

Abstract
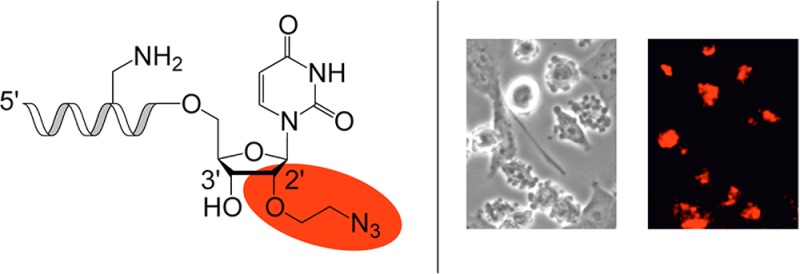
Labeled RNA becomes increasingly important for molecular diagnostics and biophysical studies on RNA with its diverse interaction partners, which range from small metabolites to large macromolecular assemblies, such as the ribosome. Here, we introduce a fast synthesis path to 3′-terminal 2′-O-(2-azidoethyl) modified oligoribonucleotides for subsequent bioconjugation, as exemplified by fluorescent labeling via Click chemistry for an siRNA targeting the brain acid-soluble protein 1 gene (BASP1). Importantly, the functional group pattern is inverse to commonly encountered alkyne-functionalized “click”-able RNA and offers increased flexibility with respect to multiple and stepwise labeling of the same RNA molecule. Additionally, our route opens up a minimal step synthesis of 2′-O-(2-aminoethyl) modified pyrimidine nucleoside phosphoramidites which are of widespread use to generate amino-modified RNA for N-hydroxysuccinimide (NHS) ester-based conjugations.
Introduction
Recently, azide-modified RNA1−5 has attracted considerable attention for being a valuable addition to the tool box of RNA bioconjugation.6,7 Of particular interest is the outreach for inverse Click labeling patterns that would create significantly more flexibility for complex labeling patterns as, for instance, needed for single-molecule fluorescence resonance energy transfer (FRET) studies.8,9 Also, azide-modified RNA will provide interesting alternatives to the existing RNA labeling concepts, such as expanding the range to Staudinger-type ligations.10 The prevalence of alkyne over azide-modified DNA and RNA stems from the straightforward integration of the alkyne functionality into the automated oligonucleotide solid-phase synthesis cycle using phosphoramidite building blocks.11−13 In contrast, azide-modified nucleoside phosphoramidites for solid-phase synthesis would encounter severe limitations because of the inherent reactivity of P(III) species with azides.14 Likewise, the rare encounter of, e.g., Staudinger ligations in the context with nucleic acids lies in the fact that neither the required azide nor the required P(III) moiety is easy to align with P(III) phosphoramidite chemistry for assembly.6,7,15−17 Hence, reported protocols for Staudinger-based conjugations on nucleic acids include inconvenient two-step procedures that attach the required N3 moiety postsynthetically onto amino group-functionalized RNA, employing N-hydroxysuccinimide (NHS) chemistry.18 Although efficient enzymatic prefunctionalization of DNA or RNA based on azide-modified nucleoside triphosphates has been reported,19−21 such a strategy would not be appropriate if single, site-specific azide modifications within nucleic acids are required.
Here, we describe the efficient preparation of a solid support for automated RNA synthesis using phosphoramidite building blocks that provides RNA with a 3′-terminal 2′-O-(2-azidoethyl) group (Figure 1). Efficient labeling with fluorescent dyes is evaluated for an siRNA application as well as the smooth transformation of the azido-labeled RNA into the corresponding amine derivative for NHS ester bioconjugation. Furthermore, potential strategies for diverse multiple label attachments are discussed. Additionally, our synthetic route opens up a minimal step synthesis of 2′-O-(2-aminoethyl) modified pyrimidine nucleoside phosphoramidites which are widely used to prepare amino-functionalized RNA.
Figure 1.

Chemical structure of 3′-end 2′-O-(2-azidoethyl) derivatized RNA. The modification allows for inverse Click labeling and selective, stepwise label attachment to RNA with diverse functional group patterns.
Results and Discussion
Chemical synthesis is the method of choice to prepare functionalized RNA with tailored properties.22 Frequently, this undertaking demands labeling with moieties that are incompatible with RNA solid-phase synthesis and, therefore, prefunctionalized RNA with tethers carrying, e.g., amino or alkyne groups is required. These anchors can then be transformed by using the classical NHS ester approach and the more recent Click conjugations, respectively.7,11,16,17 Our original efforts were driven by the motivation to equip the same RNA with an additional orthogonal anchor besides amine and alkyne groups. This goal would be amenable through azide modification that allows for selective labeling with strained cyclic alkynes,23 in the presence of both of the other attachment sites. Interestingly, not many types of chemically synthesized, azide-functionalized RNAs have been described in the literature, and for their assembly, the majority requires either phosphonate (e.g., 2′-O-[(2-azidoethoxy)methyl] RNA)3 or phosphortriester chemistry (e.g., 2′-azido RNA).4,5 Although these approaches are powerful and enable labeling of internal sequence positions, they require adjustments of standard RNA synthesis procedures which can represent a handicap for broader applications. Another recent promising approach to generate 2′-O-(2-azidoethyl) modified nucleic acids involves a convertible nucleoside, but this approach has been demonstrated thus far for DNA only.24 Here, we intended to create a fast and simple access to azide labeled RNA even if restrictions with respect to positioning of the azide group were encountered. For many applications, in particular, for multiple, specific labeling of DNA25,26 or RNA,8,9,12 3′-end azide anchors would be a major asset, provided the approach is facile and applicable to standard phosphoramidite chemistry.
We recall a previous report by Morvan and co-workers on a universal solid support for 3′-end azide labeling of DNA27 and our own studies on 3′-deoxy-3′-azido RNA28 that are compatible with the usage of nucleoside phosphoramidites. However, for the present study we aimed at an approach that keeps the 3′-OH of the oligoribonucleotide available to retain the possibility for ligations to construct larger RNA, e.g., by using in vitro selected DNA ligation enzymes.29 Hence, we focused on the ribose 2′-O position for derivatization and favored the 2′-O-(2-azidoethyl) group. Nucleosides of this type and with defined protecting group patterns have been reported as intermediates for the synthesis of 2′-O-(2-aminoethyl) modified DNA and RNA.30,31 However, applying such pathways would involve multiple steps. Here, we aimed at a one-step protecting group-free synthesis using the substrates 2,2′-anhydrouridine 1 and 2-azidoethanol (which are commercially available or can be prepared by a single transformation from the precursors uridine32 and 2-chloroethanol,33 respectively) in the presence of boron trifluoride diethyl etherate (Scheme 1). The procedure was eleborated based on reports by Egli34 and Sekine35 who demonstrated the corresponding transformation with a series of other alcohol derivatives. After careful optimization, the desired 2′-O-(2-azidoethyl) uridine 2 was achieved in acceptable yields. Compound 2 was then readily tritylated, then transformed into the corresponding pentafluorophenyl (Pfp) adipic acid ester, and finally into the functionalized solid support 3.
Scheme 1. Synthesis of the Solid Support 3 for 3′-End 2′-O-(2-azidoethyl) Modified RNA.

Reaction conditions: (a) 5 equiv HOCH2CH2N3, 2.5 equiv BF3·OEt2 in dimethylacetamide, 120 °C, 16 h, 55%; (b) 1.1 equiv DMT-Cl, in pyridine, 16 h, RT, 75%; (c) 3.5 equiv PfpOOC(CH2)4COOPfp, 1.2 equiv DMAP, in DMF/pyridine (1:1), room temperature, 1 h, 47%; (d) 3 equiv (w/w) amino-functionalized support (GE Healthcare, Custom Primer Support 200 Amino), 2 equiv pyridine, in DMF, room temperature, 48 h, loading: 60 mmol g–1.
The solid support 3 was efficiently used for automated RNA strand assembly using nucleoside phosphoramidite building blocks (Table 1). Standard cleavage and deprotection procedures resulted in high-quality crude products as exemplified in Figure 2A (top). The integrity of the azide-modified RNA was confirmed by LC-ESI mass spectrometry (Figure 2A, bottom). We also note that 2′-O-(2-azidoethyl) modified RNAs were efficiently reduced to the 2′-O-(2-aminoethyl) modified counterparts by incubation with tris(2-carboxyethyl)phosphine hydrochloride (TCEP) in aqueous solution (Figure Figure S1). Thus, the azidoethyl moiety can be used as a temporarily masked amino anchor for sequential labeling of RNA that is functionalized together with an internal 2′-O-(2-aminoethyl) or 5-aminoallyl pyrimidine modification, using NHS ester conjugation reactions only.
Table 1. Selection of Synthesized 3′-End 2′-O-(2-azidoethyl) RNAs and Corresponding Dye Label Derivatives.
| no | sequencea | amountb [nmol] | m.w.calcd [amu] | m.w.found [amu] |
|---|---|---|---|---|
| S1 | 5′-ACG UU-2′-OCH2CH2N3 | 1300 | 1599.9 | 1598.9 |
| S2 | 5′-UGU CUU AUU GGC AGA GAC CTU-2′-OCH2CH2N3 | 185 | 6724.1 | 6725.0 |
| S3 | 5′-GGU CUC UGC CAA UAA GAC ATU-2′-OCH2CH2N3 | 176 | 6717.0 | 6718.6 |
| S4 | 5′-UGU CUU AUU GGC AGA GAC CTU-2′-az-F545 | 23 | 7368.0 | 7368.8 |
| S5 | 5′-GGU CUC UGC CAA UAA GAC ATU-2′-az-F545 | 28 | 7361.7 | 7361.9 |
| S6 | 5′-AGA UGU GCC AGC AAA ACC A(Cy3–5aall-U)C UUU AAA AAA CUG GU-2′-az-ADIBO-Cy5 | 5.6 | 12826.8 | 12827.0 |
| S7 | 5′-AGA UGU GC(Cy3–5aall-U) AGC AAA ACC AUC UUU AAA AAA CUA GU-2′-az-ADIBO-Cy5 | 4.3 | 12825.8 | 12825.8 |
Figure 2.

Synthesis, labeling, and analysis of an exemplary 2′-O-(2-azidoethyl) modified RNA based on the solid support 3. (A) Anion exchange HPLC profiles of deprotected, crude (top) and purified (inset) RNA, and LC-ESI mass spectrum (bottom). (B) Reaction scheme of Click labeling with alkyne functionalized fluorescence dye (left); conditions: 5 mM CuSO4, 10 mM sodium ascorbate, 50 °C, 3 h; cRNA = 1 mM, cDye = 2 mM, H2O/CH3CN = 4/1; 60 μL total reaction volume. HPLC profiles of crude (top right) and purified (inset) labeled RNA, and LC-ESI mass spectrum (bottom); HPLC conditions: Dionex DNAPac column (4 × 250 mm), 80 °C, 1 mL min–1, 0–60% buffer B in 45 min; buffer A: Tris-HCl (25 mm), urea (6 M), pH 8.0; buffer B: Tris-HCl (25 mM), urea (6 M), NaClO4 (0.5 M), pH 8.0. For LC-ESI MS conditions, see the Experimental Procedures.
Furthermore, we demonstrated the convenience of the 2′-O-(2-azidoethyl) RNA label in a typical azide–alkyne 1,3-dipolar cycloaddition reaction (Click chemistry)6,11 (Figure 2B, Table 1). We applied the copper-catalyzed version with acetonitrile as cosolvent acting as ligand of the CuI complex, stabilizing the oxidation state.36 The labeled RNA strand at 1 mM concentration was efficiently reacted with a commercially available, alkyne-modified 5-carboxytetramethylrhodamine dye (F545) (2 mM) in the presence of sodium ascorbate, and analyzed by anion exchange chromatography (Figure 2B). For reasons of comparability, we chose the siRNA sequence system used previously to knock down the brain acid-soluble protein 1 gene (BASP1) by transient siRNA nucleofection in the chicken DF-1 cell line.4,5,37 Expression of the BASP1 gene is specifically suppressed by Myc, an evolutionary conserved oncoprotein;38 conversely, the BASP1 protein is an efficient inhibitor of Myc-induced cell transformation.37
Three dye-labeled siRNAs were annealed, one labeled at the 3′-end of the antisense strand, the second labeled at the 3′-end of the sense strand, and the third labeled at both 3′-ends (Figure 3A). All three siRNA were efficiently nucleofected into chicken DF1 cells and localized by fluorescence microscopy (Figure 3B). Not unexpectedly, due to the stringent structural requirements for antisense strand recognition within the RISC complex,39,40 efficient silencing (comparable to the unmodified reference duplex) was only observed for the sense labeled siRNA duplex, while both siRNAs with 3′-labeled antisense strands were inactive, as analyzed by Northern blot hybridization (Figure 3C). The finding that the activity of the siRNA carrying a large chemical moiety is well tolerated only when it is placed at the 3-terminus of the sense strand is in accordance with our own previous findings4 and those by others.41−43
Figure 3.
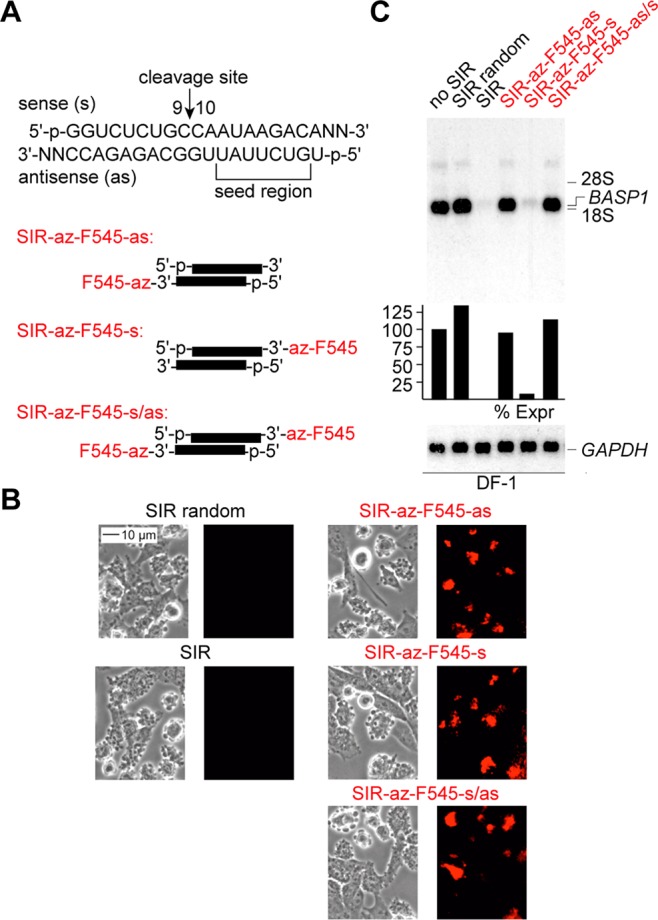
Silencing of the brain acid-soluble protein 1 gene (BASP1) by siRNA duplexes with fluorescent labels (F545) clicked to 3′-terminal 2′-O-(2-azidoethyl) anchors. (A) General organization (top) and labeling pattern of the siRNA duplex (bottom); for detailed RNA sequences see Table S1. (B) BASP1 siRNAs show cytoplasmic localization in DF1 cells visualized by fluorescence microscopy. The amounts of nucleofected siRNAs were 0.24 nmol. (C) Activities of 2′-az-F545 labeled BASP1 siRNAs and corresponding controls (random siRNA and unmodified siRNA) monitored by Northern analysis of BASP1 expression in DF1 cells. Expression of GAPDH served as loading control.
To further demonstrate the usefulness of 2′-O-(2-azidoethyl) RNA, we performed efficient dual fluorescent labeling of strands that additionally contained 5-aminoallyl uridine modifications, using NHS-chemistry and strain-promoted alkyne—azide conjugation (SPAAC).21 The sequence represents a preQ1 class-I riboswitch aptamer,44 and the obtained cyanine dye pattern is applicable for bulk FRET investigations (Table 1, Figure 4, Figure S2).
Figure 4.
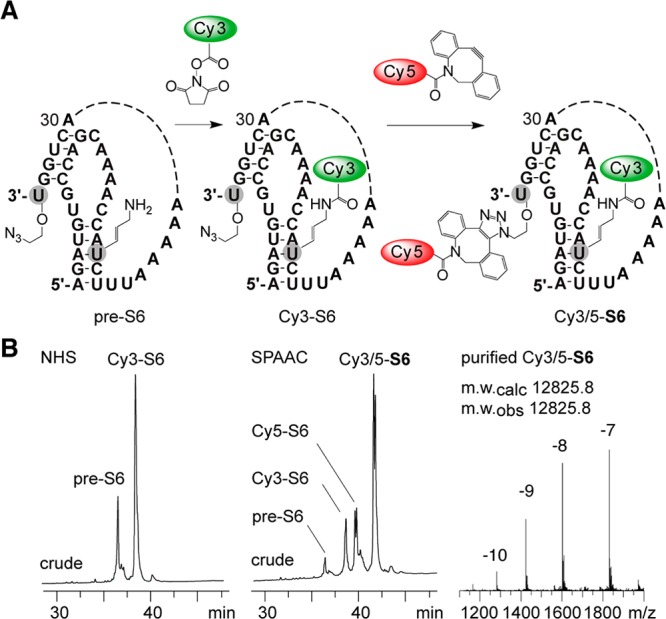
Example for double labeling of 3′-terminal 2′-O-(2-azidoethyl) modified RNA. (A) Labeling scheme for the preQ1 riboswitch RNA from Fusobacterium nucleatum.44 (B) HPLC profiles of crude reaction mixture after N-hydroxysuccinimide (NHS) ester based Cy3 conjugation (left) and subsequent strain-promoted alkyne azide conjugation (SPAAC) of Cy5 (middle), LC-ESI mass spectrum (right). For HPLC and LC-ESI mass specrometry conditions, see Figure 2 caption; for dye structures, see Figure S2.
The efficient approach to 2′-O-(2-azidoethyl) labeled RNA and their applications can be mainly attributed to the one-step synthesis of the key compound 2′-O-(2-azidoethyl) uridine 2. This derivative additionally opens up a convenient route with minimal steps to 2′-O-(2-aminoethyl) uridine phosphoramidites (Scheme 2). 2′-O-(2-Aminoethyl) modified nucleic acids have been extensively studied for various purposes,45−50 and interestingly, the reported syntheses of the building blocks usually entail initial alkylation of the ribose 2′-OH by methyl bromoacetate followed by a series of transformation reactions29,30 or involve extended protecting group concepts.48−50 The route presented here relies on tritylation of the azide 2, followed by azide to amine reduction under Staudinger conditions and trifluoroacetylation to give derivative 4. After phosphitylation,30 the corresponding uridine building block was obtained in excellent overall yield in only five steps from uridine.
Scheme 2. Short Synthesis of a 2′-O-(2-Aminoethyl) Uridine Phosphoramidite.

Reaction conditions: (a) 1.1 equiv DMT-Cl, in pyridine, 16 h, RT, 75%; (b) i. 2 equiv PPh3, 5 equiv H2O, in tetrahydrofurane, room temperature, 5 h, ii. 10 equiv CF3COOEt, 10 equiv NEt3, CH3OH, 0 °C, 14 h, 61% (over 2 steps).
Conclusions
The presented approach to 3′-terminal azide-modified RNA is significant for diverse applications in RNA biochemistry and RNA chemical biology as exemplified here for fluorescently labeled siRNAs. Another potential of this type of modification lies in the combined prefunctionalization together with amino (and, in principle, also with alkyne) moieties of the same RNA to allow for selective and stepwise attachment of sensitive moieties that cannot be directly incorporated into RNA. Efficient generation of complex labeling patterns is, e.g., required for multicolor single-molecule FRET studies and is currently undertaken in our laboratory.
Experimental Procedures
General Remarks
1H and 13C NMR spectra were recorded on a Bruker DRX 300 MHz or Avance II+ 600 MHz instrument. The chemical shifts are referenced to the residual proton signal of the deuterated solvents: CDCl3 (7.26 ppm), d6-DMSO (2.49 ppm) for 1H NMR spectra; CDCl3 (77.0 ppm) or d6-DMSO (39.5 ppm) for 13C NMR spectra (see also Figures S3–S6). 1H- and 13C-assignments were based on COSY and HSQC experiments. MS experiments were performed on a Finnigan LCQ Advantage MAX ion trap instrument. Analytical thin-layer chromatography (TLC) was carried out on Marchery-Nagel Polygram SIL G/UV254 plates. Flash column chromatography was carried out on silica gel 60 (70–230 mesh). All reactions were carried out under argon atmosphere. Chemical reagents and solvents were purchased from commercial suppliers and used without further purification. Organic solvents for reactions were dried overnight over freshly activated molecular sieves (4 Å).
2′-O-(2-Azidoethyl)uridine (2)
2,2′-Anhydrouridine 1 (565 mg, 2.5 mmol) was coevaporated with dry pyridine three times and stored over P2O5 in a desiccator for four hours before use. Then, compound 1 was suspended in DMA (4 mL) and BF3·OEt2 (785 μL, 6.25 mmol) was added under argon and heated to 120 °C. 2-Azidoethanol (1250 mg, 14.3 mmol) was injected into the solution and the mixture was refluxed for 16 h. After the reaction was finished solvents were removed in vacuo, and the oily residue was redissolved in methanol and adsorbed on silica gel. Compound 2 was purified by column chromatography on SiO2 with CHCl3/CH3OH, 95:5. Yield: 431 mg of 2 as a white solid (55%). TLC (CH2Cl2/CH3OH = 85:15): Rf = 0.51. 1H NMR (300 MHz, DMSO): δ 3.17 (m, 2H, H1–C(2″) H2–C(2″)); 3.58 (m, 2H; H1–C(5′) H2–C(5′)); 3.86 (m, 2H, H1–C(1″) H2–C(1″)); 3.88 (m, 1H, H–C(4′)); 4.04 (m, 1H, H–C(2′)); 4.60 (dd, J = 4.8 Hz, J = 9.8 Hz, 1H, H–C(3′)); 5.14 (m, 2H, HO-C(3′), HO-C(5′)); 5.72 (d, J = 8.0 Hz, 1H, H–C(5)); 5.88 (d, J = 4.8 Hz, 1H, H–C(1′)); 7.94 (d, J = 8.0 Hz, 1H, H–C(6)); 11.29 (s, 1H, NH) ppm. 13C NMR (150 MHz, DMSO): δ 49.93 (C(2″)); 60.39 (C(5′)); 68.2 (C(3′)); 68.86 (C(1″); 81.31 (C(2′); 84.93 (C(4′)); 86.15 (C(1′)); 101.79 (C(5)); 140.32 (C(6)); 150.56; 163.10 ppm. ESI-MS (m/z): [M-H]− calcd for C11H15N5O6, 312.11; found 312.46.
2′-O-(2-Azidoethyl)-5′-O-(4,4′-dimethoxytrityl)uridine (2a)
Compound 2 (372 mg, 1.19 mmol) was coevaporated with dry pyridine three times and dissolved in pyridine at room temperature and under argon atmosphere. 4,4′-Dimethoxytrityl chloride (443 mg, 1.31 mmol) was added in two portions over a period of 2 h. Stirring was continued overnight, and when TLC showed complete reaction, methanol was added and the solution was evaporated in vacuo. The residue was dissolved in CH2Cl2 and extracted with 5% citric acid, water, and saturated NaHCO3. The organic layer was dried over Na2SO4 and evaporated. The crude product was purified by column chromatography on SiO2 with CH2Cl2/CH3OH, 100:0 to 98:2. Yield: 549 mg of 2a as a white foam (75%). TLC (CH2Cl2/CH3OH = 92/8): Rf = 0.54. 1H NMR: (300 MHz, CDCl3): δ 2.58 (d, J = 9.7 Hz, 1H, HO-C(3′)); 3.49 (m, 2H, H1–C(2″), H2–C(2″)); 3.58 (m, 2H; H1–C(5′), H2–C(5′)); 3.80 (s, 6H, H3CO) 3.96 (m, 1H, H2–C(1″)); 3.96 (m, 1H, H–C(2′)); 4.04 (m,1H, H–C(4′)); 4.19 (m, 1H, H1–C(1″)); 4.51 (m, 1H, H–C(3′)); 5.30 (d, J = 8.1 Hz, 1H, H–C(5)); 5.93 (s, 1H, H–C(1′)); 6.85 (m, 4H, H–C(ar)); 7.31 (m, 9H, H–C(ar)); 8.09 (d, J = 8.1 Hz, 1H, H–C(6)); 9.16 (s, 1H, N–H) ppm. 13C NMR (150 MHz, CDCl3): δ 50.99 (C(2″)); 55.40 (CH3O); 61.03 (C(5′)); 68.43 (C(3′)); 70.09 (C(1″); 83.16 (C(2′); 87.27 (C(4′)); 87.73 (C(1′)); 102.27 (C(5)); 113.47 (C(ar)); 127.33 (C(ar)); 130.25 (C(ar));140.32 (C(6)); 144.50; 150.33; 158.91; 163.39 ppm. ESI-MS (m/z): [M+Na]+ calcd for C32H33N5O8Na, 638.21; found 638.40.
2′-O-(2-Azidoethyl)-5′-O-(4,4′-dimethoxytrityl)-3′-O-[1,6-dioxo-6-(pentafluorophenyloxy)hexyl]uridine (2b)
Compound 2a (100 mg, 162 μmol) was coevaporated with pyridine twice and dried over P2O5 for three hours, then it was dissolved in DMF/Pyr (=1/1; 4.6 mL) and DMAP (24 mg, 196 μmol), and adipinic acid pentafluorophenyl ester (275 mg, 575 μmol) was added. After one hour the reaction mixture was evaporated and coevaporated with acetone and CH2Cl2 two times. The crude product was purified by column chromatography on SiO2 with acetone/CH2Cl2, 95:5 to 85:15. Yield: 70 mg of 2b as a white foam (47%). TLC (acetone/CH2Cl2 = 8:2): Rf = 0.56. 1H NMR: (300 MHz, CDCl3): δ 1.93 (m, 4H, RO2CH2(CH2)2CH2–CO2C6F5); 2.58 (m, 2H, RO2CH2(CH2)2CH2CO2C6F5); 2.84 (t, J = 6.8, 2H, RO2CH2(CH2)2CH2CO2C6F5); 3.51 (m, 2H, H1–C(2″), H2–C(2″)); 3.58 (m, 1H; H1–C(5′)); 3.81 (m, 1H, H2–C(5′)); 3.86 (m, 1H, H2–C(1″)); 3.93 (s, 6H, H3CO); 4.11 (m, 1H, H1–C(1″)); 4.43 (m, 2H, H–C(4′)); 5.34 (dd, J1 = 7.0, J2 = 5.3, 1H, H–C(3′)); 5.51 (d, J = 8.1 Hz, 1H, H–C(5)); 5.93 (d, J = 2.55 Hz, 1H, H–C(1′), 7.00 (m, 4H, H–C(ar)); 7.40 (m, 9H, H–C(ar)); 8.14 (d, J = 8.1 Hz, 1H, H–C(6)); 9.38 (s, 1H, N–H) ppm. 13C NMR (150 MHz, CDCl3): δ 24.01, 24.20, 33.00, 33.44 (4C, RO2CH2(CH2)2CH2–CO2C6F5), 50.91 (C(2″)); 55.39 (2C, CH3O); 61.18 (C(5′)); 69.89 (C(3′)); 70.14 (C(1″); 80.90 (C(4′)); 81.23 (C(2′); 87.56 (C(1′)); 102.60 (C(5)); 113.49 (C(ar)); 127.40 (C(ar)); 130.31 (C(ar)); 135.05; 139.87 (C(6)); 144.24; 150.42; 158.95; 163.32; 169.16; 172.40 ppm. ESI-MS (m/z): [M+Na]+ calcd for C44H40N5F5O11Na, 932.24; found 932.32.
2′-O-(2-Azidoethyl)Uridine Modified Solid Support (3)
Compound 2b (70 mg, 77 μmol) was dissolved in DMF (1.7 mL) and pyridine (12 mg [12 μL], 154 μmol). Then, amino-functionalized solid support (GE Healthcare, Custom Primer Support 200 Amino, 323 mg) was added. The suspension was agitated for 48 h at room temperature and the beads were collected on a Büchner funnel. The beads were washed with N,N-dimethylformamide, methanol, and dichloromethane and dried. Capping was performed by treatment of the beads with a mixture of 3.0 mL of solution A (acetic anhydride/2,4,6-trimethylpyridine/acetonitrile, 2/3/5) and 3.0 mL of solution B (4-(N,N-dimethylamino)pyridine/acetonitrile, 0.5 M) for 5 min at room temperature. The suspension was filtrated again and the beads were washed extensively with acetonitrile, methanol, and dichloromethane and dried under vacuum. Loading of the support 3 was 60 μmol/g.
2′-O-([N-Trifluoracetyl]-2-aminoethyl)-5′-O-(4,4′-dimethoxytrityl)uridine (4)
Compound 2a (460 mg, 0.75 mmol) was dissolved in THF (7.25 mL). Water (69 μL, 3.8 mmol) and triphenylphosphine (392 mg, 1.5 mmol) was added and the solution was stirred for 5 h at room temperature. Then, ethyl trifluoroacetate (1065 mg [0.89 mL], 7.5 mmol) and triethylamine (770 mg [1.06 mL], 7.6 mmol) were added and stirring was continued overnight. The reaction mixture was evaporated and the crude product was purified by column chromatography on SiO2 with CH2Cl2/CH3OH, 100:0 to 95:5. Yield: 315 mg of 4 as a white foam (= 61%). TLC (CH2Cl2/CH3OH = 95/5): Rf = 0.4. 1H NMR (300 MHz, CDCl3): δ 2.85 (d, J =8.7 Hz, 1H, HO-C(3′)); 3.50–3.65 (m, 4H, H1–C(5′), H2–C(5′), H1–C(2″), H2–C(2″)); 3.79 (s, 6H, H3CO); 3.93–4.05 (m, 4H, H–C(2′), H–C(4′), H1–C(1″), H2–C(1″)), 4.42 (m, 1H, H–C(3′)); 5.33 (d, J =8.1 Hz, 1H, H–C(5)); 5.86 (s, 1H, H–C(1′)); 6.85 (m, 4H, H–C(ar)); 7.24–7.39 (m, 9H, H–C(ar)); 7.71 (m, 1H, HNCOCF3); 8.05 (d, J =8.1 Hz, 1H, H–C(6)); 9.95 (s, 1H, N–H) ppm. 13C NMR (150 MHz, CDCl3): δ 39.75 (C(2″)); 55.39 (CH3O); 61.08 (C(5′)); 68.55 (C(3′)); 69.37 (C(1″); 83.36 (C(2′); 83.49 (C(4′)); 87.30; 87.33 (C(1′)); 102.61 (C(5)); 113.48 (C(ar)); 127.36 (C(ar)); 130.22 (C(ar)); 135.38; 135.36; 140.01 (C(6)); 144.43; 151.13; 158.87; 158.91; 163.48 ppm. ESI-MS (m/z): [M+Na]+ calcd for C32H33N5O8Na, 708.28; found 708.21.
RNA Solid-Phase Synthesis
Standard phosphoramidite chemistry was applied for RNA strand elongation using solid support 3: for the synthesis 2′-O-TOM standard RNA nucleoside phosphoramidite building blocks were purchased from GlenResearch and ChemGenes, the polystyrene support from GE Healthcare (Custom Primer Support, 80 μmol/g; PS 200). All oligonucleotides were synthesized on a ABI 392 Nucleic Acid Synthesizer following standard methods: detritylation (80 s) with dichloroacetic acid/1,2-dichloroethane (4/96); coupling (2.0 min) with phosphoramidites/acetonitrile (0.1 M × 130 μL) and benzylthiotetrazole/acetonitrile (0.3 M × 360 μL); capping (3 × 0.4 min, Cap A/Cap B = 1/1) with Cap A: 4-(dimethylamino)pyridine in acetonitrile (0.5 M) and Cap B: Ac2O/sym-collidine/acetonitrile (2/3/5); oxidation (1.0 min) with I2 (20 mM) in THF/pyridine/H2O (35/10/5). The solutions of amidites and tetrazole, and acetonitrile were dried over activated molecular sieves (4 Å) overnight.
Deprotection of 2′-O-(2-azidoethyl) Modified RNA
The solid support was treated with MeNH2 in EtOH (33%, 0.5 mL) and MeNH2 in water (40%, 0.5 mL) for 7 h at room temperature. (For RNA containing 5-aminoallyl uridines, the column was first treated with 10% diethylamine in acetonitrile (20 mL), washed with acetonitrile (20 mL) and dried. Then, the solid support was treated with MeNH2 in EtOH (33%, 1 mL) and NH3 in H2O (28%, 1 mL) for 10 min at room temperature and 20 min at 65 °C.) The supernatant was removed from and the solid support was washed three times with ethanol/water (1/1, v/v). The supernatant and the washings were combined with the deprotection solution of the residue and the whole mixture was evaporated to dryness. To remove the 2′-silyl protecting groups, the resulting residue was treated with tetrabutylammonium fluoride trihydrate (TBAF·3H2O) in THF (1 M, 1 mL) at 37 °C overnight. The reaction was quenched by the addition of triethylammonium acetate (TEAA) (1 M, pH 7.4, 1 mL). The volume of the solution was reduced and the solution was desalted with a size exclusion column (GE Healthcare, HiPrep 26/10 Desalting; 2.6 × 10 cm; Sephadex G25) eluating with H2O; the collected fraction was evaporated to dryness and dissolved in 1 mL H2O. Analysis of the crude RNA after deprotection was performed by anion-exchange chromatography on a Dionex DNAPacPA-100 column (4 mm × 250 mm) at 80 °C. Flow rate: 1 mL/min, eluant A: 25 mM Tris·HCl (pH 8.0), 6 M urea; eluant B: 25 mM Tris·HCl (pH 8.0), 0.5 M NaClO4, 6 M urea; gradient: 0–60% B in A within 45 min or 0–40% B in 30 min for short sequences up to 15 nucleotides, UV-detection at 260 nm.
Purification of 2′-O-(2-Azidoethyl) Modified RNA
Crude RNA products were purified on a semipreparative Dionex DNAPac PA-100 column (9 mm × 250 mm) at 80 °C with flow rate 2 mL/min. Fractions containing RNA were loaded on a C18 SepPak Plus cartridge (Waters/Millipore), washed with 0.1–0.15 M (Et3NH)+HCO3–, H2O and eluted with H2O/CH3CN (1/1). RNA containing fractions were lyophilized. Analysis of the quality of purified RNA was performed by anion-exchange chromatography with same conditions as for crude RNA; the molecular weight was confirmed by LC-ESI mass spectrometry. Yield determination was performed by UV photometrical analysis of oligonucleotide solutions.
Mass Spectrometry of 2′-O-(2-Azidoethyl) Modified RNA
All experiments were performed on a Finnigan LCQ Advantage MAX ion trap instrumentation connected to an Amersham Ettan micro LC system. RNA sequences were analyzed in the negative-ion mode with a potential of −4 kV applied to the spray needle. LC: Sample (200 pmol RNA dissolved in 30 μL of 20 mM EDTA solution; average injection volume: 30 μL); column (Waters XTerraMS, C18 2.5 μm; 1.0 × 50 mm) at 21 °C; flow rate: 30 μL/min; eluant A: 8.6 mM TEA, 100 mM 1,1,1,3,3,3-hexafluoroisopropanol in H2O (pH 8.0); eluant B: methanol; gradient: 0–100% B in A within 30 min; UV-detection at 254 nm.
Copper-Catalyzed Azide–Alkyne Cycloaddition (CuAAC) Labeling
2′-O-(2-Azidoethyl) modified RNA (60 nmol) was lyophilized in a 1 mL Eppendorf tube. Then, aqueous solutions of F545 (Acetylene-Fluor 545, Click Chemistry Tools), CuSO4, and sodium ascorbate were added consecutively; acetonitrile was added as cosolvent36 to reach final concentrations of 1 mM RNA, 2 mM dye, 5 mM CuSO4, 10 mM sodium ascorbate, and a H2O/acetonitrile ratio of 4/1 in a total reaction volume of 60 μL. The reaction mixture was degassed and stirred for 3 to 4 h under argon atmosphere at 50 °C. To monitor the reaction and to purify the reaction mixtures, anion exchange HPLC as described above was used.
Double Labeling Using N-Hydroxysuccinimide Ester (NHS) Chemistry and Strain-Promoted Alkyne–Azide Cycloadditions (SPAAC)
Lyophilized 3′-end 2′-O-(2-azidoethyl) RNA (25 nmol) containing a single 5-(E-3-aminoprop-1-enyl)uridine (5-aminoallyl uridine) was dissolved in labeling buffer (25 mM phosphate buffer, pH 8.0) and DMSO (55% vol/vol) with a final concentration of 225 μM RNA and 1.125 mM Sulfo-Cy3-NHS ester in a total volume of 110 μL. The reaction mixture was shaken for 5 h at room temperature in the dark. Then, the RNA was precipitated with absolute ethanol (2.5 volumes of labeling reaction) and a 1 M aqueous solution of sodium acetate (0.2 volumes of labeling reaction), for 4 h at −20 °C. The suspension was centrifuged for 30 min at 4 °C at 13 000 × g to remove the excess of unreacted and hydrolyzed dye. The pellets were dried under high vacuum and dissolved in nanopure water and DMSO (50% vol/vol) to reach final concentrations of 312 μM RNA and 686 μM ADIBO derivatized Cy5 dye in a total volume of 80 μL. The reaction mixture was shaken for 3 h at room temperature in the dark. To monitor the reaction and to purify the reaction mixtures, anion exchange HPLC as described above was used.
RNA Interference and Northern Analysis
Delivery of siRNAs into cells and analysis of gene silencing were done essentially as described.4,5,37 Lyophilized synthetic siRNA (for sequence see Figure 3 and Table S1) targeted against the chicken BASP1 mRNA sequence 5′-CAGGUCUCUGCCAAUAAGACA-3′, were dissolved in a buffer containing 100 mM potassium acetate, 30 mM Hepes-KOH (pH 7.4), and 2 mM magnesium acetate, yielding a 40 μM siRNA solution. The solution was heated at 90 °C for 1 min, incubated at 37 °C for 1 h, and then stored at −80 °C. For transfection of siRNA, 5 × 106 cells of the chicken fibroblast line DF-1 were pelleted at 50 × g for 5 min at room temperature, suspended in 100 μL of nucleofector solution V (Lonza/Amaxa), and mixed with 12 μL of siRNA solution containing 0.24 nmol (∼3.0 μg) of duplex RNA. The mixture was subjected to electroporation (Lonza/Amaxa) using the nucleofector program U-20, and then immediately diluted with 0.5 mL of culture medium. Transfected cells were seeded onto 60-mm dishes containing 4 mL of culture medium and cultivated at 37 °C. Medium was changed after one day, and total RNA was isolated after two days with the RiboPure Kit (Ambion). Briefly, cells were homogenized in a solution containing phenol and guanidine thiocycanate. After addition of bromochloropropane, RNA was recovered from the aqueous phase by binding to a glass-fiber filter and subsequent elution using a low-salt buffer. Northern analysis using 5 μg of total RNA and specific DNA probes for detection of BASP1 or GAPDH mRNAs was performed as described previously.37
Acknowledgments
Funding by the Austrian Science Fund FWF (P21641, P23652, I1040) and the EU FP7Marie Curie ITN Project (289007) is gratefully acknowledged.
Supporting Information Available
1H and 13C NMR spectra for compounds 2, 2a, 2b, and 4; reduction of 2′-(2-azidoethyl) RNA; chemical structures of fluorescent dyes used; siRNA sequences. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Fonvielle M.; Li de La Sierra-Gallay I.; El-Sagheer A. H.; Lecerf M.; Patin D.; Mellal D.; Mayer C.; Blanot D.; Gale N.; Brown T.; van Tilbeurgh H.; Ethève-Quelquejeu M.; Arthur M. (2013) The structure of FemX(Wv) in complex with a peptidyl-RNA conjugate: mechanism of aminoacyl transfer from Ala-tRNA(Ala) to peptidoglycan precursors. Angew. Chem., Int. Ed. 52, 7278–7281. [DOI] [PubMed] [Google Scholar]
- Fonvielle M.; Mellal D.; Patin D.; Lecerf M.; Blanot D.; Bouhss A.; Santarem M.; Mengin-Lecreulx D.; Sollogoub M.; Arthur M.; Ethève-Quelquejeu M. (2012) Efficient access to peptidyl-RNA conjugates for picomolar inhibition of non-ribosomal FemX Wv aminoacyl transferase. Chem.—Eur. J. 19, 1357–1363. [DOI] [PubMed] [Google Scholar]
- Kiviniemi A.; Virta P.; Drenichev M. S.; Mikhailov S. N.; Lönnberg H. (2011) Solid-supported 2′-O-glycoconjugation of oligonucleotides by azidation and Click reactions. Bioconjugate Chem. 22, 1249–1255. [DOI] [PubMed] [Google Scholar]
- Fauster K.; Hartl M.; Santner T.; Aigner M.; Kreutz C.; Bister K.; Ennifar E.; Micura R. (2012) 2′-Azido RNA, a versatile tool for chemical biology: Synthesis, X-ray structure, siRNA applications, click labeling. ACS Chem. Biol. 7, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigner M.; Hartl M.; Fauster K.; Steger J.; Bister K.; Micura R. (2011) Chemical synthesis of site-specifically 2′-azido-modified RNA and potential applications for bioconjugation and RNA interference. ChemBioChem 12, 47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps K.; Morris A.; Beal P. A. (2011) Novel modifications in RNA. ACS Chem. Biol. 7, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lönnberg H. (2009) Solid-phase synthesis of oligonucleotide conjugates useful for delivery and targeting of potential nucleic acid therapeutics. Bioconjugate Chem. 20, 1065–1094. [DOI] [PubMed] [Google Scholar]
- Haller A.; Altman R. B.; Soulière M. F.; Blanchard S. C.; Micura R. (2013)) Folding and ligand recognition of the TPP riboswitch aptamer at single-molecule resolution. Proc. Natl. Acad. Sci. U.S.A. 110, 4188–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulière M. F.; Altman R. B.; Schwarz V.; Haller A.; Blanchard S. C.; Micura R. (2013) Tuning a riboswitch response through structural extension of a pseudoknot. Proc. Natl. Acad. Sci. U.S.A. 110, E3256–E3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa S.; Mühlberg M.; Pietrusiewicz K. M.; Hackenberger C. P. R. (2013) Traceless Staudinger acetylation of azides in aqueous buffers. Bioorg. Med. Chem. 21, 3465–3472. [DOI] [PubMed] [Google Scholar]
- Gramlich P. M. E.; Wirges C. T.; Manetto A.; Carell T. (2008) Postsynthetic DNA modification through the copper-catalyzed azide-alkyne cycloaddition reaction. Angew. Chem., Int. Ed. 47, 8350–8358. [DOI] [PubMed] [Google Scholar]
- Pradère U., Brunschweiger A., Gebert L. F. R., Lucic M., Roos M., and Hall J. (2013) Chemical synthesis of mono- and bis-labeled pre-microRNAs. Angew. Chem., Int. Ed. 52 [Online early access] doi: 10.1002/anie.201304986. [DOI] [PubMed] [Google Scholar]
- Seidu-Larry S.; Krieg B.; Hirsch M.; Helm M.; Domingo O. (2012) A modified guanosine phosphoramidite for click functionalization of RNA on the sugar edge. Chem. Commun. 48, 11014–11016. [DOI] [PubMed] [Google Scholar]
- Jawalekar A. M.; Meeuwenoord N.; Cremers J. G. O.; Overkleeft H. S.; van der Marel G. A.; Rutjes F. P. J. T.; van Delft F. L. (2008) Conjugation of nucleosides and oligonucleotides by [3 + 2] cycloaddition. J. Org. Chem. 73, 287–290. [DOI] [PubMed] [Google Scholar]
- van Berkel S. S.; van Eldijk M. B.; van Hest J. C. M. (2011) Staudinger ligation as a method for bioconjugation. Angew. Chem., Int. Ed. 50, 8806–8827. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. (2009) Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sagheer A. H.; Brown T. (2010) Click chemistry with DNA. Chem. Soc. Rev. 39, 1388–1405. [DOI] [PubMed] [Google Scholar]
- Wang C. C. Y.; Seo T. S.; Li Z.; Ruparel H.; Ju J. (2003) Site-specific fluorescent labeling of DNA using Staudinger ligation. Bioconjugate Chem. 14, 697–701. [DOI] [PubMed] [Google Scholar]
- Weisbrod S. H.; Marx A. (2008) Novel strategies for the site-specific covalent labelling of nucleic acids. Chem. Commun. 44, 5675–5685. [DOI] [PubMed] [Google Scholar]
- Winz M.-L.; Samanta A.; Benzinger D.; Jäschke A. (2012) Site-specific terminal and internal labeling of RNA by poly(A) polymerase tailing and copper-catalyzed or copper-free strain-promoted click chemistry. Nucleic Acids Res. 40, e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao H.; Sawant A. A.; Tanpure A. A.; Srivatsan S. G. (2011) Posttranscriptional chemical functionalization of azide-modified oligoribonucleotides by bioorthogonal click and Staudinger reactions. Chem. Commun. 48, 498–500. [DOI] [PubMed] [Google Scholar]
- Wachowius F.; Höbartner C. (2010) Chemical RNA modifications for studies of RNA structure and dynamics. ChemBioChem 11, 469–480. [DOI] [PubMed] [Google Scholar]
- Jewett J. C.; Bertozzi C. R. (2010) Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 39, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerowska M.; Hall L.; Richardson J.; Shelbourne M.; Brown T. (2012) Efficient reverse click labeling of azide oligonucleotides with multiple alkynyl Cy-Dyes applied to the synthesis of HyBeacon probes for genetic analysis. Tetrahedron 68, 857–864. [Google Scholar]
- Schoch J.; Staudt M.; Samanta A.; Wiessler M.; Jäschke A. (2012) Site-specific one-pot dual labeling of DNA by orthogonal cycloaddition chemistry. Bioconjugate Chem. 23, 1382–1386. [DOI] [PubMed] [Google Scholar]
- Shelbourne M.; Brown T.; El-Sagheer A. H.; Brown T. (2012) Fast and efficient DNA crosslinking and multiple orthogonal labelling by copper-free click chemistry. Chem. Commun. 48, 11184–11186. [DOI] [PubMed] [Google Scholar]
- Pourceau G.; Meyer A.; Vasseur J.-J.; Morvan F. (2009) Azide solid support for 3′-conjugation of oligonucleotides and their circularization by Click chemistry. J. Org. Chem. 74, 6837–6842. [DOI] [PubMed] [Google Scholar]
- Steger J.; Graber D.; Moroder H.; Geiermann A.-S.; Aigner M.; Micura R. (2010) Efficient access to nonhydrolyzable initiator tRNA based on the synthesis of 3′-azido-3′-deoxyadenosine RNA. Angew. Chem., Int. Ed. 49, 7470–7472. [DOI] [PubMed] [Google Scholar]
- Büttner L.; Seikowski J.; Wawrzyniak K.; Ochmann A.; Höbartner C. (2013) Synthesis of spin-labeled riboswitch RNAs using convertible nucleosides and DNA-catalyzed RNA ligation. Bioorg. Med. Chem. 21, 6171–6180. [DOI] [PubMed] [Google Scholar]
- Cuenoud B.; Casset F.; Hüsken D.; Natt F.; Wolf R. M.; Altmann K.-H.; Martin P.; Moser H. E. (1998) Dual recognition of double-stranded DNA by 2′-aminoethoxy-modified oligonucleotides. Angew. Chem., Int. Ed. 37, 1288–1291. [DOI] [PubMed] [Google Scholar]
- Jin S.; Miduturu C. V.; McKinney D. C.; Silverman S. K. (2005) Synthesis of amine- and thiol-modified nucleoside phosphoramidites for site-specific introduction of biophysical probes into RNA. J. Org. Chem. 70, 4284–4299. [DOI] [PubMed] [Google Scholar]
- McGee D. P. C.; Vaughn-Settle A.; Vargeese C.; Zhai Y. (1996) 2′-Amino-2′-deoxyuridine via an intramolecular cyclization of a trichloroacetimidate. J. Org. Chem. 61, 781–785. [DOI] [PubMed] [Google Scholar]
- Pak J. K.; Hesse M. (1998) Synthesis of penta-N-protected homocaldopentamine and its selective acylation. J. Org. Chem. 63, 8200–8204. [Google Scholar]
- Egli M.; Minasov G.; Tereshko V.; Pallan P. S.; Teplova M.; Inamati G. B.; Lesnik E. A.; Owens S. R.; Ross B. S.; Prakash T. P.; Manoharan M. (2005) Probing the influence of stereoelectronic effects on the biophysical properties of oligonucleotides: Comprehensive analysis of the RNA affinity, nuclease resistance, and crystal structure of ten 2′-O-ribonucleic acid modifications. Biochemistry 44, 9045–9057. [DOI] [PubMed] [Google Scholar]
- Saneyoshi H.; Okamoto I.; Masaki Y.; Ohkubo A.; Seio K.; Sekine M. (2007) Facile synthesis of 2′-O-cyanoethyluridine by ring-opening reaction of 2,2′-anhydrouridine with cyanoethyl trimethylsilyl ether in the presence of BF3·Et2O. Tetrahedron Lett. 48, 8554–8557. [Google Scholar]
- Paredes E.; Das S. R. (2010) Click chemistry for rapid labeling and ligation of RNA. ChemBioChem 12, 125–131. [DOI] [PubMed] [Google Scholar]
- Hartl M.; Nist A.; Khan M. I.; Valovka T.; Bister K. (2009) Inhibition of Myc-induced cell transformation by brain acid-soluble protein 1 (BASP1). Proc. Natl. Acad. Sci. U.S.A. 106, 5604–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl M.; Mitterstiller A.-M.; Valovka T.; Breuker K.; Hobmayer B.; Bister K. (2010) Stem cell-specific activation of an ancestral myc protooncogene with conserved basic functions in the early metazoan Hydra. Proc. Natl. Acad. Sci. U.S.A. 107, 4051–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Sheng G.; Juranek S.; Tuschl T.; Patel D. J. (2008) Structure of the guide-strand-containing argonaute silencing complex. Nature 456, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Juranek S.; Li H.; Sheng G.; Tuschl T.; Patel D. J. (2008) Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature 456, 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts J.; Deleavey G.; Damha M. (2008) Chemically modified siRNA: tools and applications. Drug Discovery Today 13, 842–855. [DOI] [PubMed] [Google Scholar]
- Willibald J.; Harder J.; Sparrer K.; Conzelmann K.-K.; Carell T. (2012) Click-modified anandamide siRNA enables delivery and gene silencing in neuronal and immune cells. J. Am. Chem. Soc. 134, 12330–12333. [DOI] [PubMed] [Google Scholar]
- Jayaprakash K. N.; Peng C. G.; Butler D.; Varghese J. P.; Maier M. A.; Rajeev K. G.; Manoharan M. (2010) Non-nucleoside building blocks for copper-assisted and copper-free click chemistry for the efficient synthesis of RNA conjugates. Org. Lett. 12, 5410–5413. [DOI] [PubMed] [Google Scholar]
- Rieder U.; Lang K.; Kreutz C.; Polacek N.; Micura R. (2009) Evidence for pseudoknot formation of class I preQ1 riboswitch aptamers. ChemBioChem 10, 1141–1144. [DOI] [PubMed] [Google Scholar]
- Bramsen J. B.; Laursen M. B.; Nielsen A. F.; Hansen T. B.; Bus C.; Langkjaer N.; Babu B. R.; Højland T.; Abramov M.; Van Aerschot A.; Odadzic D.; Smicius R.; Haas J.; Andree C.; Barman J.; Wenska M.; Srivastava P.; Zhou C.; Honcharenko D.; Hess S.; Müller E.; Bobkov G. V.; Mikhailov S. N.; Fava E.; Meyer T. F.; Chattopadhyaya J.; Zerial M.; Engels J. W.; Herdewijn P.; Wengel J.; Kjems J. (2009) A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 37, 2867–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollogoub M.; Dominguez B.; Fox K. R.; Brown T. (2000) Synthesis of a novel bis-amino-modified thymidine monomer for use in DNA triplex stabilisation. Chem. Commun. 23, 2315–2316. [Google Scholar]
- Cardew A. S.; Brown T.; Fox K. R. (2012) Secondary binding sites for heavily modified triplex forming oligonucleotides. Nucleic Acids Res. 40, 3753–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smicius R.; Engels J. W. (2008) Preparation of zwitterionic ribonucleoside phosphoramidites for solid-phase siRNA synthesis. J. Org. Chem. 73, 4994–5002. [DOI] [PubMed] [Google Scholar]
- Blommers M. J. J.; Natt F.; Jahnke W.; Cuenoud B. (1998) Dual recognition of double-stranded DNA by 2′-aminoethoxy-modified oligonucleotides: The solution structure of an intramolecular triplex obtained by NMR spectroscopy. Biochemistry 37, 17714–17725. [DOI] [PubMed] [Google Scholar]
- Bobkov G. V.; Mikhailov S. N.; van Aerschot A.; Herdewijn P. (2008) Phosphoramidite building blocks for efficient incorporation of 2′-O-aminoethoxy(and propoxy)methyl nucleosides into oligonucleotides. Tetrahedron 64, 6238–6251. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.