

Abstract
Aquaporin-4 (AQP4), the principal water channel in astrocytes, is involved in brain water movement, inflammation, and neuroexcitation. In this study, there was strong neuroprotection in mice lacking AQP4 in a model of global cerebral ischemia produced by transient, bilateral carotid artery occlusion (BCAO). Survival and neurological outcome were greatly improved in the AQP4−/− vs. AQP4+/+ mice after occlusion, with large and robust differences in both outbred (CD1) and inbred (C57bl/6) mouse strains without or with mechanical ventilation. Improved survival was also seen in mice lacking the scaffold protein α-syntrophin, which manifest reduced astrocyte water permeability secondary to defective AQP4 plasma membrane targeting. Intracranial pressure elevation and brain water accumulation were much reduced in the AQP4−/− vs. AQP4+/+ mice after carotid artery occlusion, as were blood–brain barrier (BBB) disruption and neuronal loss. Brain slices from AQP4−/− mice showed significantly reduced cell swelling and cytotoxicity in response to oxygen–glucose deprivation, compared with slices from AQP4+/+ mice. Our findings suggest that the neuroprotective effect of AQP4 deletion in global cerebral ischemia involves reduced astrocyte swelling and brain water accumulation, resulting in reduced BBB disruption, inflammation, and neuron death. AQP4 water transport inhibition may improve survival and neurological outcome after cardiac arrest and in other conditions associated with global cerebral ischemia.—Katada, R., Akdemir, G., Asavapanumas, N., Ratelade, J., Zhang, H., Verkman, A. S. Greatly improved survival and neuroprotection in aquaporin-4 knockout mice following global cerebral ischemia.
Keywords: water channel, brain swelling, astrocyte, neuroscience
The brain is very sensitive to ischemic injury, which limits the effectiveness of resuscitation after cardiac arrest. In adult humans, successful resuscitation is uncommon after 5 min of cardiac arrest (1). The sensitivity of brain tissue to ischemic injury is a major concern in neurological and cardiac surgical procedures that can be associated with transient ischemia, such as brain aneurysm repair, carotid endarterectomy, and cardiac surgery without bypass. Brain ischemia is thought to trigger a series of events leading to neuron death, including disruption of transmembrane ion gradients, cell swelling, excitotoxicity, inflammation, and apoptosis (2, 3).
Motivated by the idea that cell swelling and brain water accumulation are central initiating events in ischemic injury, we hypothesized that deletion of water channel aquaporin-4 (AQP4) is neuroprotective in global cerebral ischemia. AQP4 is a water-selective transporting protein expressed in astrocytes throughout the central nervous system, with concentration in foot processes at the blood–brain barrier (BBB; refs. 4, 5). We previously generated mice lacking AQP4 (AQP4−/− mice; ref. 6) and found improved neurological outcome in models of cytotoxic brain edema, including acute water intoxication and focal ischemia produced by permanent middle cerebral artery occlusion (7). Cytotoxic brain edema results from water movement into brain tissue across an initially intact BBB, which can follow from osmotic gradients (in water intoxication), cellular Na+/K+ pump dysfunction (in ischemia), or both. The improved outcomes in AQP4−/− mice were ascribed to reduced water permeability of astrocytes and of the BBB (7–9), resulting in reduced astrocyte swelling and brain water accumulation, respectively. We have also reported reduced brain inflammation in AQP4−/− mice in experimental autoimmune encephalomyelitis and after intracerebral lipopolysaccharide (LPS) administration (10), which may further confer neuroprotection in ischemic brain injury. AQP4−/− mice also manifest altered neuroexcitation during seizure activity (11), cortical spreading depression (12), and neurosensory signaling (13, 14) that may be related to altered extracellular space (ECS) K+ dynamics (15).
In this study, we investigated the potential neuroprotective effect of AQP4 gene deletion in a mouse model of global cerebral ischemia produced by transient, bilateral carotid artery occlusion (BCAO). This occlusion model has been used in mice to study mechanisms of neuronal damage and cognitive impairment after ischemia (16, 17). We found markedly improved survival and neurological outcome after BCAO in AQP4−/− mice. Mechanistic studies implicated a central role of astrocyte water permeability in the neuroprotective effect of AQP4 deficiency, which was responsible for reduced astrocyte swelling, brain edema, BBB disruption, and inflammation. The significant neuroprotection in global cerebral ischemia conferred by reduced astrocyte water permeability provides proof of concept for the development of a new class of neuroprotective therapeutics targeting AQP4 water permeability.
MATERIALS AND METHODS
Mice
Experiments were performed in male AQP4+/+ and AQP4−/− mice (age 8–12 wk; weight 25–30 g) on CD1 and C57bl/6 genetic backgrounds (6). Some experiments were performed in α-syntrophin-knockout mice on a C57bl/6 background (Jackson Laboratory, Bar Harbor, ME, USA). Animal studies were approved by the University of California–San Francisco Committee on Animal Research. Neurological scoring was assessed with a standard scoring system (18), with a maximum score of 12 (control mice) and minimum score of 0 for live but comatose mice. Scoring included 6 factors: level of consciousness, corneal reflex, respirations, righting reflex, coordination, and movement and activity; each scored 0, 1, or 2.
Transient BCAO
The mice were anesthetized with Avertin (2,2,2-tribromoethanol; 125 mg/kg, i.p.). After deep anesthesia was confirmed, a horizontal skin incision was made in the anterior neck, and the thymus and sternocleidomastoid muscles were retracted to expose the common carotid arteries. Both carotid arteries were clipped at nearly the same time with a microclip applicator with a lock (Fine Science Tools, Foster City, CA, USA) with a microserrefine clamp-curved/17 mm (jaw pressure, 100 g), at the level of the middle sternocleidomastoid muscle. After the specified occlusion time, both clips were removed at nearly the same time with clip forceps, and the neck skin was sutured. In some experiments, the mice were mechanically ventilated via a tracheostomy with a 20-gauge catheter (respiratory rate, 120/min; tidal volume, 0.6 ml, room air). Core body temperature was maintained at 36.5–37.5°C with a heated pad and overhead lamp. Cerebral blood flow (CBF) was measured in both hemispheres of the AQP4+/+ and AQP4−/− mice before and 1 min after occlusion and 5 min after reperfusion, with a noninvasive laser Doppler probe (Vasamedics, Eden Prairie, MN, USA).
Intracranial pressure (ICP) measurement
ICP was measured as described previously (19). Briefly, after deep anesthesia, the mouse was immobilized in a stereotactic frame. After a midline skull skin incision and removal of the periosteum, a craniectomy was made with a Foredom high-speed drill (45,000 rpm; 1 mm stainless-steel burr; Blackstone Industries, Bethel, CT, USA) to visualize the intact dura. Drilling was performed 0.5 mm caudal to the bregma and 1 mm right lateral to the midline. A pressure transducer (1 mm diameter, TSD104A; Biopac Systems, Santa Barbara, CA, USA) was placed into the temporal brain tissue and interfaced to a recording system (MP100A-CE; Biopac Systems). After sensor placement was confirmed, the mouse was removed from the stereotactic frame and carotid artery occlusion was performed.
Brain water content measurement
After anesthesia, the mice were killed by cervical dislocation, and the brain was removed, weighed immediately (wet weight), and dried in a vacuum oven (Model 5851; Napco, Chicago, IL, USA) at 121°C for 24 h. The dried brains were reweighed (dry weight). Brain water content was calculated as (wet weight − dry weight)/wet weight × 100.
Immunohistochemistry
Brains were removed at 24 h after 5 min of BCAO. The mice were perfused with 20 ml of PBS and then 10 ml of 4% paraformaldehyde (PFA) via the left cardiac ventricle. Paraffin-embedded sections (5 μm thick) were dewaxed twice in xylene and rehydrated in graded ethanols. After immersion in blocking buffer (1% BSA), the sections were immunostained at room temperature for 1 h with rabbit anti-AQP4 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-glial fibrillary acidic protein (GFAP; 1:200; Millipore, Temecula, CA, USA), goat anti-myelin basic protein (MBP; 1:200; Santa Cruz Biotechnology), rabbit anti-ionized calcium binding adaptor molecule 1 (Iba1; 1:200, Wako, Richmond, VA), rat anti-CD45 (1:25; Pharmingen-BD Biosciences, Oxford, UK), rabbit anti-cleaved caspase-3 (Asp175; 1:200; Cell Signaling Technology, Danvers, MA, USA), and mouse anti-NeuN (1:200; Millipore), followed by the appropriate fluorescent secondary antibody (1:200; Invitrogen-Life Technologies, Carlsbad, CA, USA) or biotinylated secondary antibody (1:500; Vector Laboratories, Burlingame, CA, USA). Sections were also stained with hematoxylin and eosin (H&E). Tissue sections were examined with a Leica DM 4000 B microscope (Leica Microsystems, Wetzlar, Germany).
Evans blue dye extravasation
Evans blue extravasation was measured as described previously (20). Briefly, 1 h before brain removal, 4% Evans blue dye in PBS (160 mg/kg; Sigma-Aldrich, St. Louis, MO, USA) was injected intravenously. At 10 min after 4.5 min of BCAO, the mouse was killed, and the left ventricle was perfused with 20 ml PBS. The brain was removed, weighed, and immersed overnight in 2 ml formamide (Sigma-Aldrich) at 55°C, to extract the Evans blue dye, which was qualified by optical absorbance at 610 nm against Evans blue/formamide standards. Data are reported as Evans blue (mg)/brain weight (g).
Cytokine assays
Brains were removed at 6 h after 5 min of BCAO, weighed, and homogenized with a Polytron benchtop homogenizer (Kinematica, Luzern, Switzerland) in 5 volumes of extraction buffer (25 mM HEPES, 0.15 M NaCl, 2 mM EDTA, 1 mM EGTA, and protease inhibitor cocktail; Sigma-Aldrich). The homogenate was centrifuged at 1000 g for 10 min at 4°C and the supernatant at 20,000 g for 40 min at 4°C. The protein concentration was assayed (BCA kit). Cytokine measurements were made with a 20-plex mouse cytokine assay (for FGF, GM-CSF, IFN-γ, IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12p40/70, IL-13, IL-17, IP-10, KC, MCP-1, MIG, MIP-1α, TNF-α, and VEGF) according to the manufacturer's instructions (Luminex kit, Biosource-Life Technologies, Camarillo, CA, USA).
Hippocampal slice measurements
Acutely prepared hippocampal slices were used for measurements of ECS volume (21). Adult mice (CD1 background, age 8–10 wk) were anesthetized and decapitated. Hippocampal slices of 300 μm thickness were cut on a vibratome (VT-1000S; Leica) in ice-cold sucrose-cerebrospinal fluid containing (in mM) 206 sucrose, 2.8 KCl, 1 CaCl2, 1 MgCl2, 2 MgSO4, 1.25 NaH2PO4, 10 d-glucose, 10 Na pyruvate, and 26 NaHCO3 (320 mosmol, pH 7.4, saturated with 5% CO2/95% O2). The slices were then incubated in artificial cerebrospinal fluid (aCSF) consisting of (in mM) 124 NaCl, 2 CaCl2, 5 KCl, 1 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose, in 5% CO2/95% O2, at 35°C for ≥1 h before measurements. To measure the ECS volume, we incubated the slices for 30 min in aCSF containing 100 μM calcein and then immobilized and perfused them with the same solution in an open bath chamber (RC-26; Warner Instruments, Hamden, CT, USA) maintained at 35°C. An optical microfiber with a micrometer-sized etched tip was placed in the CA1 area of the hippocampus, 50–100 μm beneath the surface. Oxygen-glucose deprivation (OGD) was induced by switching the perfusion to a d-glucose-deficient buffer (in mM: 124 NaCl, 2 CaCl2, 5 KCl, 1 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, 10 sucrose, and 100 μM calcein) equilibrated with 95% N2/5% CO2 for 10 min at 35°C.
To measure cytotoxicity after OGD, we generated organotypic hippocampal slice cultures from 7-d-old mice and maintained them in an interface culture in which 8 slices were cultured on each 30-mm Millicell-CM 0.4-μm-pore membrane culture insert (Millipore; ref. 22). Before OGD, the inserts were equilibrated in 1 ml aCSF saturated with 5% CO2/95% O2 for 2 h in 6-well plates. To induce OGD, we transferred the inserts to a new 6-well plate containing 1 ml of d-glucose-deficient buffer and sealed in a modular incubator hypoxia chamber (MIC-101; Billups-Rothenberg, Inc., San Diego, CA, USA) with 95% N2/5% CO2 at 37°C for 30 min. The medium was then collected for an LDH assay (Roche Diagnostics, Indianapolis, IN, USA). To test the effects of oxygen and glucose reperfusion, we returned the OGD-treated inserts to aCSF saturated with 5% CO2/95% O2 for 6 h, and the medium was assayed for LDH.
Statistical analysis
Significance in the cumulative survival studies was determined with the log-rank test. Other data are expressed as means ± se (generally, 4–6 mice/group). A significant difference was defined as P < 0.05. Group comparisons were made by 1-way ANOVA with Scheffé's post hoc test or Student's t test.
RESULTS
Improved survival and neurological outcome in AQP4−/− mice after BCAO
An initial set of BCAO studies was performed without mechanical ventilation, to compare adult AQP4+/+ and AQP4−/− mice on an outbred (CD1) genetic background. Figure 1A (left panel) summarizes percentage survival at 6 h after BCAO for different times, showing much-improved survival in the AQP4−/− mice, with 50% survival at <5 min occlusion time for the AQP4+/+ mice and at >6.5 min for the AQP4−/− mice. Neurological outcome, as assessed by a multifactorial neurological score, was much improved in the surviving AQP4−/− mice (Fig. 1A, middle panel). Figure 1A (right panel) summarizes cumulative survival as a function of time after occlusion for 4 occlusion times, showing that many of the mice that died did so soon after occlusion. CBF was measured in the AQP4+/+ and AQP4−/− CD1 mice before, during, and after occlusion. Figure 1B shows an ∼85% reduction in CBF in both the left and right brain cortex during occlusion and an increase to original levels after reperfusion. No differences in CBF were found between the AQP4+/+ and the AQP4−/− mice. We also confirmed similar vascular gross anatomy, as reported previously (7) in the AQP4+/+ and AQP4−/− mice, as visualized after intravascular ink injection (data not shown).
Figure 1.
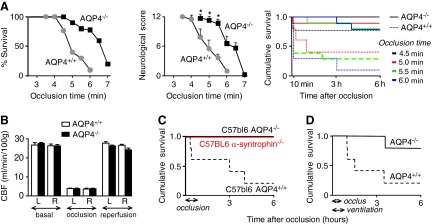
Improved survival and neurological outcome in AQP4−/− mice after BCAO. A) AQP4+/+ and AQP4−/− mice on a CD1 genetic background were subjected to BCAO (without mechanical ventilation) for between 3.5 and 7 min. Left panel: percentage survival at 6 h after occlusion: 4.5 min, P = 0.07; 5.0 min, P = 0.011; 5.5 min, P = 0.5; 6.0 min, P = 0.001. A total of 53 AQP4+/+ and 57 AQP4−/− mice were studied. Middle panel: neurological score at 6 h in surviving mice. *P < 0.05. Right panel: cumulative survival as a function of time after occlusion of 4.5, 5.0, 5.5, and 6.0 min. B) CBF in AQP4+/+ and AQP4−/− CD1 mice before and during occlusion and after reperfusion. CBF was measured in the cortex of both the left (L) and right (R) hemispheres (means±se, n=4/group). C) Cumulative survival for AQP4+/+, AQP4−/−, and α-syntrophin−/− mice on a C57bl/6 genetic background after 45 min of BCAO (n=10/group). P = 0.02. D) Survival studies in CD1 mice, as in A, for 30 min of BCAO with mechanical ventilation, during and 15 min after occlusion (n=5/group). P = 0.02.
The poor survival of AQP4+/+ mice in Fig. 1A was unexpected because reported BCAO studies in CD1 mice (23) and in inbred C57bl/6 mice (17) have shown survival after tens of minutes of occlusion. After performing preliminary survival studies in C57bl/6 mice that were in agreement with the published data, we compared survival in the AQP4+/+ and AQP4−/− mice after 45 min of BCAO. Whereas most AQP4+/+ mice died at 6 h, there was no mortality in the AQP4−/− mice (Fig. 1C).
We postulated that the poor survival of the AQP4+/+ mice in the BCAO model in Fig. 1A was related to high sensitivity of the brainstem respiratory center in CD1 mice to ischemia, which was supported by observations of respiratory distress during occlusion. After conducting preliminary survival studies in mechanically ventilated CD1 mice, we compared survival after 30 min of BCAO, in which the mice were mechanically ventilated during the occlusion, and for 15 min thereafter. Figure 1D shows reduced mortality in the AQP4−/− CD1 mice, supporting the involvement of respiratory depression in mouse mortality in Fig. 1A. (A prior BCAO occlusion study was performed in mechanically ventilated CD1 mice; ref. 23.) We conclude that, regardless of the details of the model or the mouse strain, AQP4 deletion confers strong neuroprotection after transient global ischemia, with improved mouse survival.
BCAO studies were also performed in α-syntrophin−/− mice, which were available on a C57bl/6 genetic background. These knockout mice manifest a secondary AQP4 cellular mislocalization that may be related to loss of a macromolecular complex containing AQP4, α-syntrophin, and other proteins (24) and hence represent an independent model of reduced astrocyte water permeability. Figure 1C shows greatly improved survival of the α-syntrophin−/− mice in the 45 min BCAO model, as found in the AQP4−/− mice.
Reduced brain swelling in the AQP4−/− mice after BCAO
We postulated that there would be reduced brain swelling in the AQP4−/− mice after BCAO, as prior data indicate that AQP4 is the major water transport pathway in the astrocyte plasma membrane and at the BBB (4). A group of 5 AQP4+/+ and AQP4−/− mice on a CD1 genetic background were subjected to BCAO for 4.5 min without mechanical ventilation, as in Fig. 1A, and brain water was measured 10 min later by determination of the wet-to-dry weight ratio. Figure 2A (top panel) shows similar brain water content in the control (untreated), AQP4+/+, and AQP4−/− mice, but greatly increased brain water content in the AQP4+/+ mice after BCAO. Figure 2A (bottom panel) summarizes the difference in brain water content in the BCAO treatment vs. control groups. Remarkably, brain water did not increase significantly in the AQP4−/− mice. Brain water measurements were also made in CD1 mice with the mechanical ventilation protocol (as in Fig. 1D), in which brain water was measured at 15 min after 30 min of occlusion. As found in the nonventilated mice, BCAO produced significantly greater brain water accumulation in the ventilated AQP4+/+ vs. the AQP4−/− mice.
Figure 2.
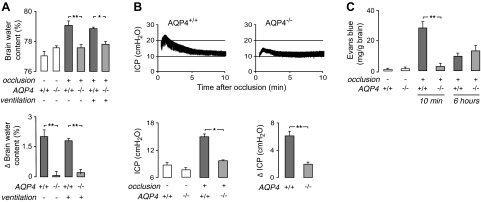
Reduced brain swelling, ICP elevation, and BBB disruption in AQP4−/− mice after BCAO. A) Brain water content was measured in nonventilated (as in Fig. 1A) and ventilated (as in Fig. 1D) CD1 mice at 10 and 15 min after 4.5 and 30 min of BCAO, respectively. Top panel: summary of brain water content for 5 mice/group. Bottom panel: difference in brain water content measured in BCAO vs. control mice. B) ICP was recorded between 0.5 and 10 min after 4.5 min of BCAO in nonventilated CD1 mice. Top panels: representative ICP recordings. Bottom left panel: mean ICP (average over 1–10 min after BCAO; n=4–5/group). Bottom right panel: difference in mean ICP (ΔICP) in BCAO vs. control mice. C) Evans blue extravasation measured in nonventilated CD1 mice at 10 min and 6 h after 4.5 min of occlusion (n=5/group). *P < 0.05, **P < 0.01.
A major consequence of excess brain water accumulation is increased ICP. ICP was measured by a pressure microtransducer inserted into the brain parenchyma. Figure 2B (top panels) shows representative ICP recordings after 4.5 min of BCAO in nonventilated AQP4+/+ and AQP4−/− mice on a CD1 genetic background. The transient elevation in ICP was significantly greater in the AQP4+/+ mice, as summarized for a series of mice in Fig. 2B (bottom panels).
Reduced BBB disruption in AQP4−/− mice after BCAO
Disruption of the BBB is another significant consequence of cerebral ischemia. BBB integrity was determined by Evans blue dye extravasation in the AQP4+/+ and AQP4−/− mice on a CD1 genetic background subjected to 4.5 min of BCAO without mechanical ventilation (as in Fig. 1A). Dye extravasation was measured at 10 min and 6 h after BCAO, in which Evans blue was injected intravenously 1 h before brain removal. Figure 2C shows minimal Evans blue extravasation in untreated mice, with greatly increased extravasation in the AQP4+/+ mice at 10 min after BCAO. There was little difference in dye extravasation in the AQP4+/+ and AQP4−/− mice at 6 h.
Reduced cell death and mildly reduced inflammation in AQP4−/− mouse brain after BCAO
An immunofluorescence assay was performed in brains from the AQP4+/+ and AQP4−/− mice on a C57bl/6 background at 24 h after 30 min of BCAO, to examine astrocyte markers (AQP4 and GFAP) and myelin (MBP). Figure 3A shows similar AQP4, GFAP, and MBP immunofluorescence in control vs. AQP4+/+ mice after BCAO, with similar GFAP and MBP immunofluorescence in the AQP4−/− mice after BCAO. Therefore, as expected, astrocyte damage and demyelination were not prominent features of the BCAO model used in this study.
Figure 3.
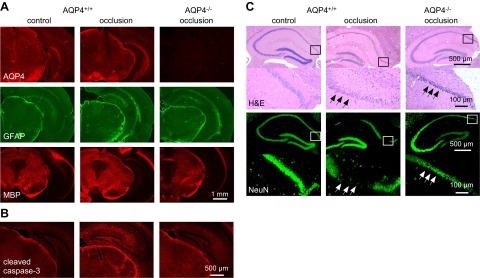
Reduced cellular damage in AQP4−/− mice after BCAO. A) AQP4, GFAP, and MBP immunofluorescence in brain sections of nonventilated C57bl/6 mice at 24 h after 30 min of BCAO. Representative of 4 mice/group. B) Immunofluorescence of cleaved caspase-3 (apoptosis marker) in control and in AQP4+/+ and AQP4−/− CD1 mice at 24 h after 5 min of BCAO (representative of 3 mice/group). C) H&E-stained hippocampal sections (top panels) in mice treated as in A, shown together with NeuN immunofluorescence (bottom panels) (representative of 3 mice/group). Higher-magnification images of boxed areas are shown under each image. Arrows indicate loss of NeuN immunofluorescence.
Apoptosis is often seen in models of cerebral ischemia in mice (25), and we therefore stained brain sections with an antibody (Asp175) against cleaved caspase-3. Figure 3B shows greater antibody staining in the brains of the AQP4+/+ vs. the AQP4−/− mice after BCAO, particularly in the hippocampus, where ischemic changes are generally seen. H&E-stained hippocampal sections showed greater loss of pyramidal neurons in the CA1 region in the AQP4+/+ than in the AQP4−/− mice after BCAO (Fig. 3C, top panels), which, as shown by the NeuN staining, represents neuronal loss (Fig. 3C, bottom panels).
The potential involvement of brain inflammation in the neuroprotective effect of AQP4 deletion was studied by inflammatory marker immunofluorescence and brain cytokine concentration. Activation of microglia was seen in the nonventilated AQP4+/+ CD1 mice at 24 h after 5 min of BCAO, as assessed by Iba1 immunofluorescence (Fig. 4A, top panels). Iba1 immunofluorescence was largely absent in the control mice and was minimal in the AQP4−/− mice after BCAO. Little or no accumulation of leukocytes was seen in the brains of the AQP4+/+ or AQP4−/− mice at 24 h after 5 min of BCAO, as assessed by CD45 immunocytochemistry (Fig. 4A, bottom panels). As a control, marked microglial activation and accumulation of CD45-positive leukocytes were seen after a brain injury produced by needle insertion (Fig. 4A, right panels).
Figure 4.

Brain inflammatory response after BCAO. A) Iba1 (marker of activated microglia) and CD45 (leukocyte marker) immunostaining of brain sections of nonventilated CD1 mice at 24 h after 5 min of BCAO. Positive controls were brain sections 3 d after intracerebral needle insertion. B) Cytokine concentrations in brain homogenates prepared 6 h after 5 min of BCAO in AQP4+/+ and AQP4−/− CD1 mice (n=5/group). *P < 0.05 vs. AQP4+/+ and AQP4−/− controls.
Cytokine concentrations were measured in brain homogenates of the AQP4+/+ and AQP4−/− mice at 6 h after 5 min of BCAO. Forty percent of mice in the AQP4+/+ group died by 6 h (no AQP4−/− mice died), and the data are therefore from the live mice. Cytokine assays showed small but significant elevations in IL-6 and IL-17 in the AQP4+/+ mice after BCAO, with no significant cytokine elevations in the AQP4−/− mice after BCAO. As a positive control, intraperitoneal injection of 10 μg LPS increased IL-6 ∼10-fold in brain homogenates (data not shown), in agreement with published data (26).
Reduced cell swelling and cytotoxicity in brain slices from AQP4−/− mice after anoxia
To further investigate AQP4-dependent mechanisms of neuroprotection in global ischemia, we measured cell swelling in acutely prepared brain slices from adult AQP4+/+ and AQP4−/− mice on a CD1 genetic background. After a stabilization period, brain slices were subjected to OGD for 10 min by exposure to a d-glucose-deficient buffer equilibrated with a 95% N2/5% CO2 (27). Cell swelling was determined by the ECS volume fraction, which was measured by a microfiberoptic method developed by our laboratory in which calcein dye fluorescence is measured with an optical microfiber with a micrometer-sized tip that is inserted into the tissue slice through the overlying solution (21). The ECS volume fraction (α) is equal to the ratio of dye molecules per unit volume in the slice to that in the external solution. Cell swelling causes ECS shrinkage and hence reduced αOGD. Measurements were performed in the CA1 area of the hippocampus, which is reported to be the most sensitive to global ischemia (28–30). Figure 5A (left panel) shows a continuous decrease in αOGD in both the AQP4+/+ and the AQP4−/− hippocampal slices during the 10 min of OGD. Little change was seen in control (not OGD-exposed) brain slices. Baseline α in the hippocampus was slightly greater in the AQP4−/− vs. AQP4+/+ slices, as reported before (21, 31). Fig. 5A (right panel) shows significantly greater relative ECS shrinkage, indicating more brain cell swelling in OGD-exposed AQP4+/+ vs. AQP4−/− brain slices.
Figure 5.
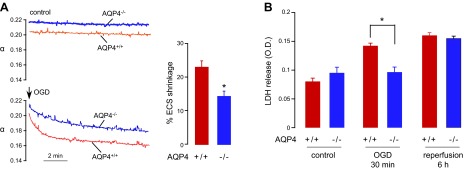
Reduced brain cell swelling and cell death in hippocampal slices from AQP4−/− mice after OGD. A) ECS volume was measured continuously in brain slices by a microfiberoptic fluorescence method. Left panel: representative data for hippocampal slices from AQP4+/+ and AQP4−/− mice without and during OGD. Right panel: percentage of ECS shrinkage, as calculated by relative ECS volume change [1 − (αOGD/αbasal)] (5 slices from 5 mice/group). *P < 0.05. B) LDH release from organotypic hippocampal slice cultures of AQP4+/+ and AQP4−/− mice after 30 min of OGD and at 6 h after return of slices to normal oxygen–glucose levels (reperfusion; 8 slices/plate insert, 4 inserts/group). *P < 0.05.
Cytotoxicity in response to OGD was measured by LDH release from organotypic hippocampal slice cultures, an approach used widely to study OGD-induced death of pyramidal neurons in the CA1 region (32, 33). After 7 d of culture, the hippocampal slices showed preserved CA1 structure and AQP4 expression (22). The slices were subjected to 30 min OGD followed by a 6 h return to normal oxygen–glucose (reperfusion). Figure 5B shows significantly greater LDH release from AQP4+/+ slices than from AQP4−/− slices after 30 min of OGD, although there was no significant difference after 6 h.
DISCUSSION
We found strong neuroprotection conferred by AQP4 gene deletion in a mouse model of global cerebral ischemia produced by transient BCAO. The differences were large and robust, as the neuroprotective effect was mouse strain independent and was seen in both mechanically ventilated and nonventilated mouse models. The neuroprotection in a mouse model of secondary AQP4 insufficiency produced by α-syntrophin knockout suggests that it is loss of AQP4-facilitated water transport that confers neuroprotection in global cerebral ischemia. As diagrammed in Fig. 6, the AQP4 water transport function is involved in water accumulation in astrocytes and whole brain during ischemia, resulting in astrocyte and brain swelling. Reduced astrocyte water permeability in AQP4 deficiency attenuates these responses. Our prior data indicate that AQP4 is the principal water transporter in the astrocyte plasma membrane (8) and at the BBB (7, 9). Secondary consequences of reduced astrocyte swelling and brain water accumulation, as found in our study, included reduced ICP elevation, BBB disruption, cytotoxicity, and inflammation, each of which can contribute to the neuroprotection conferred by AQP4 deficiency.
Figure 6.
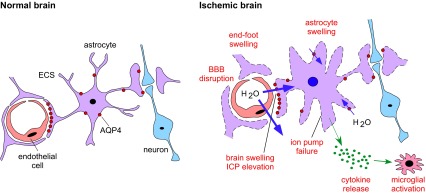
AQP4-dependent mechanisms of neuroprotection in global cerebral ischemia. In the normal brain, astrocytes occupy a strategic position between capillaries and neurons. Perivascular astrocytic end-feet at the BBB maintain brain ion and water homeostasis. In the ischemic brain, AQP4 facilitates astrocyte swelling and water movement across the BBB, producing multiple deleterious responses (red labels).
The experiments in brain slices, in which there is no BBB, support the involvement of astrocyte swelling in anoxic brain injury. Early in anoxia, cellular energy depletion leads to the failure of ATP-dependent ion transport mechanisms, resulting in neuronal excitotoxicity, with accumulation of glutamate and potassium in the ECS (2, 3). Net ion influx into astrocytes during anoxia drives water influx through AQP4, which accelerates astrocyte swelling (27, 34). Astrocyte swelling is reported to be one of the earliest morphologic changes in the ischemic brain (35). We found substantially greater reduction in ECS volume in hippocampal slices of the AQP4+/+ than from those of the AQP4−/− mice during anoxia, largely due to greater astrocyte swelling, which resulted in greater LDH release as a consequence of cell damage.
We previously reported reduced brain inflammation in AQP4−/− in models of experimental autoimmune encephalomyelitis and intracerebral LPS administration (10). Prior studies of global ischemia in rodents suggest the involvement of inflammation in the development of ischemic lesions (2, 36), with microglial activation and elevation of proinflammatory cytokines such as TNF-α (37, 38). We found in the current study modest microglial activation in mice after BCAO, which was reduced in AQP4 deficiency, although there was minimal leukocyte infiltration. Modest elevations in the brain cytokines IL-6 and IL-17 were also found in the AQP4+/+ mice, but not in the AQP4−/− mice after BCAO. Although these studies support the involvement of AQP4-dependent inflammation in the model that we used, inflammation probably plays a relatively minor role compared with AQP4-dependent astrocyte swelling and brain water accumulation.
The BCAO model has been widely used to study the mechanisms of neuron death and cognitive impairment after ischemia. Severe global ischemia is produced by 5 min of BCAO in gerbils, where the posterior communicating artery is absent or poorly developed, leading to neuronal death primarily in the hippocampus, astrocyte activation, and later, myelin loss (39). In rats, global ischemia has been induced by occlusion of both common carotid arteries and both vertebral arteries (usually after 10–20 min of occlusion), producing neuronal damage and oligodendrocyte apoptosis (40). In mice, a transient BCAO model was used in the C57bl/6 strain, which is reported to be more susceptible to ischemia than are other strains (41), because of poor anastomosis between the carotid and vertebrobasilar circulation (42). Similar to our current data, 20–30 min of BCAO in C57bl/6 mice produced extensive hippocampal cell death (41). Oligodendrocyte injury and myelin loss were also seen, but only several days after BCAO (43). We found in the current study that CD1 mice were also quite sensitive to BCAO, in agreement with a prior report of hippocampal injury in ventilated CD1 mice after 10 min of BCAO (44).
Our results suggest the utility of AQP4 water transport inhibition or down-regulation of AQP4 protein expression at the astrocyte cell surface for neuroprotection in cerebral ischemia. Although a variety of compounds have been reported to inhibit AQP4 water permeability, including carbonic anhydrase inhibitors, antiepileptics, salts such as tetraethylammonium, and even anti-AQP4 autoantibodies in neuromyelitis optica (45–47), subsequent studies did not confirm bona fide inhibition by these compounds or antibodies (48, 49). It was speculated that the apparent inhibition was an artifact of the oocyte swelling assay. Most toxic insults to the brain, such as ischemia, trauma, and infection, cause up-regulation of AQP4 expression (4). Inhibitors of AQP4 water permeability or of AQP4 expression in astrocytes may provide neuroprotection in cerebral ischemia.
In summary, we found marked neuroprotection in mice, conferred by AQP4 deficiency after transient global cerebral ischemia. The low water permeability of the astrocyte cell plasma membrane and the BBB in AQP4 deficiency resulted in reduced astrocyte and brain swelling, which attenuated pathogenic secondary responses, such as BBB disruption and inflammation, leading to neuronal dysfunction and death. Our results suggest the possibility of AQP4-targeted therapeutics to extend the duration of cerebral ischemia associated with favorable neurological outcomes.
Acknowledgments
This work was supported by grants DK35124, EY13574, EB00415, and DK72517 from the U.S. National Institutes of Health (Bethesda, MD, USA) and grants from the Guthy-Jackson Charitable Foundation.
Footnotes
- aCSF
- artificial cerebrospinal fluid
- AQP4
- aquaporin-4
- BBB
- blood–brain barrier
- BCAO
- bilateral carotid artery occlusion
- CBF
- cerebral blood flow
- ECS
- extracellular space
- GFAP
- glial fibrillary acidic protein
- H&E
- hematoxylin and eosin
- Iba1
- ionized calcium binding adaptor molecule 1
- ICP
- intracranial pressure
- LPS
- lipopolysaccharide
- MBP
- myelin basic protein
- OGD
- oxygen–glucose deprivation
REFERENCES
- 1. Fujioka M., Okuchi K., Sakaki T., Hiramatsu K., Miyamoto S., Iwasaki S. (1994) Specific changes in human brain following reperfusion after cardiac arrest. Stroke 25, 2091–2095 [DOI] [PubMed] [Google Scholar]
- 2. Dirnagl U., Iadecola C., Moskowitz M. A. (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 22, 391–397 [DOI] [PubMed] [Google Scholar]
- 3. Moskowitz M. A., Lo E. H., Iadecola C. (2010) The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Papadopoulos M. C., Verkman A. S. (2013) Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 14, 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nielsen S., Nagelhus E. A., Amiry-Moghaddam M., Bourque C., Agre P., Ottersen O. P. (1997) Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 17, 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma T., Yang B., Gillespie A., Carlson E. J., Epstein C. J., Verkman A. S. (1997) Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J. Clin. Invest. 100, 957–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Manley G. T., Fujimura M., Ma T., Noshita N., Filiz F., Bollen A. W., Chan P., Verkman A. S. (2000) Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 6, 159–163 [DOI] [PubMed] [Google Scholar]
- 8. Solenov E., Watanabe H., Manley G. T., Verkman A. S. (2004) Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. Am. J. Physiol. Cell Physiol. 286, C426–C432 [DOI] [PubMed] [Google Scholar]
- 9. Thiagarajah J. R., Papadopoulos M. C., Verkman A. S. (2005) Noninvasive early detection of brain edema in mice by near-infrared light scattering. J. Neurosci. Res. 80, 293–299 [DOI] [PubMed] [Google Scholar]
- 10. Li L., Zhang H., Varrin-Doyer M., Zamvil S. S., Verkman A. S. (2011) Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. 25, 1556–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Binder D. K., Yao X., Zador Z., Sick T. J., Verkman A. S., Manley G. T. (2006) Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 53, 631–636 [DOI] [PubMed] [Google Scholar]
- 12. Padmawar P., Yao X., Bloch O., Manley G. T., Verkman A. S. (2005) K+ waves in brain cortex visualized using a long-wavelength K+-sensing fluorescent indicator. Nat. Methods 2, 825–827 [DOI] [PubMed] [Google Scholar]
- 13. Lu D. C., Zhang H., Zador Z., Verkman A. S. (2008) Impaired olfaction in mice lacking aquaporin-4 water channels. FASEB J. 22, 3216–3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li J., Verkman A. S. (2001) Impaired hearing in mice lacking aquaporin-4 water channels. J. Biol. Chem. 276, 31233–31237 [DOI] [PubMed] [Google Scholar]
- 15. Jin B. J., Zhang H., Binder D. K., Verkman A. S. (2013) Aquaporin-4-dependent K+ and water transport modeled in brain extracellular space following neuroexcitation. J. Gen. Physiol. 141, 119–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tajiri S., Oyadomari S., Yano S., Morioka M., Gotoh T., Hamada J. I., Ushio Y., Mori M. (2004) Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 11, 403–415 [DOI] [PubMed] [Google Scholar]
- 17. Soares L. M., Schiavon A. P., Milani H., de Oliveira R. M. (2013) Cognitive impairment and persistent anxiety-related responses following bilateral common carotid artery occlusion in mice. Behav. Brain Res. 249, 28–37 [DOI] [PubMed] [Google Scholar]
- 18. Abella B. S., Zhao D., Alvarado J., Hamann K., Vanden Hoek T. L., Becker L. B. (2004) Intra-arrest cooling improves outcomes in a murine cardiac arrest model. Circulation 109, 2786–2791 [DOI] [PubMed] [Google Scholar]
- 19. Yang B., Zador Z., Verkman A. S. (2008) Glial cell aquaporin-4 overexpression in transgenic mice accelerates cytotoxic brain swelling. J. Biol. Chem. 283, 15280–15286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ratelade J., Bennett J. L., Verkman A. S. (2011) Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS One 6, e27412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang H., Verkman A. S. (2010) Microfiberoptic measurement of extracellular space volume in brain and tumor slices based on fluorescent dye partitioning. Biophys. J. 99, 1284–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang H., Bennett J. L., Verkman A. S. (2011) Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann. Neurol. 70, 943–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhen G., Dore S. (2007) Optimized protocol to reduce variable outcomes for the bilateral common carotid artery occlusion model in mice. J. Neurosci. Methods 166, 73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neely J. D., Amiry-Moghaddam M., Ottersen O. P., Froehner S. C., Agre P., Adams M. E. (2001) Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. U. S. A. 98, 14108–14113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiwa N. S., Garrard P., Hainsworth A. H. (2010) Experimental models of vascular dementia and vascular cognitive impairment: a systematic review. J. Neurochem. 115, 814–828 [DOI] [PubMed] [Google Scholar]
- 26. Datta S. C., Opp M. R. (2008) Lipopolysaccharide-induced increases in cytokines in discrete mouse brain regions are detectable using Luminex xMAP technology. J. Neurosci. Methods 175, 119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duffy S., MacVicar B. A. (1996) In vitro ischemia promotes calcium influx and intracellular calcium release in hippocampal astrocytes. J. Neurosci. 16, 71–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ouyang Y. B., Voloboueva L. A., Xu L. J., Giffard R. G. (2007) Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J. Neurosci. 27, 4253–4260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kirino T., Tamura A., Sano K. (1985) Selective vulnerability of the hippocampus to ischemia: reversible and irreversible types of ischemic cell damage. Prog. Brain Res. 63, 39–58 [DOI] [PubMed] [Google Scholar]
- 30. Pulsinelli W. A. (1985) Selective neuronal vulnerability: morphological and molecular characteristics. Prog. Brain Res. 63, 29–37 [DOI] [PubMed] [Google Scholar]
- 31. Yao X., Hrabetova S., Nicholson C., Manley G. T. (2008) Aquaporin-4-deficient mice have increased extracellular space without tortuosity change. J. Neurosci. 28, 5460–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cho S., Liu D., Fairman D., Li P., Jenkins L., McGonigle P., Wood A. (2004) Spatiotemporal evidence of apoptosis-mediated ischemic injury in organotypic hippocampal slice cultures. Neurochem. Int. 45, 117–127 [DOI] [PubMed] [Google Scholar]
- 33. Finley M., Fairman D., Liu D., Li P., Wood A., Cho S. (2004) Functional validation of adult hippocampal organotypic cultures as an in vitro model of brain injury. Brain Res. 1001, 125–132 [DOI] [PubMed] [Google Scholar]
- 34. Chen M., Simard J. M. (2001) Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J. Neurosci. 21, 6512–6521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Garcia J. H., Kalimo H., Kamijyo Y., Trump B. F. (1977) Cellular events during partial cerebral ischemia, I: electron microscopy of feline cerebral cortex after middle-cerebral-artery occlusion. Virchows Arch. B Cell Pathol. 25, 191–206 [DOI] [PubMed] [Google Scholar]
- 36. Magnus T., Wiendl H., Kleinschnitz C. (2012) Immune mechanisms of stroke. Curr. Opin. Neurol. 25, 334–340 [DOI] [PubMed] [Google Scholar]
- 37. Murakami Y., Saito K., Hara A., Zhu Y., Sudo K., Niwa M., Fujii H., Wada H., Ishiguro H., Mori H., Seishima M. (2005) Increases in tumor necrosis factor-alpha following transient global cerebral ischemia do not contribute to neuron death in mouse hippocampus. J. Neurochem. 93, 1616–1622 [DOI] [PubMed] [Google Scholar]
- 38. Kim D. H., Kim J. M., Park S. J., Lee S., Yoon B. H., Ryu J. H. (2010) Early-activated microglia play a role in transient forebrain ischemia-induced neural precursor proliferation in the dentate gyrus of mice. Neurosci. Lett. 475, 74–79 [DOI] [PubMed] [Google Scholar]
- 39. Mickel H. S. (1990) Effect of hyperoxia differs during ischemia and reperfusion. Stroke 21, 1641–1642 [PubMed] [Google Scholar]
- 40. Petito C. K., Olarte J. P., Roberts B., Nowak T. S., Jr., Pulsinelli W. A. (1998) Selective glial vulnerability following transient global ischemia in rat brain. J. Neuropathol. Exp. Neurol. 57, 231–238 [DOI] [PubMed] [Google Scholar]
- 41. Yamamoto Y., Shioda N., Han F., Moriguchi S., Nakajima A., Yokosuka A., Mimaki Y., Sashida Y., Yamakuni T., Ohizumi Y., Fukunaga K. (2009) Nobiletin improves brain ischemia-induced learning and memory deficits through stimulation of CaMKII and CREB phosphorylation. Brain Res. 1295, 218–229 [DOI] [PubMed] [Google Scholar]
- 42. Yang G., Kitagawa K., Matsushita K., Mabuchi T., Yagita Y., Yanagihara T., Matsumoto M. (1997) C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res. 752, 209–218 [DOI] [PubMed] [Google Scholar]
- 43. Walker E. J., Rosenberg G. A. (2010) Divergent role for MMP-2 in myelin breakdown and oligodendrocyte death following transient global ischemia. J. Neurosci. Res. 88, 764–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murakami K., Kondo T., Kawase M., Chan P. H. (1998) The development of a new mouse model of global ischemia: focus on the relationships between ischemia duration, anesthesia, cerebral vasculature, and neuronal injury following global ischemia in mice. Brain Res. 780, 304–310 [DOI] [PubMed] [Google Scholar]
- 45. Huber V. J., Tsujita M., Yamazaki M., Sakimura K., Nakada T. (2007) Identification of arylsulfonamides as aquaporin 4 inhibitors. Bioorg. Med. Chem. Lett. 17, 1270–1273 [DOI] [PubMed] [Google Scholar]
- 46. Huber V. J., Tsujita M., Kwee I. L., Nakada T. (2009) Inhibition of aquaporin 4 by antiepileptic drugs. Bioorg. Med. Chem. 17, 418–424 [DOI] [PubMed] [Google Scholar]
- 47. Hinson S. R., Romero M. F., Popescu B. F., Lucchinetti C. F., Fryer J. P., Wolburg H., Fallier-Becker P., Noell S., Lennon V. A. (2012) Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc. Natl. Acad. Sci. U. S. A. 109, 1245–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rossi A., Ratelade J., Papadopoulos M. C., Bennett J. L., Verkman A. S. (2012) Neuromyelitis optica IgG does not alter aquaporin-4 water permeability, plasma membrane M1/M23 isoform content, or supramolecular assembly. Glia 60, 2027–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang B., Zhang H., Verkman A. S. (2008) Lack of aquaporin-4 water transport inhibition by antiepileptics and arylsulfonamides. Bioorg. Med. Chem. 16, 7489–7493 [DOI] [PMC free article] [PubMed] [Google Scholar]