG:35–55-induced EAE, only the antigen-specific Th1/Th17 response in the spinal cord (but not in the spleen) was associated with the clinical disease severity. While, in CIA, lower Th1/Th17 frequencies were observed in the target organ (i.e. the joint) after anti-TNF-α treatment and clinical disease was ameliorated, we observed no difference in the antigen-specific Th1/Th17 response in the spinal cord. In accordance, clinical EAE severity was unaltered in our study. On this basis we hypothesize that TNF-α blockade has the dual effect of inhibiting inflammation, but enhancing pathogenic T cell responses, such that the net effect on pathology is minimal.
One of the possible explanations as to why TNF-α blockade had no immunosuppressive effect on the Th1/Th17 response in the CNS could be that TNF-α may not be required for Th1/Th17 function in this target organ. While T cell co-stimulation is certainly necessary for T cell effector functioning, this co-stimulation may be mediated by molecules other than TNF-α during MOG:35–55-induced EAE, e.g. CD28 (compare with 28). In addition, TNF-α may have no unbalanced proinflammatory properties in the CNS during EAE. As described above, TNF-α binding can induce apoptosis. In a recent report oligodendrocyte apoptosis was identified to be a crucial event occurring during early EAE, and to contribute to axonal degeneration and neuronal loss 29–30. The assumption that oligodendrocyte apoptosis paves the way for axonal pathologies suggests that anti-TNF-α treatment may reduce the amount of neurodegeneration. Because the effect of anti-TNF-α treatment on neurodegeneration has not been delineated clearly in the past, we have performed ultrastructural analysis of PBS-, Humira®- and Enbrel®-treated mice. Having analysed various parameters of neurodegeneration, we observed no reduction in axonal pathology after TNF-α blockade. We therefore conclude that inhibition of TNF-α conveys no histoprotective effect in MOG:35–55-induced EAE. We further postulate that the amount of neurodegeneration is independent of apoptosis, and that TNF-α is not involved directly in axonal damage. Rather, TNF-α might function as an inflammatory mediator, stimulating chemokine production, which indirectly attracts more phagocytes into the CNS 31.
Although disease severity of established EAE was unaltered after TNF-α blockade, onset and incidence of the disease were reduced significantly despite the enhanced Th1/Th17 responses observed in spleen cells. The enhanced splenic response implies that TNF-α blockade could have an indirect anti-inflammatory effect that was not due to a reduced response to immunization. Rather, TNF-α blockade may have retained the pathogenic Th1/Th17 cells in the spleen and prevented their egress towards the CNS. It is tempting to speculate that a treatment stop leads to a sudden egress of these pathogenic T helper cells from the spleen into the CNS, and thus causes clinical deterioration. Indeed, our preliminary results indicate a significant reduction in the Th1 response in the spleen after treatment stop, which appeared to be accompanied by an increase in the clinical score (data not shown). However, further experiments are needed to confirm this notion.
In conclusion, although TNF-α blockade exerts protective effects in some autoimmune diseases, it is not capable of down-regulating neither the antigen-specific Th1/Th17 response nor histopathology in the CNS in EAE. Furthermore, the expansion of Th1/Th17 cells observed in Enbrel®-treated mice may have deleterious consequences when the TNF-α inhibitor is withdrawn, providing one possible explanation for the failure of TNF-α inhibition in human MS.
Author contributions
H. B. and M. S. R. designed the study, performed experiments, analysed the data and wrote the manuscript. F. O. H. and F. T. performed experiments. R. O. W. helped to design the study and write the manuscript. S K. designed the study, performed experiments, analysed data and wrote the manuscript. All authors have read and approved the final manuscript.
Acknowledgments
We would like to thank Christian Hoffmann, Evelyn Janßen and Jolanta Kozlowski for expert technical assistance. This work was supported by research grants from the German Research Foundation (DFG) (project KU 2760/2-1), the Köln Fortune Programm and the Imhoff-Stiftung (grants to S. K.).
Disclosure
The authors declare that they have no competing interests.
References
- 1.Dopp JM, Sarafian TA, Spinella FM, Kahn MA, Shau H, de Vellis J. Expression of the p75 TNF receptor is linked to TNF-induced NFkappaB translocation and oxyradical neutralization in glial cells. Neurochem Res. 2002;27:1535–1542. doi: 10.1023/a:1021608724117. [DOI] [PubMed] [Google Scholar]
- 2.Cheng X, Yang L, He P, Li R, Shen Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer's disease and non-demented patients. J Alzheimers Dis. 2010;19:621–630. doi: 10.3233/JAD-2010-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov. 2010;9:482–493. doi: 10.1038/nrd3030. [DOI] [PubMed] [Google Scholar]
- 4.Kollias G, Kontoyiannis D. Role of TNF/TNFR in autoimmunity: specific TNF receptor blockade may be advantageous to anti-TNF treatments. Cytokine Growth Factor Rev. 2002;13:315–321. doi: 10.1016/s1359-6101(02)00019-9. [DOI] [PubMed] [Google Scholar]
- 5.Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–196. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- 6.Martínez F, Nos P, Benlloch S, Ponce J. Hidradenitis suppurativa and Crohn's disease: response to treatment with infliximab. Inflamm Bowel Dis. 2001;7:323–326. doi: 10.1097/00054725-200111000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Heiberg MS, Koldingsnes W, Mikkelsen K, et al. The comparative one-year performance of anti-tumor necrosis factor alpha drugs in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: results from a longitudinal, observational, multicenter study. Arthritis Rheum. 2008;59:234–240. doi: 10.1002/art.23333. [DOI] [PubMed] [Google Scholar]
- 8.Reich K, Nestle FO, Papp K, et al. EXPRESS study investigators. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet. 2005;366:1367–1374. doi: 10.1016/S0140-6736(05)67566-6. [DOI] [PubMed] [Google Scholar]
- 9.Lichtenstein GR, Yan S, Bala M, Blank M, Sands BE. Infliximab maintenance treatment reduces hospitalizations, surgeries, and procedures in fistulizing Crohn's disease. Gastroenterology. 2005;128:862–869. doi: 10.1053/j.gastro.2005.01.048. [DOI] [PubMed] [Google Scholar]
- 10.The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. Neurology. 1999;53:457–465. [PubMed] [Google Scholar]
- 11.Kodama S, Davis M, Faustman DL. The therapeutic potential of tumor necrosis factor for autoimmune disease: a mechanistically based hypothesis. Cell Mol Life Sci. 2005;62:1850–1862. doi: 10.1007/s00018-005-5022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mix E, Meyer-Rienecker H, Hartung HP, Zettl UK. Animal models of multiple sclerosis – potentials and limitations. Prog Neurobiol. 2010;92:386–404. doi: 10.1016/j.pneurobio.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992;89:9784–9788. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Notley CA, Inglis JJ, Alzabin S, McCann FE, McNamee KE, Williams RO. Blockade of tumor necrosis factor in collagen-induced arthritis reveals a novel immunoregulatory pathway for Th1 and Th17 cells. J Exp Med. 2008;205:2491–2497. doi: 10.1084/jem.20072707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baker D, Butler D, Scallon BJ, O'Neill JK, Turk JL, Feldmann M. Control of established experimental allergic encephalomyelitis by inhibition of tumor necrosis factor (TNF) activity within the central nervous system using monoclonal antibodies and TNF receptor-immunoglobulin fusion proteins. Eur J Immunol. 1994;24:2040–2048. doi: 10.1002/eji.1830240916. [DOI] [PubMed] [Google Scholar]
- 16.Taoufik E, Tseveleki V, Chu SY, et al. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-kappaB. Brain. 2011;134:2722–2735. doi: 10.1093/brain/awr203. [DOI] [PubMed] [Google Scholar]
- 17.Kruglov AA, Lampropoulou V, Fillatreau S, Nedospasov SA. Pathogenic and protective functions of TNF in neuroinflammation are defined by its expression in T lymphocytes and myeloid cells. J Immunol. 2011;187:5660–5670. doi: 10.4049/jimmunol.1100663. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Marino MW, Wong G, et al. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat Med. 1998;4:78–83. doi: 10.1038/nm0198-078. [DOI] [PubMed] [Google Scholar]
- 19.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 20.Rottlaender A, Villwock H, Addicks K, Kuerten S. Neuroprotective role of fibroblast growth factor-2 in experimental autoimmune encephalomyelitis. Immunology. 2011;133:370–378. doi: 10.1111/j.1365-2567.2011.03450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Oppenheim JJ. Contrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunity. FEBS Lett. 2011;585:3611–3618. doi: 10.1016/j.febslet.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma HL, Napierata L, Stedman N, et al. Tumor necrosis factor alpha blockade exacerbates murine psoriasis-like disease by enhancing Th17 function and decreasing expansion of Treg cells. Arthritis Rheum. 2010;62:430–440. doi: 10.1002/art.27203. [DOI] [PubMed] [Google Scholar]
- 23.Kleijwegt FS, Laban S, Duinkerken G, et al. Critical role for TNF in the induction of human antigen-specific regulatory T cells by tolerogenic dendritic cells. J Immunol. 2010;185:1412–1418. doi: 10.4049/jimmunol.1000560. [DOI] [PubMed] [Google Scholar]
- 24.Ablamunits V, Bisikirska B, Herold KC. Acquisition of regulatory function by human CD8(+) T cells treated with anti-CD3 antibody requires TNF. Eur J Immunol. 2010;40:2891–2901. doi: 10.1002/eji.201040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Housley WJ, Adams CO, Nichols FC, et al. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J Immunol. 2011;186:6779–6787. doi: 10.4049/jimmunol.1003868. [DOI] [PubMed] [Google Scholar]
- 26.Eliçabe RJ, Cargnelutti E, Serer MI, et al. Lack of TNFR p55 results in heightened expression of IFN-γ and IL-17 during the development of reactive arthritis. J Immunol. 2010;185:4485–4495. doi: 10.4049/jimmunol.0902245. [DOI] [PubMed] [Google Scholar]
- 27.O'Connor W, Jr, Kamanaka M, Booth CJ, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunnlaugsdottir B, Skaftadottir I, Ludviksson BR. Naive human T-cells become non-responsive towards anti-TNFalpha (infliximab) treatment in vitro if co-stimulated through CD28. Scand J Immunol. 2008;68:624–634. doi: 10.1111/j.1365-3083.2008.02181.x. [DOI] [PubMed] [Google Scholar]
- 29.Mc Guire C, Volckaert T, Wolke U, et al. Oligodendrocyte-specific FADD deletion protects mice from autoimmune-mediated demyelination. J Immunol. 2010;185:7646–7653. doi: 10.4049/jimmunol.1000930. [DOI] [PubMed] [Google Scholar]
- 30.Mc Guire C, Beyaert R, van oo G. Death receptor signalling in central nervous system inflammation and demyelination. Trends Neurosci. 2011;34:619–628. doi: 10.1016/j.tins.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Sheng WS, Hu S, Ni HT, Rowen TN, Lokensgard JR, Peterson PK. TNF-alpha-induced chemokine production and apoptosis in human neural precursor cells. J Leukoc Biol. 2005;78:1233–1241. doi: 10.1189/jlb.0405221. [DOI] [PubMed] [Google Scholar]