

Abstract
Hereditary angioedema (HAE) and acquired angioedema (AAE) are rare life-threatening conditions caused by deficiency of C1 inhibitor (C1INH). Both are characterized by recurrent unpredictable episodes of mucosal swelling involving three main areas: the skin, gastrointestinal tract and larynx. Swelling in the gastrointestinal tract results in abdominal pain and vomiting, while swelling in the larynx may be fatal. There are limited UK data on these patients to help improve practice and understand more clearly the burden of disease. An audit tool was designed, informed by the published UK consensus document and clinical practice, and sent to clinicians involved in the care of HAE patients through a number of national organizations. Data sets on 376 patients were received from 14 centres in England, Scotland and Wales. There were 55 deaths from HAE in 33 families, emphasizing the potentially lethal nature of this disease. These data also show that there is a significant diagnostic delay of on average 10 years for type I HAE, 18 years for type II HAE and 5 years for AAE. For HAE the average annual frequency of swellings per patient affecting the periphery was eight, abdomen 5 and airway 0·5, with wide individual variation. The impact on quality of life was rated as moderate or severe by 37% of adult patients. The audit has helped to define the burden of disease in the UK and has aided planning new treatments for UK patients.
Keywords: acquired angioedema; C1 esterase inhibitor, complement; hereditary angioedema; primary immunodeficiency; secondary immunodeficiency; SERPING1
Introduction
Hereditary angioedema (HAE) is a rare disease due to C1 inhibitor deficiency with autosomal dominant inheritance caused by mutations in SERPING1. These result in either low levels of C1 inhibitor (C1INH) (type I HAE) or normal levels with reduced C1 inhibitor function (type II HAE) 1. A third type of HAE is now recognized (type III HAE), or HAE with normal C1INH due in some cases to mutations in Factor XII (FXII) 2–3. Acquired angioedema (AAE) may be caused by anti-C1INH antibodies and tends to be associated with haematological malignancy or, more rarely, autoimmune disease 4–5. Surveys suggest that HAE affects one in 50–100 000 of the population 6–7 and a recent study underlined the importance of diagnosis and appropriate treatment, as the mortality of HAE patients who had not been diagnosed was 29% compared to 3% in those who had been diagnosed 8.
The mechanism causing angioedema in HAE is the generation of increased levels of bradykinin, and is distinct from allergic angioedema due to mast cell activation where the key mediator is histamine. Mutations in SERPING1 result in reduced levels or reduced function of C1INH, which is an inhibitor of the contact system. Lack of inhibition allows activated FXIIa to promote the conversion of prekallikrein to kallikrein which, in turn, enhances the conversion of high molecular weight kininogen (HMWK) to bradykinin (Fig. 1). Bradykinin, a potent vasoactive peptide, mediates increased capillary permeability and oedema by binding to the bradykinin2 receptor (BK2R) 9–12.
Figure 1.
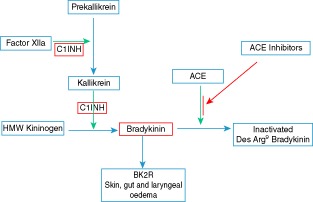
Pathways leading to the generation of bradykinin. C1 inhibitor (C1INH), in red, blocks the conversion of HMW kininogen to bradykinin while the effect of ACE inhibitors is to impede the breakdown of bradykinin. High molecular weight kininogen (HMW kininogen), bradykinin B2 receptor, angiotensin converting enzyme (ACE), angiotensin converting enzyme inhibitors (ACE inhibitors).
There are a number of treatments available for HAE. For long-term prophylaxis of frequent attacks, oral therapies such as attenuated androgens (danazol, stanozolol, oxandrolone and tibolone) 13,14 or anti-fibrinolytics (tranexamic acid and aminocaproic acid) may be used. Regular intravenous infusions of C1 esterase inhibitor are an additional therapeutic option for prophylaxis 16.
Treatment options for acute attacks have increased recently and include plasma-derived C1 inhibitors (Berinert and Cinryze), recombinant C1 inhibitor (Ruconest), a kallikrein inhibitor (Ecallantide licensed in the United States but not in the United Kingdom) and a bradykinin B2 receptor antagonist (Icatibant). Antihistamines, steroids and adrenaline are not effective in HAE. In acquired angioedema, treatment of the underlying haematological malignancy may result in improvement in terms of the swellings.
There have been a number of surveys of HAE in other countries 6–18 detailing the numbers of patients, diagnoses, attack frequency and diagnostic delay, but there is limited information regarding UK practice and patients. Given the recent increase in the number of therapeutic options for HAE patients as well as new guidelines and consensus documents 14–21, this audit aimed to provide more detailed information on UK patients and practice to help inform planning decisions and raise awareness of this condition.
Materials and methods
An audit tool (available as online additional information) to gather anonymized patient data was designed in Microsoft Excel based on the UK HAE Consensus guidelines 21 and clinical practice. The spread sheet included 93 data points per patient entry covering seven main areas: demographics, diagnosis and diagnostic delay, biochemistry and monitoring, family history, clinical, socioeconomic and impact on quality of life. Within the clinical section, additional information was included to define the sites of attacks to help ensure that the data were comparable. Peripheral attacks included facial, genital and extremities, while airway attacks included intraoral and laryngeal. The protocol and audit tool were reviewed by the University Hospital of Wales (UHW) Research and Development (R&D) Department and an opinion obtained from the local Ethics Committee Chair. There was agreement that the project fulfilled criteria for an audit and confirmation was issued by the UHW R&D Department. Ethical approval was therefore not required.
The audit tool was sent electronically to a range of organizations to ensure that clinicians most likely to care for HAE patients could be involved. These included the British Society for Immunology Clinical Immunology and Allergy Section (BSI-CIAS), the British Association of Allergy and Clinical Immunology (BSACI), the British Association of Dermatologists (Audit Group) and the Immunology Consultants Travellers Group. The Excel template was then sent from there to individual clinicians in centres across the United Kingdom.
Data on current patients were collected for the previous 12 months by clinicians with information from the patient, medical notes and pathology results systems. Anonymized data sets gathered in the period 2010–12 were returned to the Immunology Department in Cardiff for collation and analysis. Summary statistics (summed values, means, medians, standard error of the mean, percentages overall and for disease and group subsets where appropriate) were calculated for quantitative and qualitative variables using Microsoft Excel and Graphpad Prism version 6·0 and rounded to whole numbers or a single decimal place.
Results
Patient characteristics
Data were returned from 14 centres (Fig. 2) [Birmingham, Brighton and Sussex, Cardiff, Glasgow, Guildford, Liverpool, London, Manchester (adult), Manchester (paediatrics), Newcastle, Oxford, Preston, Salford and Swansea] covering mainly adults and some children.
Figure 2.
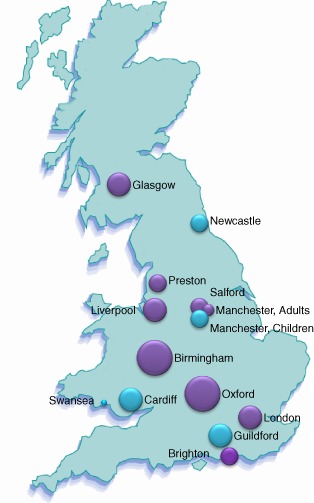
Distribution of responses from UK centres. The locations of the 14 centres across the United Kingdom are shown and the sizes of the circles are proportional to the numbers of data sets from each centre. Purple circles show centres returning adult patient data and blue where data from children were also submitted.
Types 1 and II HAE diagnoses were made based on biochemical and functional levels of C1INH. Type III angioedema diagnoses were confirmed by sequencing of factor XII (FXII), an assay which became available in the United Kingdom only towards the latter part of data collection. Type III, now known as ‘hereditary angioedema with normal C1 inhibitor’, has two subgroups – with or without FXII mutations. Three female patients with type III HAE were confirmed on sequencing; their ages were 28, 30 and 53 years. Diagnoses categorized as ‘other’ represent cases which were not fully worked-up or were being reinvestigated.
A total of 376 patients were identified: 59% females and 41% males. There was a smaller percentage of type II HAE (6%) (Fig. 3) diagnoses compared to 15% in other reports 1.
Figure 3.
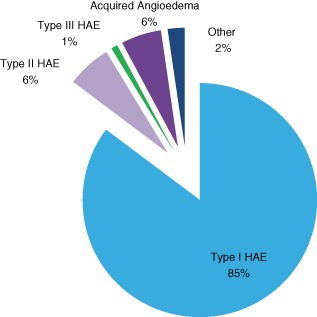
Hereditary angioedema (HAE) and acquired angioedema (AAE) diagnoses. The percentages of 376 patients within the different diagnostic categories.
Diagnostic delay
Data collected on diagnostic delay in 249 patients reveal a huge variation in time from onset of symptoms to diagnosis (Fig. 4). A minority of patients (3%) have negative values, corresponding to a diagnosis being made prior to the onset of symptoms, due usually to a diagnosis being made in another family member. Excluding these cases, the average time to diagnosis was 10 years for the group as a whole, with a median of 5 years.
Figure 4.
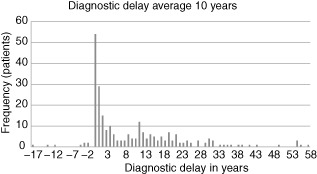
Diagnostic delay. Diagnostic delay is shown with the number of years from the onset of symptoms to the time of diagnosis on the x-axis and the frequency in numbers of patients on the y-axis. Where patients have negative values this is due to a diagnosis being made before the onset of symptoms, usually because the diagnosis had been made previously in another family member, which was the case in 3·2% of the 249 patients.
Considerable variation in diagnostic delay was observed between the different diagnostic categories when these were analysed separately; type I HAE (10 years), type II HAE (18 years) and AAE (5 years). Diagnostic delay in children was inevitably shorter at 2 years.
However, the full distribution of diagnostic delays is highly skewed. While almost half of all patients were diagnosed within 5 years of the onset of symptoms, the remaining 52% of patients experienced delays of up to 58 years. Such delays are of particular importance, as the risk of death from HAE has been shown to be three- to ninefold higher in undiagnosed patients 8.
Monitoring
Complement C3 and C4 levels were generally performed at clinic visits or annually, and 78% (40 of 51) of normal C4 results were from patients on either attenuated androgens or, in one case, C1INH prophylaxis. This leaves a small overall percentage of patients (3%) who were not on attenuated androgens and had a normal C4 recorded. Liver function tests were measured in the majority and lipids in a lower proportion, probably reflecting the use of attenuated androgens. Autoantibody testing was not routine; testing revealed positive anti-nuclear antibodies (ANA) in eight patients, thyroid peroxidase antibodies in five patients, with individual patients positive for adrenal antibodies, glutamic acid decarboxylase (GAD) antibodies and anti-neutrophil cytoplasmic antibodies [with a perinuclear indirect immunofluorescence pattern (pANCA) on a background of Crohn's disease]. Hepatitis serology testing was variable and incomplete.
Treatment
Information on acute treatment on 343 patients (Fig. 5a) showed that the majority, 62%, had C1INH available at home, with 8% receiving prophylactic C1INH and 30% attending accident and emergency departments for C1INH acute treatment. Small numbers of patients (6%) were given icatibant, due perhaps to its relatively recent availability, and the majority of these also had access to C1INH.
Figure 5.
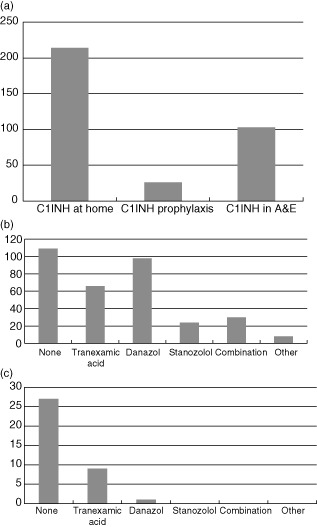
Acute treatment and long-term prophylaxis. (a) Numbers of patients who have C1 inhibitor (C1INH) available at home, in accident and emergency departments or are taking C1INH prophylaxis (total 343). (b) Numbers of adult patients taking each of the currently available oral prophylactic medications (total 335). (c) Long-term prophylaxis in children. Numbers of paediatric patients taking each of the currently available oral prophylactic medications (total 37).
Treatment with oral agents for long-term prophylaxis demonstrates a clear and expected difference in the use of this form of medication between adults (total 335 patients) and children (total 37 patients) (Fig. 5b,c). Children were less likely to need long-term prophylaxis and attenuated androgens are contraindicated, except in exceptional circumstances. The majority of children (73%) were on no regular medication and those who required therapy were treated with tranexamic acid. Sixty-seven per cent of adults received long-term prophylaxis with oral medication, the majority taking attenuated androgens.
Analysis of frequency and site of attacks
Data on attack frequency were available for 323 patients; overall analysis showed that peripheral attacks are the most frequent form of attack in HAE and constitute 58% of all swellings. There was considerable variability in the numbers of peripheral attacks per year between patients, with an overall mean of eight peripheral swellings annually (Fig. 6a).
Figure 6.
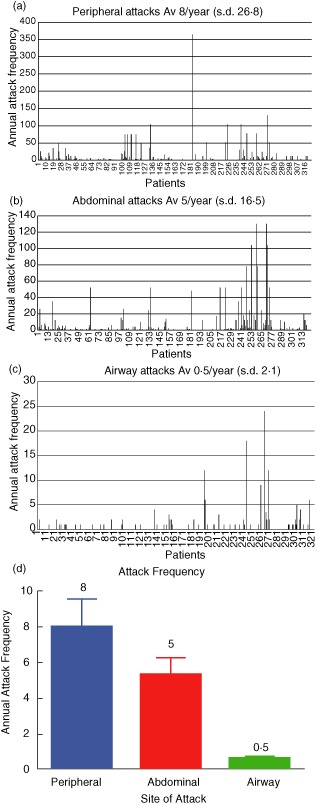
Analysis of annual peripheral, abdominal and airway attack frequency (total 323). (a) Peripheral attack frequency. (b) Abdominal attack frequency. (c) Airway attack frequency. The mean annual attack frequency is shown with individual patients on the x-axis and the frequency of attacks on the y-axis; s.d. (d) Average annual attack frequency. The mean overall attack frequency per patient per year at the three main sites of swelling.
Patients have, on average, 5 attacks of abdominal pain per year, and these constitute 38% of all attacks. The huge variability in mean annual attack frequency is again highlighted (Fig. 6b).
Attacks affecting the airway are the least frequent, at 4% of all attacks; however, 19% of patients (n = 62) experienced an airway attack during the 12 previous months, with some having up to two per month (Fig. 6c).
Figure 6d shows the average annual attack frequency at the three main sites of swelling. The percentages of attacks (peripheral 58, abdominal 38 and airway 4%) are similar to that described in 235 patients in a previous study (peripheral 46, abdominal 33 and oral 6%) 17. In a subanalysis (data not shown), the differences in attack frequency did not appear to be accountable to differences in prescribing of attenuated androgens. When attack frequency at the main three sites was compared for types I and II HAE, no differences were observed. The variation in attack frequency ranged from 30% of patients who had had no attacks during the year to others with daily attacks.
Socioeconomic and quality of life data
Information on the employment status of 213 patients shows that 76% were in employment (full- or part-time), were homemakers or students. Seven percent were unemployed and the remainder retired. The percentage in paid employment (full- and part-time) was 48% (Fig. 7). Information on days lost from work/school or where activities of daily living could not be performed was available on 116 patients, with an annual mean of 9 days per patient [standard deviation (s.d.) 24]; however, this is almost certainly an underestimate, as it was not possible to analyse non-numerical data (e.g. yes: +++, plenty, very few, etc.) in 11 patients.
Figure 7.
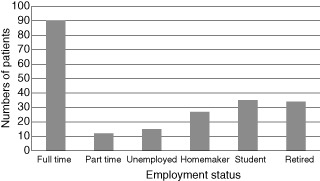
Employment status. The numbers of patients represented in the different employment categories (total 213).
The impact of HAE on quality of life was assessed by asking patients to rate the overall impact of their disease on their quality of life as either none, mild, moderate or severe. Information was available on 223 patients and the impact was noted to be moderate or severe in 37% of adult patients (Fig. 8a). While this approach is straightforward, detail and sensitivity is likely to be improved significantly using a validated disease-specific quality-of-life tool for HAE 22.
Figure 8.
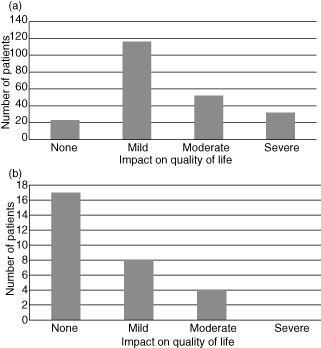
Comparison of the impact on quality of life in adults and children. (a) Impact on quality of life in adults. Patient's rating of the impact of the disease on their quality of life for adults (total 223). (b) Impact on quality of life in children. Parent's rating of the impact of the disease on their quality of life for children (total 29).
Paediatric data
Swellings are generally rare before the age of 2 years and are relatively uncommon before adolescence, with a mean age of onset of swellings of 8–12 years 23. The reported impact on quality of life in children available in 29 patients was rated as moderate in 14%, with no patients in the severe category. The annual frequency of swellings in children was peripheral, six; abdominal, six; and airway, 0·2 (Fig. 8b). Interpretation of attack frequency and impact upon quality of life in children is complicated by the fact that this information is reported by the parents. Furthermore, as there may be an increase in swellings during adolescence, consideration of childhood as less than 16 years of age may not capture important information.
Family history
The questions on family history included one on deaths in the family related directly to an HAE attack. When multiple entries for the same family members (and two entries stating ‘uncertain’ or ‘possible’) were excluded there was a total of 55 deaths in 33 families, ranging from one to three deaths per family. This clearly underpins the lethal potential of this condition. It should be noted that errors in either omission of deaths or inclusion of duplicate deaths are possibilities, although every effort was made to ensure the accuracy of the analysis. A subgroup analysis of all 57 patients who had had a death in the family showed that these were type I HAE in all but one case, and there was a slightly longer diagnostic delay of 12 years in this group compared to the overall diagnostic delay of 10 years. This appears to argue against a death in the family resulting in a clear reduction in diagnostic delay for other family members. When analysed separately, the average annual frequency of swellings in families with one or more deaths was: peripheral 14, abdominal two and airway 0·6. However, drawing firm conclusions from these frequencies is difficult, given the small size of the group. There was a minor increase in airway swellings above the overall average, but it is likely that factors other than the specific SERPING1 mutations modify swelling frequency, severity and site. Data from two patients' swellings in whom peripheral swellings were described as ‘too many’ rather than giving a numerical value were excluded.
Acquired angioedema (AAE)
Acquired angioedema (AAE) accounted for 6% of cases (n = 19) of angioedema. The average age of onset was 68 years, with equal numbers of males and females. The underlying diagnoses, where available, were haematological [chronic lymphocytic leukaemia (CLL) in three cases, and the following diagnoses were all reported in individual patients: non-Hodgkin lymphoma (NHL), B cell lymphoma, marginal zone lymphoma (MZL), follicular lymphoma, Waldenström's macroglobulinaemia and an immunoglobulin (Ig)M kappa paraprotein, in order of frequency]. There was no report of AAE associated with connective tissue or autoimmune disease.
Although the numbers of patients reported with acquired angioedema is small (n = 19), there was the suggestion of a difference in the frequency of swellings compared with hereditary angioedema, with mean values of peripheral 0·7, abdominal one and airway 0·9 per patient per year. The overall frequency of swellings appears lower – particularly peripheral and abdominal – with a more even spread of sites and the possibility that airway swellings occur at a higher rate (60% higher than HAE). Any differences should, however, be interpreted with caution due to the smaller numbers of patients and clear variability between individuals. In addition, 45% of AAE patients did not have a swelling during the previous year. Anti-C1 esterase inhibitor antibodies were not tested routinely and reported as positive in only two patients, perhaps reflecting the lack of availability of this assay at the time of data collection. Thirteen patients were taking long-term prophylaxis: six tranexamic acid, five danazol, one on both tranexamic acid and danazol and one on prophylactic C1INH.
Discussion
This study describes the first National Audit of patients with hereditary and acquired C1 inhibitor deficiency in the United Kingdom, capturing detailed information from 376 patients attending 14 centres in England, Scotland and Wales.
The results demonstrate clearly that HAE remains a potentially lethal disorder. The 55 reported deaths signify under-recognition of HAE in the United Kingdom, emphasized further by the very long diagnostic delays.
At 10 years overall this is shorter than the times reported in some earlier surveys, with an apparent gradual decline in diagnostic delay from the 1970s at 21 years in the United States to 13 years in a Spanish study from 2005, and more recently 10 years in a Danish study in 2009 6,7. The diagnostic delay, however, remains longer than has been shown for other primary immunodeficiency disorders, such as common variable immunodeficiency (CVID), at 6–8 years 24. The variability is very wide, from more than 50 years in some cases (maximum 58 years) and in others, particularly those with a known family history, the diagnosis may be made a number of years before their first attack. The overall data show that 13% of patients had a diagnostic delay of more than 25 years.
The differences in the diagnostic delay for types I and II HAE are difficult to explain, although the availability of robust functional C1INH testing may have had an impact and it is noteworthy that the frequency of type II diagnoses at 6% is somewhat lower than has been reported in some other series at 15% 18; it is, however, the same as that reported in a Danish survey at 6% 6. The relatively recent availability in the United Kingdom of genetic testing for a subset of type III HAE (hereditary angioedema with normal C1 inhibitor) and its rarity may also explain the low frequency of diagnoses at 1%. Acquired angioedema (AAE) has a much shorter diagnostic delay, which may be due to better recognition in patients attending secondary care for haematological malignancy.
Attack frequency shows the most frequent swellings to be cutaneous followed by abdominal swellings, with considerable variation between individuals and centres. Attacks threatening the airway are least frequent, with an overall mean of 0·5 per patient per year. It is possible with this information to perform modelling in terms of the likely requirement for treatment for acute attacks, and this data has already informed applications for HAE treatments to the All Wales Medicines Strategy Group (AWMSG). In a further analysis, however (not shown), the huge variation in attack frequency did not appear related to the different levels of use of attenuated androgens at different reporting centres. One potential explanation may be a reduction in attack frequency following the introduction of attenuated androgens for selected patients with a higher initial frequency of attacks. Groups of patients at either end of the severity spectrum may constitute informative candidates for the study of co-factors that might help to explain these differences. In those patients with no attacks for 12 months and who hold a home supply for acute treatment, there may be merit in providing those therapies with the longest possible shelf-life to minimize waste.
Information presented by this audit helps to characterize the experience of HAE patients in the United Kingdom and may be used to inform health policy decision-making at a national level. In particular, modelling exercises performed to evaluate the potential impact of new therapies for the treatment of HAE [either performed by or presented to Health Technology Assessment (HTA) agencies, such as AWMSG, SMC and NICE] will benefit from the data collected, where there is a paucity of available evidence relating to the burden of disease of this rare condition in the United Kingdom.
There are limitations to this audit, in that data have not been obtained on every patient with HAE in the United Kingdom. It is possible that there may be centres where the patient characteristics or medical practice are different, which might thus influence the findings. The paediatric data set is small, and analysis of a larger data set in children would be helpful.
The audit has established a baseline for a wide range of parameters for HAE patients in the United Kingdom. Areas for improvement in practice were identified when compared with the original consensus documents, such as monitoring of lipids, liver function tests and hepatitis serology. There has been rapid progress in the development of guidelines, and as practice may change with the availability of more effective therapies it will thus be important to re-audit to investigate possible improvements for patients. There are also a range of therapies at different stages of development which may also impact upon how HAE is treated in the future. The area of quality of life assessment would be optimized with the use of a disease-specific tool. The use of existing and developing databases as well as, potentially, smartphone applications may also facilitate real-time data entry and analysis.
Lessons were also learned as to how best to obtain clear high-quality data. Questionnaires should be simple and quick to complete, given the pressures on clinical time. Where possible, data should be numerical to make analysis more straightforward and linked to stated guideline criteria. Adults and children need to be assessed separately, recognizing the many differences in practice, disease severity (children reaching adolescence may experience increased attack frequency), development and impact on family life that exist between these groups.
The future for patients with HAE and AAE, however, looks bright not only with the current range of treatments available but with an intense focus of research into angioedema. New therapies currently under clinical investigation include subcutaneous C1INH (CSL Behring, King of Prussia, PA, USA), hyaluronidase-facilitated subcutaneous C1INH (Viropharma, Exton, PA, USA), an oral kallikrein inhibitor Bcx 4161 (Biocryst, Durham, NC, USA), cyclomimetic cyclic peptides inhibiting plasma kallikrein (Ra Pharma, Cambridge, MA, USA), monoclonal anti-kallikrein antibodies Dx 2930 (Dyax, Burlington, MA, USA) and even, in the future, gene therapy for C1INH deficiency. As well as these new developments, there also appears to be a protective role for women taking progestogen-only birth control pills, particularly those with anti-gonadotrophic activity such as norethisterone 25. In summary, it is hoped that this and future audits will serve to help inform the decision-making process in planning future care for patients with bradykinin-mediated angioedema.
Acknowledgments
The British Society for Immunology Clinical Immunology and Allergy Section (BSI-CIAS) received an unrestricted grant of £5000 from Shire to support data entry. S. J. is supported by an NISCHR Fellowship.
Disclosure
S Jolles – Consulting, speaker, meeting support from Shire, CSL Behring, Viropharma and SOBI.
P Williams – No disclosure
E Carne – Meeting support CSL Behring and Shire.
H Mian – No Disclosure
A Huissoon – Meeting support CSL Behring, Shire and Viropharma. Consulting Viropharma.
G Wong – No Disclosure
S Hackett – Meeting support CSL Behring
J Lortan – No disclosure
V Platts – No Disclosure
H Longhurst and S Grigoriadou and members of their department have received funding to attend conferences and other educational events, have acted as medical advisor or speaker, have received donations to her departmental fund, have received financial and other assistance with patient care projects and/or have participated in clinical trials with the following companies: CSL Behring, Pharming/Swedish Orphan, Jerini/Shire, Dyax, Viropharma, Baxter and Grifols.
J Dempster – Performed consultancy work for Virophrama, Shire and CSL Behring
S Deacock – No disclosure
S Kahn – No Disclosure
J Darroch – Meeting support Shire
C Simon – No Disclosure
M Thomas–No Disclosure
V Pavaladurai – No disclosure
H Alachkar – No Disclosure
A Herwadkar – No Disclosure
M Abinun – No Disclosure
P Arkwright – No Disclosure
M Tarzi – Speaker and travel support CSL Behring and Shire.
M Helbert – Speaker, consulting, conference support CSL Behring, consulting and conference support Shire and consulting Viropharma.
C Bangs – No Disclosure
C Pastacaldi – No Disclosure
C Phillips – Consulting for Viropharma
H Bennett – Consulting for Viropharma
T El-Shanawany – Consulting and meeting support from Shire, CSL Behring and Viropharma.
References
- 1.Rosen FS, Pensky J, Donaldson V, Charache P. Hereditary angioneurotic edema: two genetic variants. Science. 1965;148:957–958. doi: 10.1126/science.148.3672.957. [DOI] [PubMed] [Google Scholar]
- 2.Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 2000;356:213–217. doi: 10.1016/S0140-6736(00)02483-1. [DOI] [PubMed] [Google Scholar]
- 3.Zuraw BL, Bork K, E Binkley K, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc. 2012 doi: 10.2500/aap.2012.33.3627. [DOI] [PubMed] [Google Scholar]
- 4.Cicardi M, Zanichelli A. Acquired angioedema. Allergy Asthma Clin Immunol. 2010;6:14. doi: 10.1186/1710-1492-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cicardi M, Zanichelli A. The acquired deficiency of C1-inhibitor: lymphoproliferation and angioedema. Curr Mol Med. 2010;10:354–360. doi: 10.2174/156652410791317066. [DOI] [PubMed] [Google Scholar]
- 6.Bygum A. Hereditary angio-oedema in Denmark: a nationwide survey. Br J Dermatol. 2009;161:1153–1158. doi: 10.1111/j.1365-2133.2009.09366.x. [DOI] [PubMed] [Google Scholar]
- 7.Roche O, Blanch A, Caballero T, Sastre N, Callejo D, Lopez-Trascasa M. Hereditary angioedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain. Ann Allergy Asthma Immunol. 2005;94:498–503. doi: 10.1016/S1081-1206(10)61121-0. [DOI] [PubMed] [Google Scholar]
- 8.Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol. 2012;130:692–697. doi: 10.1016/j.jaci.2012.05.055. [DOI] [PubMed] [Google Scholar]
- 9.Bossi F, Fischetti F, Regoli D, et al. Novel pathogenic mechanism and therapeutic approaches to angioedema associated with C1 inhibitor deficiency. J Allergy Clin Immunol. 2009;124:1303–10 e4. doi: 10.1016/j.jaci.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A. Plasma bradykinin in angio-oedema. Lancet. 1998;351:1693–1697. doi: 10.1016/S0140-6736(97)09137-X. [DOI] [PubMed] [Google Scholar]
- 11.Nussberger J, Cugno M, Cicardi M, Agostoni A. Local bradykinin generation in hereditary angioedema. J Allergy Clin Immunol. 1999;104:1321–1322. doi: 10.1016/s0091-6749(99)70030-8. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan AP, Joseph K. The bradykinin-forming cascade and its role in hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104:193–204. doi: 10.1016/j.anai.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Bowen T, Cicardi M, Farkas H, et al. 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol. 2010;6:24. doi: 10.1186/1710-1492-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cicardi M, Bork K, Caballero T, et al. Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy. 2012;67:147–157. doi: 10.1111/j.1398-9995.2011.02751.x. [DOI] [PubMed] [Google Scholar]
- 15.Longhurst H, Cicardi M. Hereditary angio-oedema. Lancet. 2012;379:474–481. doi: 10.1016/S0140-6736(11)60935-5. [DOI] [PubMed] [Google Scholar]
- 16.Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363:513–522. doi: 10.1056/NEJMoa0805538. [DOI] [PubMed] [Google Scholar]
- 17.Agostoni A, Cicardi M. Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Balt) 1992;71:206–215. doi: 10.1097/00005792-199207000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Frank MM, Gelfand JA, Atkinson JP. Hereditary angioedema: the clinical syndrome and its management. Ann Intern Med. 1976;84:580–593. doi: 10.7326/0003-4819-84-5-580. [DOI] [PubMed] [Google Scholar]
- 19.Caballero T, Farkas H, Bouillet L, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. 2012;129:308–320. doi: 10.1016/j.jaci.2011.11.025. [DOI] [PubMed] [Google Scholar]
- 20.Craig T, Pursun EA, Bork K, et al. WAO guideline for the management of hereditary angioedema. World Allergy Organ J. 2012;5:182–199. doi: 10.1097/WOX.0b013e318279affa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gompels MM, Lock RJ, Abinun M, et al. C1 inhibitor deficiency: consensus document. Clin Exp Immunol. 2005;139:379–394. doi: 10.1111/j.1365-2249.2005.02726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weller K, Groffik A, Magerl M, et al. Development and construct validation of the angioedema quality of life questionnaire. Allergy. 2012;67:1289–1298. doi: 10.1111/all.12007. [DOI] [PubMed] [Google Scholar]
- 23.Farkas H, Varga L, Szeplaki G, Visy B, Harmat G, Bowen T. Management of hereditary angioedema in pediatric patients. Pediatrics. 2007;120:e713–722. doi: 10.1542/peds.2006-3303. [DOI] [PubMed] [Google Scholar]
- 24.Cunningham-Rundles C. How I treat common variable immune deficiency. Blood. 2010;116:7–15. doi: 10.1182/blood-2010-01-254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saule C, Boccon-Gibod I, Fain O, et al. Benefits of progestin contraception in non-allergic angioedema. Clin Exp Allergy. 2013;43:475–482. doi: 10.1111/cea.12055. [DOI] [PubMed] [Google Scholar]