

Abstract
Transplantation of endothelial cells (ECs) for therapeutic vascularization or tissue engineering is a promising method for increasing tissue perfusion. Here, we report on a new approach for enhanced EC transplantation using targeted nanoparticle transfection to deliver proangiogenic microRNA-132 (miR-132) to cultured ECs before their transplantation, thereby sensitizing cells to the effects of endogenous growth factors. We synthesized biodegradable PLGA polymer nanoparticles (NPs) that were loaded with miR-132 and coated with cyclic RGD (cRGD) peptides that target integrin αvβ3 expressed on cultured human umbilical vein ECs (HUVECs), increasing NP uptake through clathrin-coated pits. Unlike previously reported NPs for miR delivery, these NPs slowly release RNA for several weeks. The endocytosed NPs remain in clathrin-coated vesicles from which they mediate intracellular delivery of siRNA or miRNA. Transfection of HUVECs with miR-132 enhances growth factor-induced proliferation and migration in 2D culture, producing a 1.8- and 5-fold increase, respectively. However, while the effects of conventional transfection were short-lived, NP transfection produced protein knockdown and biological effects that were significantly longer in duration (≥6 d). Transfection of HUVECs with miR-132 NP resulted in a 2-fold increase in the number of microvessels per square millimeter compared to lipid after transplantation into immunodeficient mice and led to a higher number of mural cell-invested vessels than control transfection. These data suggest that sustained delivery of miR-132 encapsulated in a targeted biodegradable polymer NP is a safe and efficient strategy to improve EC transplantation and vascularization.—Devalliere, J., Chang, W. G., Andrejecsk, J. W., Abrahimi, P., Cheng, C. J., Jane-wit, D., Saltzman, W. M., Pober, J. S. Sustained delivery of proangiogenic microRNA-132 by nanoparticle transfection improves endothelial cell transplantation.
Keywords: nanotechnology, neovascularization, controlled release, tissue engineering
Microvessels perform functions essential for cell and tissue function and survival, such as delivering oxygen and nutrients and removing by-products and waste. The ability to induce the formation of new microvessels could have a wide range of applications, including the treatment of myocardial ischemia (1), the repair of tissue defects induced by acute traumas (burns; ref. 2) or chronic wounds (nonhealing ulcers; ref. 3), and engineering 3-dimensional (3D) thick tissues (4, 5). One strategy to create new microvessels is to allow self-assembly from transplantation of either mature endothelial cells (ECs) or committed endothelial progenitor cells. Once they are placed in a host, the fate of the transplanted cells cannot be controlled, and vessels may accumulate in areas where vascularization is irrelevant. To limit this, vessel formation can be initiated ex vivo within a scaffold that can be implanted at the desired site, allowing vessel maturation and inosculation to be completed in situ. We have previously explored this approach using fully differentiated human umbilical vein ECs (HUVECs) suspended within a hydrated collagen-fibronectin gel. During the first 18–24 h, HUVECs assemble themselves into cords that begin to form lumens. Once implanted into the anterior abdominal wall or hind limb of a C.B-17 severe combined immunodeficiency (SCID)/beige mouse, HUVEC cords evolve into microvascular conduits (6).
The formation of human EC-lined vessels within the engrafted construct and inosculation with mouse microvessels occurs slowly over a matter of weeks (3, 6, 7). To enhance the utility of this approach, it is necessary to accelerate the rate of vessel formation and maturation. We previously reported that gels containing HUVECs that had been retrovirally transduced with the antiapoptotic protein Bcl-2 display more mature microvessels compared to gels containing untransduced or control vector-transduced HUVECs. Furthermore, HUVECs transduced by Bcl-2 induce a host arteriogenic response that increases perfusion to the implantation site (7). Vessel formation using transplanted Bcl-2 cells can be further accelerated and therapeutic outcome enhanced by delivering exogenous growth factors. Specifically, delivery of vascular endothelial growth factor (VEGF) or VEGF combined with MCP-1 increased functional vessel formation and led to a higher number of pericyte/smooth muscle cell-invested vessels (8–10). In this study, we propose an alternative to delivery of therapeutic VEGF protein, namely enhancing the responsiveness of the EC to its proangiogenic effects. MicroRNAs (miRNAs) have emerged as essential regulators of gene expression and actively participate in various aspects of angiogenesis, including proliferation, migration, and morphogenesis of endothelial cells (11). miRNA-132, in particular, has been described as an angiogenic switch targeting p120RasGAP in the endothelium, permitting increased activation of Ras and leading to neovascularization in response to growth factor (12). On the basis of this recent discovery, we hypothesized that delivering miRNA-132 to HUVECs before their incorporation into protein gel constructs would support their transplantation and enhance the vascularization process. Ideally, the effect of the exogenous miRNA should be of sufficient duration to affect the formation and maturation of new microvessels but then abate once a stable vasculature has been established. The ideal duration might be for 1–2 wk, typically longer than that achieved with conventional lipid-based transfection reagents.
Nanoparticles (NPs) composed of poly(lactic-co-glycolic acid) (PLGA), a biodegradable and nontoxic polymer, have been shown to be efficient and chemically modifiable carriers of short interfering RNA (siRNA; refs. 13, 14) and miRNA (15). Further, we formulated PLGA NPs with surface-bound cyclo Arg-Gly-Asp-D-Phe-Lys (cRGD) peptide for specific delivery of miRNA into cultured human ECs, targeting αvβ3 integrin, which is highly expressed by HUVECs in vitro. Previous reports have described lipid-based NPs for RGD-mediated delivery of miR after intravenous injection (12, 16): Those systems were not suitable for this study because they cannot provide sustained release of the encapsulated miR. Our results demonstrate that ex vivo miRNA-132 (miR-132) delivery to HUVECs via PLGA NPs enhances and accelerates the vascularization process after HUVEC transplantation, potentially improving therapeutic efficacy. Moreover, the duration of the effect due to sustained release of miR-132 is considerably greater than that obtained with conventional lipid transfection and could further increase vessel formation in vivo after implantation into mouse hosts.
MATERIALS AND METHODS
Endothelial cell culture and treatments
HUVECs were isolated from umbilical veins by treatment with collagenase and serially passaged 2–6 times in gelatin-coated tissue culture plastic using M199 medium supplemented with 20% FBS, 2% l-glutamine, 1% penicillin, and streptomycin and endothelial cell growth supplement [i.e., fibroblast growth factor 1 (FGF-1), 1:100 dilution; BD Biosciences, San Jose, CA, USA] complexed to 50 μg/ml porcine intestinal heparin (Sigma-Aldrich, St. Louis, MO, USA) at 37°C in a 5% CO2 humidified air incubator. Where indicated, HUVECs were pretreated for 30 min with nystatin (5μM; Sigma-Aldrich), chlorpromazine (2 μg/ml; Sigma-Aldrich) or dynasore (80 μM; Sigma-Aldrich) in M199 medium prior to addition of NPs, Alexa Fluor 488-labeled transferrin (15 μg/ml, Molecular Probes, Carlsbad, CA, USA) Alexa Fluor 488-labeled acetylated low-density lipoproteins (15 μg/ml; Molecular Probes) or Alexa Fluor 488-labeled cholera toxin subunit B (1 μg/ml, Molecular Probes). Where indicated, cells were treated with mouse anti-human integrin αvβ3 Ab (10 μg/ml, Millipore, Bedford, MA, USA) for 30 min prior to NP incubation.
miRNAs, siRNAs, and transfections
The miRNA 132 (UAACAGUCUACAGCCAUGGUCG) and siRNAs targeting survivin (GUCCGGUUGCGCUUUCCUUUC; J-003459-08), caveolin 1 (J-003467-06), clathrin (J-004001-09), and AP2M1 (J-008170-05) were prepared by Dharmacon (Thermo Scientific, Lafayette, CO, USA). Fluorescently labeled 5′-carboxyfluorescein (FAM)-siRNAs were purchased from Integrated DNA Technologies (Coralville, IA, USA). HUVECs were transfected at 70–90% confluence with specific miRNAs/siRNAs or nontargeting miRNA/siRNA (CN-001000-01/D-001810-01), using Lipofectamine RNAiMAX (Invitrogen, Grand Island, NY, USA) to the indicated final concentration. The cells were analyzed 48 h after transfection by RT-PCR or Western blot analysis.
NP synthesis
PLGA with terminal ester group [poly(D,L-lactide-co-glycolide); 50:50 monomer ratio and 0.55–0.75 dl/g inherent viscosity] was purchased from Lactel (Birmingham, AL, USA). Polyvinyl alcohol and coumarin 6 (C6) were obtained from Sigma-Aldrich. C6-loaded NPs were prepared using an oil-in-water single-emulsion technique (15). miR-loaded-NPs and siRNA-loaded-NPs were fabricated using a modified water-in-oil-in-water (w/o/w) double-emulsion method (13). In brief, miRNA or siRNA (50–100 nmol) was complexed with spermidine in pH 7.4 Tris-EDTA buffer (10 mM Tris–HCl, 1 mM EDTA) at an 8:1 polyamine nitrogen to nucleotide phosphate (N:P) ratio. This aqueous solution was added dropwise to the polymer solution of PLGA dissolved in dichloromethane (2 ml) and sonicated to form the first emulsion. This mixture was then added dropwise to 2 ml of 5% polyvinyl alcohol (PVA) and sonicated to form the double emulsion. NP surfaces were coated with the αVβ3 integrin-binding cRGD peptide or the control nonbinding cyclo Arg-Ala-Asp-D-Phe-Lys (cRAD) peptide (Peptide International, Louisville, KY, USA) via a PEGylated phospholipid linker 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethyleneglycol)-2000] (DSPE-PEG; Nanocs, Boston, MA, USA), as described previously (14).
NP characterization
The morphology, percentage loading, and kinetics of controlled release were evaluated after synthesis of the particles. Dry NPs were visualized using scanning electron microscopy (SEM; XL30 ESEM, FEI Co., Hillsboro, OR, USA) to assess NP morphology and size distribution. The amount of miRNA/siRNA encapsulated in NPs was determined using a modified organic extraction method, as described previously (13). Briefly, NPs were dissolved in dichloromethane (DCM) for 1 h. Two volumes of TE buffer was added to the organic solution, vortexed, and centrifuged (16,100 g for 10 min at 4°C) to extract miRNA/siRNA into the aqueous phase. The amount of miRNA/siRNA in the aqueous fraction was measured using the QuantIT PicoGreen assay (Invitrogen), according to the manufacturer's instructions. Loading of C6 was determined by dissolving NPs in DMSO for 1 h, and C6 content was quantified by spectrofluorescence using a Molecular Devices SpectraMax M5 (excitation/emission: 458/505 nm; Molecular Devices, Sunnyvale, CA, USA).
To characterize the release profile, NPs were suspended in PBS and incubated at 37°C in a rotary shaker. Release of miRNA/siRNA was determined at several time intervals over 20 d. At each sampling time, the NP suspension was centrifuged for 5 min at 16,100 g and the supernatant was collected and analyzed for determination of miRNA/siRNA content. An equal volume of PBS was replaced for continued monitoring of miRNA/siRNA release. The amount of miRNA/siRNA was determined using the organic extraction method.
RT-PCR
Total RNA and miRNAs were isolated using an RNeasy plus kit or an miRNeasy kit (Qiagen, Valencia, CA, USA), and reverse transcription was performed using the QuantiTect reverse transcription kit (Qiagen) or TaqMan MicroRNA reverse transcription kit (Applied Biosystems, Life Technologies, Grand Island, NY, USA), respectively. Transcript levels were quantified by qRT-PCR using the following probes from Applied Biosystems: survivin (Hs00977611_g1), hypoxanthine-guanine phosphoribosyl transferase 1 (HPRT-1; Hs99999909_m1), miR-132 (000457), and RNU48 (001006). For quantification, triplicates were normalized by the concomitant quantification of HPRT or small-nucleolar RNAs RNU48 for miRNAs. Relative expression was calculated, according to the 2−ΔΔCt method, as described previously (17).
Western blot analysis
Cells were lysed in RIPA buffer containing protease and phosphatase inhibitors (Sigma-Aldrich). Cell lysates were resolved by SDS–PAGE, and proteins were transferred to Immobilon-P PVDF (Millipore) and blocked in 5% BSA solution. Where indicated, membranes were subjected to analysis by Western blot analysis using primary antibodies against survivin (1:400 dilution; R&D Systems, Minneapolis, MN, USA), RasGAP (1:200 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or GAPDH (1:1000 dilution; Chemicon, Temecula, CA, USA) and appropriate peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA). Antibody-bound proteins were detected using Pico West Chemiluminescent substrate (Thermo Scientific). Image analysis and blot quantification was performed with ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
FACS and DNA content analysis
NP uptake and internalization were monitored using flow cytometry. Cells were grown to 80% confluency, then C6-loaded NPs were added and incubated for the indicated time at 37°C. Cells were washed with PBS and harvested using trypsin. To isolate internalized NPs, cells were incubated with 0.1% (w/v) trypan blue for 5 min to quench any extracellular fluorescence of live cells (i.e., surface-associated NPs). Cells were washed with 1% BSA in PBS and then analyzed on a FACSCalibur (BD Biosciences).
Cellular DNA content analyses were performed by flow cytometry as follows: ECs were harvested, washed twice in PBS, fixed in ice-cold 70% ethanol, and incubated for 24 h at 4°C. Fixed cells were then stained with 20 μg/ml propidium iodide, 10 μg/ml RNase A (Sigma-Aldrich), and 1 mg/ml glucose in PBS (1 ml/2×105 cells). Results were analyzed with FlowJo 7.2.5 software (TreeStar Inc., Ashland, OR, USA).
Assessment of toxicity
To assess apoptosis or necrosis, HUVEC monolayers were allowed to take up NPs for the indicated times, after which cytotoxicity assays were performed on suspended cells using an annexinV/PI apoptosis kit from eBiosiences (San Diego, CA, USA) according to the manufacturer's instructions. Labeled cells were then analyzed by flow cytometry on a BD Biosciences FACSCalibur using FlowJo 7.2.5 software. To assess autophagic injury, HUVECs were grown on glass coverslips (microscope analysis) or 96-well plate, treated with NP or tamoxifen (10 μM, a known inducer of autophagy), for 24 or 72 h. Cells were incubated with monodansylcadaverine (MDC; Abcam, Cambridge, UK) for 10 min at 37°C and were washed; formation of autophagic vacuoles was assessed using a fluorescence microscopy viewed through a 512-nm filter.
Confocal fluorescence microscopy
NP association and intracellular fate were visualized by confocal fluorescence microscopy. ECs were grown on glass coverslips, and C6-loaded NPs were added to cells at a final concentration of 500 μg/ml for 18 h. After treatment, cultures were extensively washed with PBS to remove unassociated NPs, fixed for 10 min in PBS containing 1% paraformaldehyde and permeabilized with PBS containing 4% BSA (Sigma-Aldrich) and 0.1% saponin (Sigma-Aldrich) for 20 min. Cells were washed with PBS, blocked with 10% donkey serum diluted in PBS-4% BSA-0.1% saponin for 1 h. Cells were then incubated with anti-LAMP1 (1:50 dilution; Santa Cruz Biotechnology), anti-caveolin-1 (1:100 dilution; Cell Signaling Technology, Danvers, MA, USA) or anti-clathrin-1 (1:100 dilution; Cell Signaling Technology) antibodies overnight at 4°C. Cells were rewashed and incubated for 1 h with donkey anti-mouse Alexa Fluor 546 or anti-rabbit Alexa Fluor 546 (1:2000 dilution; Invitrogen). Cells were washed in PBS containing 1% BSA, and mounted with ProLong antifade DAPI reagent (Molecular Probes). Cell viability was assessed using Live/Dead Cells Viability Assay (Invitrogen). Protein gels were incubated at room temperature for 30 min with PBS-containing red-fluorescent ethidium homodimer 1 (4 μM) and green-fluorescent calcein-AM (2 μM). Gels were washed 3 times with PBS prior to visualization. In this assay, viable cells display green fluorescence, whereas dead cells lose membrane integrity and display red fluorescence. All specimens were examined using a Leica TCS SP5 spectral confocal microscope (Leica Microsystems, Bannockburn, IL, USA).
Spheroid sprouting model
EC spheroids were generated by incubating HUVECs in medium containing 0.25% methylcellulose in complete M199 medium in 96-well nonadherent U-bottomed plates, as described previously (18). Spheroids were harvested, washed, and suspended within a collagen-fibronectin gel containing VEGF (50 ng/ml). After 18 h incubation at 37°C, the numbers of sprouts were quantified by microscopy using a Zeiss Axiovert 200 M inverted fluorescence microscope (Carl Zeiss, Oberkochen, Germany) with a Hamamatsu ORCA-AG high-resolution camera (Hamamatsu Photonics, Hamamatsu, Japan) and ImageJ software.
Protein gel implants
All experiments were performed under protocols approved by the Yale University Institutional Review Board and Institutional Animal Care and Use Committee. Collagen-fibronectin gels were prepared as described previously (7). HUVECs were transfected with miR-132 using lipofection or treated with miR-132-NP 24 h prior being suspended in collagen-fibronectin solution (1×106 cells/300 μl gel). Solutions were incubated at 37°C for 20 min to allow for collagen polymerization, after which an equal volume of complete M199 was overlaid, and gels were incubated for 18 h at 37°C in 5% CO2 air. The gels were then implanted into bluntly dissected abdominal wall subcutaneous pockets of C.B-17 SCID/beige female mice (10–12 wk) under anesthesia. At the time points indicated, the animals were euthanized, and the gels were harvested, fixed with 2% paraformaldehyde, paraffin-embedded, and sectioned. Vessel counts were assessed on hematoxylin and eosin (H&E)-stained sections.
Statistical analysis
Results are expressed as means ± se for replicate experiments. Statistical analysis was performed using GraphPad Prism software (GraphPad Software, San Diego, CA, USA) by the parametric analysis of variance test as appropriate. A value of P < 0.05 was considered statistically significant.
RESULTS
Synthesis and characterization of unconjugated, cRGD, and cRAD NPs
NPs, with or without cRGD or cRAD peptides on their surface, were prepared using a modified double-emulsion solvent evaporation process, permitting encapsulation of siRNA or miRNA, as described previously (13, 19). PEGylated phospholipids have been utilized with success to coat ligands on the surface of colloidal NPs (14). We exploited the amphipathic properties of DSPE-PEG to functionalize the surface of PLGA NPs. During formulation, DSPE-PEG is dissolved in the aqueous phase of the second emulsion, where it is available to associate with the hydrophobic water-in-oil droplets of the primary emulsion. After solvent evaporation, the acyl chains remained associated with the polymer, tethering PEG to the surface of the NPs. Ligand-coated particles (Fig. 1A) were formulated by coupling DSPE-PEG-NHS to the lysine primary amine of cRGD or cRAD peptide before addition to the second emulsion. NP morphology and size were measured from representative SEM micrographs (Fig. 1B). Unmodified and modified NPs are similar in size (148±55 and 157±61 nm, respectively) and spherical shape. To optimize methods for RNA delivery, we encapsulated a siRNA that targets survivin. The loading of this siRNA was 174 pmol siRNA/mg NP for unmodified NPs and 154 pmol siRNA/mg NP for DSPE-PEG-cRGD NPs, a difference that does not reach statistical significance. The incorporation of DSPE-PEG-cRGD onto PLGA NPs did not noticeably alter siRNA release profile; a burst release was observed during the first hours (∼40% of the NP load was released in the first 24 h), which was followed by sustained linear release over several days (Fig. 1C). After 48 h of continuous incubation, unmodified NPs and DSPE-PEG-cRGD NPs released 65 and 61% of their siRNA content, respectively.
Figure 1.
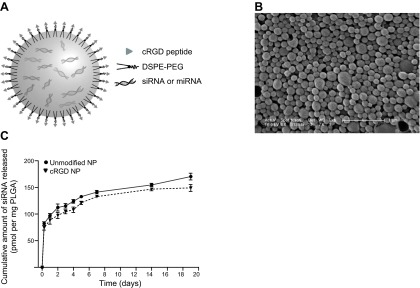
Characteristics of targeted cRGD NPs. A) Schematic of cRGD-coated NPs. The oligonucleotide-loaded NPs comprised a spherical PLGA core coated with phospholipid-linked PEGylated cRGD ligands. B) Representative scanning electron micrograph of oligonucleotide-loaded NPs. Scale bar = 1 μm. C) Controlled-release profile for unmodified and cRGD NPs loaded with survivin siRNA.
Uptake of NPs by cultured human ECs
To follow uptake of NPs into cells, NPs were loaded with the fluorescent dye coumarin 6 (C6) instead of RNA. Confocal fluorescence microscopy of HUVECs revealed that NP uptake is time and concentration-dependent for both formulations (Fig. 2A, B). The uptake was linear for several hours before reaching a saturation limit after 24 h of incubation. Only 50% of cells exhibited uptake after 24 h with a 500 μg/ml dose of unmodified NPs, whereas all cells were positive for the C6 marker using cRGD formulation, demonstrating an increase in NP internalization induced by the cRGD coating. The uptake and total number of NP events in cells were proportional to NP dose and tended to be greater for cRGD NPs (10.1±2.1 unmodified NPs/cell vs. 18.5±4.8 cRGD NPs/cell after 18 h of incubation; Fig. 2C, D). Flow cytometry was used to quantify cells with internalized NPs (Fig. 2E, F). NP cellular accumulation increased over time and was proportional to NP dose. The amount of internalized NPs by flow cytometry showed a 2.9-fold increase of cRGD C6 NP uptake compared to unmodified C6 NPs 18 h postincubation (1316±184 MFI for cRGD NPs vs. 444±55 MFI for unmodified NPs, P < 0.05). To assess the specificity of RGD-mediated targeting, NPs were coated with a control cyclic peptide, cRAD. In contrast to the results observed with cRGD NPs, NPs with cRAD did not exhibit significantly more NP uptake than unmodified NPs (Fig. 2E, F), consistent with the interpretation that cRGD enhances internalization of NPs through its specific binding with αvβ3 integrin.
Figure 2.

Microscopy and FACS analysis showing the effect of cRGD ligand coating on NP uptake. HUVECs were treated with C6-labeled unmodified, cRAD, or cRGD NPs for indicated period of time (1–24 h) with 500 μg NPs/ml (A, C, E) or different doses (50–500 μg NPs/ml) for 24 h (B, D, F). A, B) Quantitative comparison of the number of C6-positive cells, based on confocal microscopy analysis, results are expressed as percentage of cells transduced (means± sem. *P < 0.05. C, D) Quantitative comparison of total NP events per EC, based on confocal fluorescence microscopic analysis (means±sem). *P < 0.05, **P < 0.005. E, F) FACS analysis of internalized NPs; results are expressed as mean fluorescent intensity (means±sem); *P<0.05).
Silencing efficiency of NPs is clathrin dependent in ECs
We next assessed NP intracellular fate and ability to deliver the encapsulated therapeutic agents into the cytoplasm. Immunofluorescence microscopy of C6 NP-positive cells showed an absence of colocalization for both control and targeted NPs with caveolin 1, whereas there was extensive colocalization of NPs and clathrin (Fig. 3). The fluorescence colocalization patterns for both types of NPs with clathrin were found at the plasma membrane and intracellularly, presumably because of internalized clathrin-coated vesicles. The use of Lamp1 protein as a lysosomal marker showed that only a limited number of unmodified or targeted NPs were in lysosomes (Fig. 3). To further investigate the endocytosis pathway involved in NP internalization, we used pharmacological inhibitors, namely nystatin, known to interfere with caveolin-dependent endocytosis, chlorpromazine, which inhibits clathrin-mediated endocytosis, and dynasore, a cell-permeable inhibitor of dynamin 1 and 2 that affects both pathways. We first ruled out cytotoxicity and then established specificity of these agents using labeled endocytic probes, namely, transferrin and acLDL, which are known to enter cells via clathrin-coated pits, and cholera toxin B (CTB), which is internalized through caveolae. None of these agents produced significant cytotoxity in these assays, as assessed by annexin V surface staining or loss of PI exclusion (Supplemental Fig S1A). As expected, transferrin and acLDL uptake were decreased by chlorpromazine by 40 and 73%, respectively, with no significant effect from nystatin; however, CTB internalization was diminished in the presence of nystatin by 72% compared to untreated cells and was unaffected by chlorpromazine (Fig. 4A). Dynasore blocked both clathrin-mediated and caveolar endocytic mechanisms, as demonstrated by decreases in transferrin, acLDL, and CTB uptake (Fig. 4A). Chlorpromazine decreased unmodified and cRGD NP endocytosis by 55 and 41%, respectively, whereas nystatin did not consistently impair uptake of either NP in ECs, suggesting that NP internalization occurred largely through a clathrin-mediated pathway. Dynamin inhibition of NP uptake further supports the involvement of endocytic vesicle processes (Fig. 4B). We also utilized siRNA knockdown to provide a more precise identification of the endocytic pathway used for uptake of NP (Supplemental Fig. S1B). The uptake of transferrin and acLDL molecules decreased as a result of clathrin heavy-chain down-regulation (61 and 43%, respectively) or silencing of AP2M1, an adaptor protein essential in clathrin-mediated endocytosis (79 and 71% compared to siRNA control treatment, respectively). In contrast, the use of a siRNA specific to caveolin 1 diminished CTB uptake by 60% without significantly affecting transferrin and acLDL endocytosis (Fig. 4C), which further validated our siRNA strategy. The inhibition of the clathrin pathway, using si-clathrin or si-AP2M1, clearly prevented NP entry in HUVECs, as assessed by FACS analysis, showing a ∼60% reduction of unmodified and cRGD NP uptake compared with siRNA-treated control cells. In contrast, si-caveolin did not affect NP internalization, corroborating that NP uptake does not involve caveolin-mediated uptake in cultured HUVECs (Fig. 4D).
Figure 3.
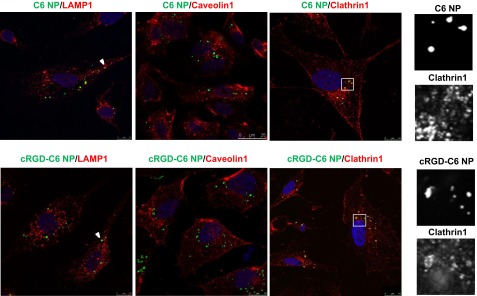
NPs escape endolysosomal degradation and markedly colocalize with clathrin vesicles. Cells were incubated with unmodified or cRGD NPs for 18 h, fixed, and stained for Lamp-1, caveolin1, and clathrin1 (red). Each panel represents 1 experiment of 3, showing a single confocal slice; nuclei stained using DAPI (blue) and NPs loaded with C6 (green) are merged with red staining.
Figure 4.

NP internalization and related knockdown efficiency are prevented by inhibition of the clathrin-mediated pathway. A, B) Cells were pretreated with nystatin (5 μM), chlorpromazine (2 μg/ml) or dynasore (80 μM), and endocytosis of Alexa Fluor 488 transferrin (15 μg/ml), acetylated low-density lipoproteins (15 μg/ml), and cholera toxin subunit B (1 μg/ml) (A) or C6 unmodified or cRGD NPs (500 μg/ml) (B) were assessed by flow cytometry. Results are expressed as a percentage of MFI based on endocytosis observed in control cells treated with vehicle (means±sem). *P < 0.05, **P < 0.005, ***P < 0.0005. C, D) Cells were transduced with siRNA against clathrin1, AP2M1, or caveolin1 2 d prior to incubation with endocytosis markers (C) or C6 NPs (D). E) Cells knocked down for clathrin 1, AP2M1, or caveolin 1 were incubated with unmodified or cRGD NPs loaded with survivin siRNA. Silencing of survivin was determined by qRT-PCR at 2 d post-treatment and normalized to HPRT-1. Results are expressed as a percentage of down-regulation (means±sem). ns, not significant. **P < 0.01, ***P < 0.0005.
Gene silencing by NP transfection of siRNA
To test the capacity of our NPs to transfect cells with RNA, we administered siRNA-survivin NPs to cultured HUVECs and measured target mRNA levels normalized to a housekeeping control gene (HPRT) as a function of time (Fig. 5A). Inhibition of gene expression produced by unmodified NPs and cRGD NPs was compared with siRNA delivered using a conventional lipid transfection agent, Lipofectamine RNAiMAX. Relative to samples treated with a siRNA control, lipid-based transfection of siRNA-survivin produced 67% knockdown 2 d after transfection. The gene inhibition decreased rapidly over time with only 20% knockdown on d 3. Survivin mRNA levels returned to the normal basal level at 4 d post-transfection. We next tested the effect of incorporation of siRNA molecules into NP. To do so, we prepared siRNA-survivin-labeled NPs, extracted the siRNA-survivin from the NP, demonstrated its integrity on a nondenaturing PAGE gel (Supplemental Fig. S2A), and then established that it was as efficient in silencing survivin expression in cultured cells as siRNA not subjected to NP incorporation and extraction (Supplemental Fig. S2B). As reported in earlier studies using other siRNAs (13, 14), the formulation process does not alter the physicochemical or functional properties of the siRNA.
Figure 5.

Gene silencing by siRNA-loaded cRGD NPs compared to uncoated NPs and lipid-based transfection. A) HUVECs were incubated with survivin siRNA-loaded unmodified or cRGD NPs (100 nM) or transfected with the survivin-specific siRNA (100 nM) using Lipofectamine RNAiMax. Silencing of survivin was determined by qRT-PCR at 2, 3, 4, and 6 d post-treatment. Survivin mRNA level was normalized to HPRT-1. Results are expressed as a percentage of down-regulation based on survivin basal level obtained with siRNA-Ctl conditions (means±sem). ns, not significant. *P < 0.05, **P < 0.005. B) Representative Western blot analysis comparing survivin expression after siRNA delivery using Lipofectamine RNAiMax (Lipo) or cRGD NPs after 2 or 6 d post-transfection. Blots were reprobed with an anti-GAPDH antibody to ensure equal loading. C) Survivin levels were quantified after normalization to GAPDH by densitometry; results are expressed as a percentage of basal expression. D) HUVECs were either transfected with FAM-siRNA (100 nM) using Lipofectamine RNAiMax or incubated with FAM-siRNA-loaded cRGD NPs (100 nM) for 24 h. Cells were washed, fixed, and observed under confocal fluorescence microscope.
We then tested the capacity of the siRNA NP to knock down target genes. Unmodified siRNA-survivin NPs and cRGD NPs produced 24 and 47% knockdown, respectively, after 2 d. However, in contrast to lipid-based transfection, in which siRNA-mediated knockdown disappears by d 4, we observed a prolonged gene silencing in HUVECs using NPs, with 21 and 36% knockdown at d 4 for unmodified NPs and cRGD NPs, respectively. We established that siRNA-survivin-targeted NPs exhibited 20% less knockdown efficiency compared with the lipid-based transfection during the first days post-transfection but induced a significant and more sustained gene silencing (>6 d for cRGD NPs vs. 3d for Lipofectamine). We confirmed this result by assessing survivin protein level by Western blot analysis. The decrease in protein level induced by targeted NP was less pronounced than with lipid-based transfection (52±1.1% decrease for cRGD NPs vs. 82±5.2% decrease for lipid-based transfection as compared to basal level, P<0.01), but more sustained with significant protein inhibition at least 6 d after treatment in cell culture (41±1.7% decrease for cRGD NPs vs. −10±6.1% decrease for lipid-based transfection as compared to basal level, P<0.01; Fig. 5B, C). Thus, we demonstrated that NPs are effective oligonucleotide delivery vehicles to ECs, with more sustained effects, in vitro. cRGD NPs generated almost 2-fold greater knockdown than unmodified NPs, confirming that cRGD peptide, by targeting αvβ3 integrin, enhances siRNA delivery to cultured HUVECs and further demonstrates the utility of modifying the NP surfaces with functional ligands. Finally, we confirmed that blocking uptake of NPs also reduced the efficacy of the siRNA delivery by using siRNA-survivin NPs. As shown in Fig. 4E, inhibition of the clathrin pathway impaired almost completely the survivin down-regulation mediated by siRNA-survivin unmodified NPs (28±3.1% survivin mRNA decrease for siRNA control (siRNA-Ctl) vs. 3.7±2.9% for si-RNA clathrin 1 and 3±2.1% for siRNA-AP2M1, P<0.01) or siRNA-survivin cRGD NPs (46.6±1.1% survivin mRNA decrease for siRNA-Ctl vs. 9.3±3.5% for siRNA-clathrin1 and −2.3±0.9% for siRNA-AP2M1, P<0.001). As expected, inhibiting the caveolin-mediated endocytosis pathway using siRNA-caveolin 1 did not affect NP-mediated siRNA delivery: the down-regulation of the survivin target in the presence of siRNA-caveolin1 was comparable to control (23.3±1.2% survivin mRNA decrease for unmodified NPs and 41.6±5% for cRGD NPs). Cumulatively, these results show that cRGD NPs are more effective at entering cells compared to naked NPs, but both types of NPs are largely taken up through clathrin-mediated endocytosis and seem to have similar intracellular fates that largely avoid endosomal-lysosomal degradation.
A likely explanation of the sustained effect of NP-delivered siRNA compared to siRNA delivered to the cytoplasm by lipid-based transfection is that NPs serve as depots within an intracellular vesicular compartment that slowly releases siRNA into the cytosol, protecting it from degradation. To confirm that siRNA remains associated with NPs following endocytosis, we used a fluorescently tagged siRNA (FAM-siRNA) and compared the intracellular location of the fluorescence signal in cells transfected with NPs vs. a lipid-based transfection regent. Confocal fluorescence microscopy of HUVECs revealed that different methods of transfection resulted in distinct subcellular localization patterns (Fig. 5D). HUVECs transfected with Lipofectamine display a diffuse cytoplasmic distribution of the FAM-siRNA 24 h after treatment, whereas cRGD NP cells exhibit bright intracellular clusters comparable to those seen using C6-NPs, indicating that as expected, the bulk of the siRNA remains associated with the NP. The cytosol of cells treated with the cRGD NP became faintly fluorescent consistent with the gradual release of FAM-siRNA from the particles present within clathrin-coated endosomes.
miR-132 delivery using cRGD NP allows significant and sustained proliferative and migratory effects in vitro
miR-132 is a critical regulator of the downstream events that control endothelial proliferation, tube formation and angiogenesis in vivo (12). Since cRGD PLGA NPs could safely and effectively silence genes in EC when loaded with siRNA, we hypothesized that targeted NP transfection of miRNA into cultured HUVECs could improve microvessel formation. We first established that cRGD NPs were able to efficiently enter and deliver miR-132. While miR-132 is barely detectable in quiescent ECs in vitro, cRGD-NP elicited an ∼650-fold increase in miR-132 level (Supplemental Fig. S2A). The bioactivity of miR-132 was tested by measuring its ability to significantly decrease one of its known targets, the RasGAP protein. We found that cRGD NPs loaded with miR-132 produced equal RasGAP inhibition compared with miR-132 delivered using Lipofectamine RNAiMAX (Supplemental Fig. S2B). We next examined whether the exogenous expression of miR-132 was sufficient to increase endothelial proliferation and tube formation induced by protein growth factors, such as VEGF-A and basic FGF (bFGF or FGF-2). Cell cycle analysis performed 2 d after transfection showed that both miR-132 delivery systems promoted cell proliferation in response to VEGF-A and bFGF in a dose-dependent manner, with an ∼1.7- and 1.5-fold increase, respectively, compared to cells transfected with a miRNA control (miR-Ctl, Fig. 6A, B). When the same assays were conducted 6 d after transfection, HUVECs transfected using Lipofectamine reverted to a similar proliferation rate as control cells, whereas miR-132 NP-transfected cells still displayed a significantly amplified response to growth factors (1.4-fold increase with NP miR-132 vs. NP miR-Ctl for a 80 ng/ml VEGF-A dose, 1.8-fold increase with NP miR-132 vs. NP miR-Ctl for a 5% bFGF dose) (Fig. 6C, D). In a different assay of angiogenic potential, we assessed EC sprouting from cell spheroids in collagen gels and measured cumulative sprout length (CSL) induced by VEGF-A at various times after miR-132 transfection (3, 4, and 6 d). After 3 d, lipofection and NP delivery systems led to similar levels of sprouting, with a 5-fold increase of CSL induced by miR-132 expression. But the enhancement of CSL following Lipofectamine transfection decreased rapidly over the first few days and reached a basal level after 4 d. In contrast, transfection using NP delivery again showed an extended biological effect up to 6 d in vitro (Fig. 6E). These results confirmed that miR-132 NPs produced significant and sustained release of miRNA, generating a prolonged effect on proliferation and sprout formation. Finally, viability of transfected cells was evaluated after inclusion within the gel at various time points (18, 24 and 36 h) in vitro. We have previously noticed that nontransformed HUVECs died rapidly after inclusion in 3D gel (20) if not implanted in mice. We showed that HUVEC overexpressing miR-132 tolerated cultured 3D in the gel better than control cells, with a marked increase of cell viability after 18 and 24 h of incubation (Supplemental Fig. S3). Moreover, these results demonstrated that these types of NPs are not cytotoxic in these settings.
Figure 6.
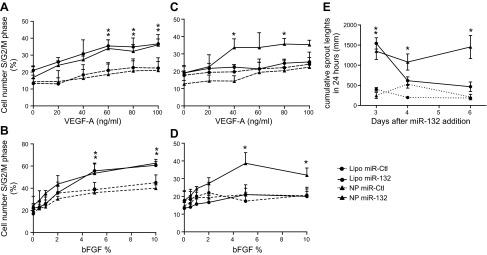
NP-based overexpression of miR-132 promotes proliferation and migration in vitro. HUVECs were either transfected with miR-Ctl (circles and dashed trace) or miR-132 (circles and solid trace) (100 nM) using Lipofectamine RNAiMax or incubated with miR-Ctl-loaded cRGD NPs (triangles and dashed trace) or miR-132-loaded cRGD NPs (100 nM) (triangles and solid trace) A, B) At 2 d after transfection, cells were treated with indicated dose of VEGF-A (A) or bFGF (B) for 18 h, DNA content analyses were performed, and percentage of cells in proliferative state (S/G2/M phase) was quantified (means± sem). **P < 0.01. C, D) Same experiments were repeated at 5 d after transfection/NP incubation (means± sem). *P < 0.05. E) At 3, 4, or 6 d after miR treatments, EC spheroids were generated and embedded in collagen, and cumulative sprout lengths were quantified 24 h later (means±sem). *P < 0.05, **P < 0.01.
Ex vivo delivery of miR-132 promotes vascularization in gels implanted in SCID/Bg mice
To evaluate the effectiveness of our delivery system in enhancing vessel formation and maturation by HUVECs, we analyzed human EC-lined microvessels in gels containing HUVECs transfected with miR-Ctl or miR-132 using either NP or Lipofectamine transfection after implantation into the abdominal wall of SCID/bg mice. Vascular density was assessed by evaluation of H&E-stained sections from gel implants retrieved after 2 wk (Fig. 7A–D). miR-132 therapy yielded a statistically significant increase in vessel density and in total lumen area compared to control EC gels (P<0.001; Fig. 7E, G). Moreover, NP delivery displayed a 2-fold increase in the number of microvessels per square millimeter compared to Lipofectamine (60±9.7 for miR-132 Lipo vs. 116±21 for miR-132 NPs), suggesting that the prolonged release of miR-132 further enhanced the vascularization process. Analysis of vessel morphometry revealed that smaller-diameter vessels were formed in miR-132 grafts than in miR-Ctl gels (Fig. 7F), as measured by a decrease in average vessel diameter between the conditions (P<0.01). We have previously shown that recruitment of pericytes to the vessel wall correlates with reduced diameter of the vessels formed, and many of the human-EC-lined vessels stained positive for mouse-specific smooth muscle α-actin (SMA), indicating that gels were infiltrated by host cells that invested the EC-lined microvessels (21). We assessed the number of vessels invested with SMA-expressing cells (Fig. 8B–E) and found that miR-132 resulted in a significant increase in SMA+ vessels relative to gels containing miR-Ctl cells (Fig. 8A). However, we did not observe a statistically significant difference in the number of vessels that had recruited mural cells between transfections mediated by NPs and Lipofectamine, consistent with the similar reduction in average lumen size observed in these groups.
Figure 7.

Vessel formation within gels is increased by miR-132 delivery. HUVECs were transfected, suspended in a gel, and then implanted into abdominal wall of C.B-17 SCID/beige mice. A–D) H&E-stained sections of implants, retrieved after 2 wk with Lipofectamine miR-Ctl (A), Lipofectamine miR-132 (B), NP miR-Ctl (C), NP miR-132 (D). Scale bar = 50 μm. E) Quantitation of the number of microvessels per square millimeter. F) Measurement of vessel diameter (μm). G) Quantification of the lumen area (percentage of total gel area; means± sem); n = 5. *P < 0.05, **P < 0.01, ***P < 0.0005.
Figure 8.
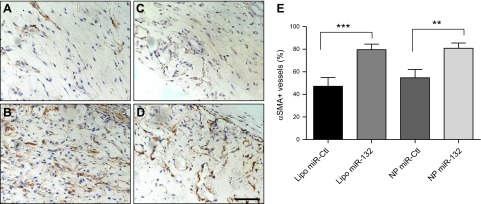
miR-132 delivery results in increased number of SMA-invested vessels. HUVECs were transfected, suspended in gel, and implanted into abdominal wall of C.B-17 SCID/beige mice. Implants were retrieved after 2 wk, and sections were stained with mouse-specific smooth muscle α-actin (SMA) antibody. A–D) Lipofectamine miR-Ctl (A), Lipofectamine miR-132 (B), NP miR-Ctl (C), and NP miR-132 (D). Scale bar = 100 μm. E) Quantitation of positive SMA vessels (percentage of total vessel number; means±sem); n = 5. *P < 0.05, **P < 0.01, ***P < 0.0005.
DISCUSSION
In this study, we delivered miRNA-132 to cultured ECs using cRGD-targeted NP to determine whether this approach could enhance new blood vessel network formation after EC transplantation. The use of collagen-fibronectin-based scaffold to support EC engraftment allows us to control the vascularization site and localizes biological effects to the area of the implant. We showed that PLGA cRGD-NPs densely loaded with miR-132 lead to efficient and sustained gene silencing in ECs of the intended target, RasGAP-1. We showed that cRGD increases the efficiency of NP uptake and that both targeted and nontargeted NPs are endocytosed through a clathrin-dependent mechanism. Interestingly, endocytosed NPs in HUVECs persist in clathrin-coated vesicles, from which they appear to mediate sustained intracellular delivery of functional oligonucleotides. We further demonstrated that a major effect of miR-132 transfection in vitro is to enhance the response of the ECs to protein growth factors and that this effect correlates with enhanced vessel-forming potential of transplanted ECs, judged by the number of human EC-lined vessels that form, when compared to EC transplantation using control miRNA transfection. Transfection with miR-132 also increases vessel maturation, as assessed by a higher number of pericyte-invested vessels and a narrowing of the mean diameter of the vessels, indicative of functional pericyte-EC interactions. Notably, the in vivo effects generated by NP-based delivery are more pronounced than those observed with conventional lipid-based transfection using an optimized lipid reagent. We attribute the advantage of the NP-based approach to the more sustained release of miRNA within the transfected ECs, allowing biological effects, such as growth factor-induced proliferation and migration, to remain augmented for a longer period of time.
An understanding of the dynamics of uptake and intracellular trafficking provide insight into the mechanism of delivery of oligonucleotides and can serve as a baseline for a rational design of new targeted particle systems (22). Using a combination of endocytic inhibitors, we found that NPs entered HUVECs principally via clathrin-mediated endocytosis. To avoid the pitfalls of using pharmacological inhibitors to study the endocytic route (lack of specificity on the part of the inhibitor itself, efficacy of inhibition and viability of cells after exposure to inhibitors highly cell dependent; ref. 23), we complemented the pharmacological approach using an RNAi strategy. Both approaches have limitations such as toxicity of the drugs, incomplete knockdown with siRNA, and off-target effects for both. Although both approaches to limit clathrin-mediated uptake failed to completely block NP endocytosis, all of the agents tested reduced NP uptake comparable to their inhibitory effects on known cargo proteins for these pathways. On the basis of this observation, we hypothesize that residual uptake of NPs in the presence of inhibitory drugs could still involve clathrin-mediated uptake, but we cannot exclude the possibility that other endocytic mechanisms, such as phagocytosis, macropinocytosis, or less-defined clathrin- and caveolae-independent pathways (24), may also be involved in NP internalization. The lack of specific agents for studying those pathways prevented us from further characterizing NP entry (25).
After their internalization, vesicles containing cargoes taken up by clathrin-mediated mechanisms typically uncoat and are then channeled to early endosomes that can undergo acidification and fuse with other vesicular structures, giving rise to late endosome. Late endosomes then fuse with lysosomes, typically leading to the degradation of the internalized structures (26). Therefore, it is surprising that targeted PLGA NPs avoid lysosomal degradation by remaining in clathrin-coated vesicles for several days (<72 h). This may contribute to the prolonged release of siRNA or miRNA intracellularly. Confocal images of FAM-siRNA highlight the different internalization mechanisms involved in lipid transfection and NP delivery. Lipofectamine is a cationic liposome that forms complexes by associating with negatively charged oligonucleotides. The lipid-siRNA complexes fuse with plasma membrane of cells, and oligonucleotides rapidly disassociate from the lipid carrier, allowing rapid cytoplasmic delivery of the oligonucleotides. In contrast, the bulk of transfected siRNA remains associated with PLGA NP, and only a small amount appears in the cytosol, consistent with a model in which siRNA liberation from NPs after internalization into cells is gradual but sustained. This discrepancy between the two approaches of delivering oligonucleotides can explain the different kinetics of silencing. It is unknown at this time the mechanism by which oligonucleotide is transferred from within the clathrin-coated vesicles that contain the NP into the cytosol where siRNA or miRNA act.
We targeted our NPs with cRGD peptide in order to enhance NP uptake by cultured ECs. The cRGD compound used has been shown to be a specific ligand for the αvβ3 integrin (27). Although the αvβ3 integrin is barely detectable on ECs lining quiescent vessels, this receptor is highly expressed in proliferating endothelium in vivo serving as a marker of angiogenesis, tumor development, and metastasis (28–30). It is also highly expressed on HUVECs cultured in the presence of protein growth factors. In this study, we compared uptake and delivery efficiency of naked NPs with cRGD NPs in cultured HUVECs. We showed that cRGD targeting leads to a 3-fold net increase in the amount of NP uptake and generated almost 2-fold greater knockdown when loaded with siRNA. The incubation of an antibody against αvβ3 integrin prior to NP addition decreased cRGD NP uptake by 50% without affecting naked NP endocytosis (Supplemental Fig. S4), demonstrating that cRGD amplifies association of NPs with cells through its binding with αvβ3 integrin. These results demonstrate that in addition to improving selective delivery to tissues or cells, coating NPs with ligands can enhance uptake and therapeutic effectiveness in cultured cells.
Several previous studies have exploited αvβ3 integrin expression to selectively target tumor endothelium (12, 31, 32) or diseased retinal endothelium (due to age-related macular degeneration and diabetic retinopathy; ref. 33) with RGD-grafted NPs loaded with oligonucleotides (16). Promising results for delivery of miRNA were obtained using cRGD-targeted liposomes (12, 16). We also note a recent abstract describing RGD-targeted ultrasmall magnetic NPs (34). The PLGA NPs used in the present study have a property that is found in none of the other formulations: PLGA provides a sustained release of the miRNA (Fig. 1C). While we have shown that NPs similar to the ones we describe in this report are effective for in vivo targeting (35), we have not attempted to optimize transfection of ECs in an intact host here. Instead, we designed our NPs for in vitro treatment of cultured ECs, as these are likely to be the type of cells used in the engineering of tissues. This is the first report that we know of in which a NP system providing sustained miR release was used to treat cells ex vivo, to produce a sustained effect after cell transplantation.
The potential value of miRNAs as therapeutic agents or targets is now widely recognized, and several candidates have already progressed into clinical development (36–40). Individual miRNAs regulate numerous mRNA targets, often encoding multiple proteins of complex intracellular networks. Thus, the ability to therapeutically manipulate miRNA expression or function can have a profound impact on cellular phenotypes. A serious obstacle for the translation of miRNA therapies into the clinic is the lack of effective and safe delivery systems. Although viral vectors can be highly efficient at delivering nucleic acids, the potential for mutagenicity, limited loading capacities, and most importantly, safety risks caused by their inflammatory and immunogenic effects, severely limit the clinical applicability of viruses. These concerns have led to the pursuit of nonviral alternatives. Lipid-mediated delivery is the most widely used. However, a well-known obstacle for clinical applications is its unsatisfactory efficiency for many cell types. Moreover, cationic lipids have been documented to cause dose-dependent toxicity in cultured cells and in mice (41–43). They can also elicit immune activation (44, 45). Biodegradable NPs represent a promising alternative approach. The polymer used in the present study, PLGA, is well known to be safe and is used in many implantable and injectable products that have been approved by the U.S. Food and Drug Administration. As we show here, PLGA NPs have the capacity to encapsulate large amounts of genetic material and can readily be surface-modified. Moreover, we found that PLGA NPs allow a more sustained effect than that obtained with conventional lipid transfection due to prolonged release of oligonucleotides. Because duration of oligonucleotide release can be altered by changing the PLGA formulation process, we believe that it should be possible to design NPs that provide biological effects over time periods that are controllable.
Many studies have suggested that actions on multiple angiogenic pathways may have a better, possibly synergistic, effect on neovascularization compared to actions that target a single growth factor-dependent pathway (46). Emerging evidence suggests that targeting miRNA provides unique advantages by being downstream of multiple growth factor pathways (47). For example, miR-132 acts as an activating switch for quiescent endothelium (12). One of the identified targets of miR-132 is RASA1, which encodes RasGAP protein, a known negative regulator of the Ras/Raf pathway of ERK-1,2 activation. The decreased expression of RasGAP mediated by miR-132 increases Ras activity, thereby enhancing neovascularization induced by ERK-activating protein growth factors (48). This may not be the only effect exerted on ECs by this particular miRNA, but evaluation of decreases in RASA1 expression serves as a convenient assay for quantitating the efficacy of mi-R132 transfection. We report that delivery of synthetic miR-132 mediated by NPs, increases cell proliferation and tube-forming capacity induced by growth factors (12, 48, 49). Furthermore, NP transfection exerts a beneficial effect on vessel formation in vivo following implantation into mouse hosts. Specifically, vessels induced by miR-132 treatment displayed smaller diameter than controls. We attribute this change in vessel morphology to a more rapid maturation process. This supposition is supported by the fact that smaller-diameter vessels correlate with greater recruitment of mural cells. We propose that pericyte coverage of newly formed vessels reduces their diameter. These data agree with previous observations showing that reduced EC-pericyte associations increase EC vessel width in both in vitro (3D collagen matrices) and in vivo (quail embryos) models (50).
The principal factor released by ECs to recruit pericytes to the microvessel wall is PDGF-B. We quantified PDGF-B secretion by HUVECs overexpressing miR-132 but did not observe any rise compared to control cells. The mechanism by which miR-132 affects vessel maturation may be related to its action on some other signal for pericyte recruitment.
In the past years, various strategies have been proposed for improving neovascularization, including growth factor delivery, material-, gene- and cellular-based therapies. Material-based approaches and cell therapy are attractive methods for stimulating vascular network formation, but clinical trials have not resulted in consistent benefit, despite promising results in animal models (51–54). The use of synthetic polymer scaffolds has limitations due to low-EC viability and engraftment causing a rapid thrombus formation in small-diameter vessels and lack of physiological functionality of transplanted cells (55, 56). Clinical trials using transplantation of progenitor cell populations have revealed that the majority of cells reimplanted in patients rapidly died (2). The present study demonstrates the value of combining different approaches for enhancing the vessel-forming potential of transplanted ECs. To address this need, we have developed a safe and effective miRNA delivery system that can enhance the efficacy of cell-based therapeutic vascularization. This strategy of delivery of miRNA-132 to HUVECs before their incorporation into implants may be used for the development of primary treatment strategies for critical tissue ischemia or in the production of bioengineered solid organs, which require rapid, sustained tissue perfusion.
Supplementary Material
Acknowledgments
The authors thank Gwen Arrington and Louise Benson (Yale School of Medicine, Yale University) for provision of HUVECs and Amos Brooks (Research Histology, Department of Pathology, Yale University) for immunohistochemistry.
This work was supported by grants from the U.S. National Institutes of Health (HL108684 and HL085416). W.G.C. was supported by a Clinical and Translational Science Award (KL2-RR024138) from the National Center for Advancing Translational Science, and P.A. was supported by a Paul and Daisy Soros Fellowship for New Americans.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 3D
- 3-dimensional
- bFGF
- basic fibroblast growth factor
- cRAD
- cyclo Arg-Ala-Asp-D-Phe-Lys
- cRGD
- cyclo Arg-Gly-Asp-D-Phe-Lys
- DSPE
- 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethyleneglycol)-2000]
- FAM
- 5′-carboxyfluorescein
- EC
- endothelial cell
- FGF
- fibroblast growth factor
- H&E
- hematoxylin and eosin
- HPRT-1
- hypoxanthine-guanine phosphoribosyl transferase 1
- HUVEC
- human umbilical vein endothelial cell
- miR-Ctl
- microRNA control
- miRNA
- microRNA
- NP
- nanoparticle
- PLGA
- poly(lactic-co-glycolic acid)
- SCID
- severe combined immunodeficiency
- SEM
- scanning electron microscopy
- SMA
- smooth muscle α-actin
- siRNA
- short interfering RNA
- siRNA-Ctl
- short interfering RNA control
- VEGF
- vascular endothelial growth factor
REFERENCES
- 1. Rosenzweig A. (2006) Cardiac cell therapy—mixed results from mixed cells. N. Engl. J. Med. 355, 1274–1277 [DOI] [PubMed] [Google Scholar]
- 2. Gibot L., Galbraith T., Huot J., Auger F. A. (2010) A preexisting microvascular network benefits in vivo revascularization of a microvascularized tissue-engineered skin substitute. Tissue Eng. A 16, 3199–3206 [DOI] [PubMed] [Google Scholar]
- 3. Shepherd B. R., Enis D. R., Wang F., Suarez Y., Pober J. S., Schechner J. S. (2006) Vascularization and engraftment of a human skin substitute using circulating progenitor cell-derived endothelial cells. FASEB J. 20, 1739–1741 [DOI] [PubMed] [Google Scholar]
- 4. Johnson P. C., Mikos A. G., Fisher J. P., Jansen J. A. (2007) Strategic directions in tissue engineering. Tissue Eng. 13, 2827–2837 [DOI] [PubMed] [Google Scholar]
- 5. Jain R. K., Au P., Tam J., Duda D. G., Fukumura D. (2005) Engineering vascularized tissue. Nat. Biotechnol. 23, 821–823 [DOI] [PubMed] [Google Scholar]
- 6. Schechner J. S, Nath A. K., Zheng L., Kluger M. S., Hughes C. C., Sierra-Honigmann M. R., Lorber M. I., Tellides G., Kashgarian M., Bothwell A. L., Pober J. S. (2000) In vivo formation of complex microvessels lined by human endothelial cells in an immunodeficient mouse. Proc. Natl. Acad. Sci. U. S. A. 97, 9191–9196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Enis D. R, Shepherd B. R., Wang Y., Qasim A., Shanahan C. M., Weissberg P. L., Kashgarian M., Pober J. S., Schechner J. S. (2005) Induction, differentiation, and remodeling of blood vessels after transplantation of Bcl-2-transduced endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 102, 425–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jay S. M., Shepherd B. R., Bertram J. P., Pober J. S., Saltzman W. M. (2008) Engineering of multifunctional gels integrating highly efficient growth factor delivery with endothelial cell transplantation. FASEB J. 22, 2949–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jay S. M., Saltzman W. M. (2009) Controlled delivery of VEGF via modulation of alginate microparticle ionic crosslinking. J. Control. Release 134, 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jay S. M, Shepherd B. R., Andrejecsk J. W., Kyriakides T. R., Pober J. S., Saltzman W. M. (2010) Dual delivery of VEGF and MCP-1 to support endothelial cell transplantation for therapeutic vascularization. Biomaterials 31, 3054–3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suárez Y., Sessa W. C. (2009) MicroRNAs as novel regulators of angiogenesis. Circ. Res. 104, 442–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anand S., Majeti B. K., Acevedo L. M., Murphy E. A., Mukthavaram R., Scheppke L., Huang M., Shields D. J., Lindquist J. N., Lapinski P. E., King P. D., Weis S. M., Cheresh D. A. (2010) MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat. Med. 16, 909–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woodrow K. A, Cu Y., Booth C. J., Saucier-Sawyer J. K., Wood M. J., Saltzman W. M. (2009) Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat. Mater. 8, 526–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng C. J., Saltzman W. M. (2011) Enhanced siRNA delivery into cells by exploiting the synergy between targeting ligands and cell-penetrating peptides. Biomaterials 32, 6194–6203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cartiera M. S., Johnson K. M., Rajendran V., Caplan M. J., Saltzman W. M. (2009) The uptake and intracellular fate of PLGA nanoparticles in epithelial cells. Biomaterials 30, 2790–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu X. Q., Song W. J., Sun T. M., Zhang P. Z. and, Wang J. (2011) Targeted delivery of antisense inhibitor of miRNA for antiangiogenesis therapy using cRGD-functionalized nanoparticles. Mol. Pharm. 8, 250–259 [DOI] [PubMed] [Google Scholar]
- 17. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 18. Chang W. G., Andrejecsk J. W., Kluger M. S., Saltzman W. M., Poner J. S. (2013) Pericytes modulate endothelial sprouting. [E-pub ahead of print] Cardiovasc. Res. 10.1093/cvr/cvt215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Babar I. A, Cheng C. J., Booth C. J., Liang X., Weidhaas J. B., Saltzman W. M., Slack F. J. (2012) Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. U. S. A. 109, E1695–E1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ilan N., Mahooti S., Madri J. A. (1998) Distinct signal transduction pathways are utilized during the tube formation and survival phases of in vitro angiogenesis. J. Cell Sci. 111, 3621–3631 [DOI] [PubMed] [Google Scholar]
- 21. Maier C. L., Shepherd B. R., Yi T., Pober J. S. (2010) Explant outgrowth, propagation and characterization of human pericytes. Microcirculation 17, 367–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nimesh S. (2012) Recent patents in siRNA delivery employing nanoparticles as delivery vectors. Recent Patents DNA Gene Seq. 6, 91–97 [DOI] [PubMed] [Google Scholar]
- 23. Vercauteren D., Vandenbroucke R. E., Jones A. T., Rejman J., Demeester J., De Smedt S. C., Sanders N. N., Braeckmans K. (2010) The use of inhibitors to study endocytic pathways of gene carriers: optimization and pitfalls. Mol. Ther. J. Am. Soc. Gene Ther. 18, 561–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El-Sayed A., Harashima H. (2013) Endocytosis of gene delivery vectors: from clathrin-dependent to lipid raft-mediated endocytosis. Mol. Ther. 21, 1118–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ivanov A. I. (2008) Pharmacological inhibition of endocytic pathways: is it specific enough to be useful? Methods Mol. Biol. 440, 15–33 [DOI] [PubMed] [Google Scholar]
- 26. Conner S. D., Schmid S. L. (2003) Regulated portals of entry into the cell. Nature 422, 37–44 [DOI] [PubMed] [Google Scholar]
- 27. Ruoslahti E., Pierschbacher M. D. (1987) New perspectives in cell adhesion: RGD and integrins. Science 238, 491–497 [DOI] [PubMed] [Google Scholar]
- 28. Brooks P. C., Clark R. A., Cheresh D. A. (1994) Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 264, 569–571 [DOI] [PubMed] [Google Scholar]
- 29. Costouros N. G., Diehn F. E., Libutti S. K. (2002) Molecular imaging of tumor angiogenesis. J. Cell. Biochem. Suppl. 39, 72–78 [DOI] [PubMed] [Google Scholar]
- 30. Desgrosellier J. S., Cheresh D. A. (2010) Integrins in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer 10, 9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Danhier F., Pourcelle V., Marchand-Brynaert J., Jérôme C., Feron O., Préat V. (2012) Targeting of tumor endothelium by RGD-grafted PLGA-nanoparticles. Methods Enzymol. 508, 157–175 [DOI] [PubMed] [Google Scholar]
- 32. Murphy E. A, Majeti B. K., Barnes L. A., Makale M., Weis S. M., Lutu-Fuga K., Wrasidlo W., Cheresh D. A. (2008) Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc. Natl. Acad. Sci. U. S. A. 105, 9343–9348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pollinger K., Hennig R., Ohlmann A., Fuchshofer R., Wenzel R., Breunig M., Tessmar J., Tamm E. R., Goepferich A. (2013) Ligand-functionalized nanoparticles target endothelial cells in retinal capillaries after systemic application. Proc. Natl. Acad. Sci. U. S. A. 110, 6115–6120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yigit M., Ghosh S., Zdravka Medarova Z., Moore A. V. (2011) Abstract 2952: Image-guided miRNA therapy for metastatic breast cancer. Cancer Res. 72, 2952–2952 [Google Scholar]
- 35. Zhou J., Patel T., Fu M., Bertram J. B., Saltzman W. M. (2012) (2012) Octa-functional PLGA nanoparticles for targeted and efficient siRNA delivery to tumors. Biomaterials 33, 583–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wiggins J. F., Ruffino L., Kelnar K., Omotola M., Patrawala L., Brown D., Bader A. G. (2010) Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 70, 5923–5930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson S. M., Grosshans H., Shingara J., Byrom M., Jarvis R., Cheng A., Labourier E., Reinert K. L., Brown D., Slack F. J. (2005) RAS is regulated by the let-7 microRNA family. Cell 120, 635–647 [DOI] [PubMed] [Google Scholar]
- 38. Trang P., Medina P. P., Wiggins J. F., Ruffino L., Kelnar K., Omotola M., Homer R., Brown D., Bader A. G., Weidhaas J. B., Slack F. J. (2010) Regression of murine lung tumors by the let-7 microRNA. Oncogene 29, 1580–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lanford R. E., Hildebrandt-Eriksen E. S., Petri A., Persson R., Lindow M., Munk M. E., Kauppinen S., Ørum H. (2010) Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327, 198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jopling C. L., Yi M., Lancaster A. M., Lemon S. M., Sarnow P. (2005) Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309, 1577–1581 [DOI] [PubMed] [Google Scholar]
- 41. Breunig M., Lungwitz U., Liebl R., Goepferich A. (2007) Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. Proc. Natl. Acad. Sci. USA 104, 14454–14459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lv H., Zhang S., Wang B., Cui S., Yan J. (2006) Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control Release 114, 100–109 [DOI] [PubMed] [Google Scholar]
- 43. Khazanov E., Simberg D., Barenholz Y. (2006) Lipoplexes prepared from cationic liposomes and mammalian DNA induce CpG-independent, direct cytotoxic effects in cell cultures and in mice. J. Gene Med. 8, 998–1007 [DOI] [PubMed] [Google Scholar]
- 44. Szebeni J., Muggia F., Gabizon A., Barenholz Y. (2011) Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: prediction and prevention. Adv. Drug Deliv. Rev. 63, 1020–1030 [DOI] [PubMed] [Google Scholar]
- 45. Conwell C. C., Liu F., Huang L. (2007) Several serum proteins significantly decrease inflammatory response to lipid-based non-viral vectors. Mol. Ther. 16, 370–377 [DOI] [PubMed] [Google Scholar]
- 46. Bergers G., Hanahan D. (2008) Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Anand S., Cheresh D. A. (2011) MicroRNA-mediated regulation of the angiogenic switch. Curr. Opin. Hematol. 18, 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Katare R, Riu F., Mitchell K., Gubernator M., Campagnolo P., Cui Y., Fortunato O., Avolio E., Cesselli D., Beltrami A. P., Angelini G., Emanueli C., Madeddu P. (2011) Transplantation of human pericyte progenitor cells improves the repair of infarcted heart through activation of an angiogenic program involving micro-RNA-132. Circ. Res. 109, 894–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gomes R. S. M., das Neves R. P., Cochlin L., Lima A., Carvalho R., Korpisalo P., Dragneva G., Turunen M., Liimatainen T., Clarke K., Ylä-Herttuala S., Carr C., Ferreira L. (2013) Efficient pro-survival/angiogenic miRNA delivery by an MRI-detectable nanomaterial. ACS Nano 7, 3362–3372 [DOI] [PubMed] [Google Scholar]
- 50. Stratman A. N., Schwindt A. E., Malotte K. M., Davis G. E. (2010) Endothelial-derived PDGF-BB and HB-EGF coordinately regulate pericyte recruitment during vasculogenic tube assembly and stabilization. Blood 116, 4720–4730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bramfeldt H., Sabra G., Centis V., Vermette P. (2010) Scaffold vascularization: a challenge for three-dimensional tissue engineering. Curr. Med. Chem. 17, 3944–3967 [DOI] [PubMed] [Google Scholar]
- 52. Assmus B., Honold J., Schächinger V., Britten M. B., Fischer-Rasokat U., Lehmann R., Teupe C., Pistorius K., Martin H., Abolmaali N. D., Tonn T., Dimmeler S., Zeiher A. M. (2006) Transcoronary transplantation of progenitor cells after myocardial infarction. N. Engl. J. Med. 355, 1222–1232 [DOI] [PubMed] [Google Scholar]
- 53. Lunde K., Solheim S., Aakhus S., Arnesen H., Abdelnoor M., Egeland T., Endresen K., Ilebekk A., Mangschau A., Fjeld J. G., Smith H. J., Taraldsrud E., Grøgaard H. K., Bjørnerheim R., Brekke M., Müller C., Hopp E., Ragnarsson A., Brinchmann J. E., Forfang K. (2006) Intracoronary injection of mononuclear bone marrow cells in acute myocardial infarction. N. Engl. J. Med. 355, 1199–1209 [DOI] [PubMed] [Google Scholar]
- 54. Fischer-Rasokat U., Assmus B., Seeger F. H., Honold J., Leistner D., Fichtlscherer S., Schächinger V., Tonn T., Martin H., Dimmeler S., Zeiher AM. (2009) A pilot trial to assess potential effects of selective intracoronary bone marrow-derived progenitor cell infusion in patients with nonischemic dilated cardiomyopathy: final 1-year results of the transplantation of progenitor cells and functional regeneration enhancement pilot trial in patients with nonischemic dilated cardiomyopathy. Circ. Heart Fail. 2, 417–423 [DOI] [PubMed] [Google Scholar]
- 55. Gui L., Muto A., Chan S. A., Breuer C. K., Niklason L. E. (2009) Development of decellularized human umbilical arteries as small-diameter vascular grafts. Tissue Eng. A 15, 2665–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Motlagh D., Allen J., Hoshi R., Yang J., Lui K., Ameer G. (2007) Hemocompatibility evaluation of poly(diol citrate) in vitro for vascular tissue engineering. J. Biomed. Mater. Res. A 82, 907–916 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.