

Abstract
Activation of adenosine A2A receptor (A2AR) promotes fibrosis and collagen synthesis. However, the underlying mechanism is still unclear, not least because cAMP, its principal effector, has been found to inhibit TGFβ1-induced collagen synthesis. Here, we show that in primary normal human dermal fibroblasts, A2AR stimulation with CGS21680 elicits a modest cAMP increase (150±12% of control; EC50 54.8 nM), which stimulates collagen1 (Col1) and collagen3 (Col3), but maximal cAMP resulting from direct activation of adenylyl cyclase by forskolin (15,689±7038% of control; EC50 360.7 nM) inhibits Col1 and increases Col3. Similar to Col1 expression, fibroblast proliferation increased following physiological cAMP increases by CGS21680 but was inhibited by cAMP increases beyond the physiological range by forskolin. The A2AR-mediated increase of Col1 and Col3 was mediated by AKT, while Col3, but not Col1, expression was dependent on p38 and repressed by ERK. TGFβ1 induced phosphorylation of Smad2/3 and increased Col3 expression, which was prevented by Smad3 depletion. In contrast, CGS21680 did not activate Smad2/3, and Smad2/3 knockdown did not prevent CGS21680-induced Col1 or Col3 increases. Our results indicate that cAMP is a concentration-dependent switch for collagen production via noncanonical, AKT-dependent, Smad2/3-independent signaling. These observations explain the paradoxical effects of cAMP on collagen expression.—Perez-Aso, M., Fernandez, P., Mediero, A., Chan, E. S., and Cronstein, B. N. Adenosine 2A receptor promotes collagen production by human fibroblasts via pathways involving cyclic AMP and AKT but independent of Smad2/3.
Keywords: fibrosis, intracellular signaling
adenosine is an endogenous purine nucleoside, extracellular levels of which are dramatically increased under stressful conditions, and which acts as a potent endogenous modulator of inflammation and tissue repair (1, 2). There are 4 adenosine receptors (A1R, A2AR, A2BR, and A3R), and they all belong to the G-protein-coupled receptor (GPCR) superfamily (3, 4). The adenosine A2A receptor (A2AR) plays a significant role in wound healing and fibrosis (5–9). Interestingly, the A2AR promotes wound healing by increasing collagen production directly and indirectly by stimulating connective tissue growth factor (CTGF) production. In addition to promoting wound closure (5, 10), A2AR stimulation promotes scar progression and dermal fibrosis, both of which are characterized by increased collagen type III (Col3) deposition out of proportion to collagen type I (Col1). Moreover, A2AR promotion of collagen synthesis is also detrimental in pathological conditions characterized by fibrosis, such as dermal fibrosis, scleroderma, and cirrhosis (6–9), and A2AR blockade or deletion prevents dermal fibrosis in mice treated with bleomycin (11) and hepatic fibrosis in mice treated with CCl4 and thioacetamide (9, 12). Similarly, A2AR blockade prevents scarring by reducing collagen content and misalignment (8).
A2AR increases Col1 and Col3 via different intracellular pathways, as previously shown in human stellate cells, in the LX-2 hepatic stellate cell line (13), and in normal human dermal fibroblasts (NHDFs; ref. 14), where the A2AR modulates the Col1:Col3 balance, a determinant of the collagen quality in wounded vs. normal skin, so that the Col1:Col3 ratio decreases as A2AR activation increases (14). Moreover, decreased Col1:Col3 ratio in the scar is prevented by A2AR pharmacological blockade (8). Thus, in normal skin, where adenosine concentration varies from 30 to 300 nM, there is a Col1:Col3 ratio of 4:1, but in hypertrophic and immature scars, where adenosine concentration is likely to be present at higher concentrations, the ratio decreases to 2:1 (15). These data indicate that A2AR promotes Col1 and Col3 synthesis with different sensitivities. Similarly, different cAMP-dependent signaling systems also demonstrate different sensitivities to cAMP concentrations. For example, the reduction of intracellular levels of cAMP in a narrow range of 10–50 nM switches the turning response from attraction to repulsion of the axon growth cone (16) and alters the cytosolic cAMP switches between attractant and repellent cone growth responses to netrin 1 (17).
The well-known profibrotic agent transforming growth factor β1 (TGF-β1) promotes collagen synthesis via the canonical Smad pathway (18–20), as well as Smad-independent pathways (21–23) dependent on AKT (24, 25). The underlying mechanism of A2AR-mediated collagen synthesis needs further investigation since paradoxically, the A2AR signals mainly via cAMP (26, 27), but exchange protein directly activated by cAMP 1 (Epac1), a principle cAMP effector, has been reported to decrease collagen production, among other antifibrotic actions (28, 29).
The most well-known effect of adenosine A2AR receptor stimulation is increasing intracellular cAMP (26, 30, 31), and the A2AR has been shown not to couple to the Gq/PLC/PKC pathway (26, 27). In fact, we have previously shown that direct activation of PKC by PMA dramatically represses both Col1 and Col3 in NHDFs (14). In support of the role of cAMP in signaling for A2AR-mediated Col1 and Col3 expression, we recently reported that activation of downstream signaling for A2AR-mediated increases in Col1 and Col3 expression proceeds via two distinct pathways with varying sensitivities to cAMP activation. More highly cAMP-sensitive PKA activation stimulates Col1 expression and less cAMP-sensitive Epac activation promotes both Col1 and Col3 expression (14). However, the signaling cascade from A2AR stimulation to increased collagen production is complex, not least because cAMP and its downstream molecules PKA and Epac1 have been reported to inhibit collagen production (28), and others have suggested that stimulating increased cAMP levels or activating Epac inhibits fibrosis (29), and the precise mechanisms of the antifibrotic effects of cAMP are not clearly defined (reviewed in ref. 29). To further understand this paradox, in the present work, we analyzed the intracellular signaling pathways for collagen synthesis on A2AR activation with CGS21680 or on direct AC activation with forskolin, which were compared to the well-known fibrotic agent TGF-β1. We found that A2AR increased cAMP within a physiological range that promotes collagen synthesis in an AKT-dependent, Smad-independent pathway.
MATERIALS AND METHODS
Antibodies, reagents, and cell line
CGS21680 was purchased from Tocris Bioscience (Ellisville, MO, USA). Antibodies to Col1 and Col3 were purchased from SouthernBiotech (Birmingham, AL, USA), and the p-Smad3, p-ERK, ERK, p38, p-AKT, and AKT antibodies were obtained from Cell Signaling Technology (Boston, MA, USA). The p-p38 antibody was obtained from Promega (Madison, WI, USA), and the p-Smad2 and Smad6/7 antibodies and the inhibitors SB203850, wortmannin, and LY294002 were obtained from Calbiochem (San Diego, CA. USA). The inhibitor U0126, the RIPA buffer, forskolin, and the protease and the phosphatase inhibitor cocktails were purchased from Sigma-Aldrich (St. Louis, MO, USA). The Smad2/3, CTGF, and actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and secondary antibodies were obtained from LI-COR Biosciences (Lincoln, NE, USA). TGF-β1 was from R&D Systems (Minneapolis, MN, USA) and NHDFs were purchased from Lonza (Walkersville, MD, USA).
Stimulation, preparation of cellular extracts, and Western blot analysis
As described previously (14), passage 1–5 of 75% confluent NHDF cells were stimulated during 24 h with the CGS21680 or forskolin at different concentrations. When indicated, the inhibitors U0126, SB203850 (1 μM), LY294002 (10 μM), or wortmannin (0.1 μM) were added 30 min before CGS21680. For all the experiments with inhibitors/siRNAs, a dose-response from 0.1 to 10 μM of CGS21680 was run in parallel. After stimulation, cells were washed with cold PBS and lysed with RIPA buffer containing protease inhibitor cocktail and phosphatase inhibitor cocktail. The protein content was measured by the BCA protein assay (Thermo Fisher Scientific, Pittsburgh, PA, USA). Protein extract (1 μg) for Col1 and 10 μg for the rest of antibodies were separated on SDS-polyacrylamide gels. Then, proteins were transferred to nitrocellulose membranes for immunoblotting. Prior to antibody incubation, membranes were blocked in Tris-buffered saline with 0.1% Tween 20 (TBST) plus 3% BSA (Sigma-Aldrich). Primary antibodies were incubated overnight at 4°C at 1:500 dilution for Col1, Col3, and Smad2/3, while at 1:1000 for p-ERK, ERK, p-p38, p38, p-AKT, AKT, p-Smad2, p-Smad3, Smad6/7, and actin. Membranes were then washed with TBST, incubated with secondary antibodies (1:10000 diluted) for 1 h at room temperature, and images were captured and analyzed by the LI-COR imaging system and Image Studio 2.0 software (LI-COR Biosciences). Band quantification was first normalized to actin, ERK, p38, AKT, or to Smad2/3, and then the percentage was calculated against the nonstimulated control blotted in the same membrane. For Col1, the upper band corresponding to Col1α1 (32, 33) was quantified.
RNA interference (siRNA)
Double-stranded siRNAs to SMAD2 (s8397), SMAD3 (s8400), ERK1 (s11141), ERK2 (s11137), AKT1 (632), AKT2 (103305), and AKT3 (110901), and negative siRNA (4390844) used as a control, were purchased from Ambion (Life Technologies, Grand Island, NY, USA). Cells were transfected with siRNA (50 pmol for single knockdown, 25 pmol of each for double knockdown, and 16.7 pmol for AKT1/2/3 triple knockdown), using Lipofectamine RNAiMAX (Invitrogen Life Technologies). After 24 h, CGS21680 or TGF-β1 was added for 24 h, and cellular extracts were prepared as described above.
cAMP measurements
Intracellular cAMP was measured with the direct cAMP ELISA kit from Enzo Life Sciences (Plymouth Meeting, PA, USA). Briefly, 50% confluent NHDFs were starved for 24 h and incubated at concentrations and with stimuli as indicated over 20 min, and cAMP levels were analyzed according to manufacturer's protocol.
Proliferation and cytotoxicity assays
Sixty percent confluent NHDFs were stimulated with 0.01, 0.1, 1, and 10 μM of CGS21680 and forskolin during 24 h, and the MTS proliferation assay or the LDH-release CytoTox nonradioactive cytotoxicity assay from Promega was performed according to manufacturer's protocol.
Statistical analysis
Statistical differences were determined using 1- or 2-way ANOVA or Student's t test carried out using GraphPad 6.00 software (GraphPad Software, San Diego, CA, USA) on a PC. The α nominal level was set at 0.05 in all cases. A value of P < 0.05 was considered significant.
RESULTS
A2AR and AC activation regulates fibroblast proliferation and Col1 and Col3 synthesis
It has previously been reported that the second messenger cAMP decreases collagen production, based on experiments in which high (50 μM) concentrations of membrane-permeable cAMP analogues are applied to cells (28, 29). Nonetheless, A2AR stimulation increases collagen expression, and these receptors signal principally via increases in cAMP. We examined the effect of the specific A2AR agonist CGS21680 and direct adenylyl cyclase (AC) activation by forskolin on cell proliferation and Col1 and Col3 production. We found that 10 μM CGS21680 increased intracellular cAMP levels (basal cAMP levels 0.173±0.062 pmol/ml) within the physiological range (150±12% of control; EC50 54.8 nM), whereas direct AC activation by forskolin promoted a more dramatic cAMP increase beyond the physiological range (15,689±7038% of control; EC50 360.7 nM) (Fig. 1A). Fibroblast proliferation was increased by A2AR activation with CGS2180, but inhibited by forskolin 10 μM (Fig. 1B). As we have recently reported (14), A2AR activation with CGS21680 increased both Col1 and Col3 in a dose-dependent fashion with different sensitivities (Fig. 1C); Col3 increased only at micromolar concentrations of CGS21680, whereas Col1 was increased by both nanomolar and micromolar concentrations of the A2AR agonist. The direct AC activator forskolin inhibited Col1 expression in a dose-dependent fashion, but Col3 expression was increased at all of the tested concentrations of agonist. None of the described effects were due to cytotoxicity, as determined by LDH release (Supplemental Fig. S1).
Figure 1.

A2AR and adenylyl cyclase activation differentially regulate proliferation and Col1 and Col3. A, B) cAMP intracellular levels (basal levels, 0.173±0.062 pmol/ml; A) and fibroblast proliferation (B) were quantified after incubation with increasing concentrations of CGS21680 and forskolin. C) Col1 and Col3 expression was examined by Western blot after 24 h of stimulation with CGS21680 or forskolin at the indicated concentrations, and bands were quantified. Data represent means ± se of 4 independent experiments. Statistical analysis was performed by 1-way ANOVA with post hoc Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 for CGS21680 vs. nonstimulated control; #P < 0.05, ##P < 0.01 for forskolin vs. nonstimulated control.
These results indicate that physiological levels of cAMP resulting from pharmacologic A2AR stimulation promote fibroblast proliferation and collagen production, but that the high levels of cAMP, beyond the physiological range that can be achieved by direct and irreversible activation of AC by forskolin, inhibit fibroblast proliferation and Col1 production. Col3 is increased at both low and supraphysiological levels of intracellular cAMP. These findings suggest that the previously described effects of cell-permeable cAMP analogues on Col1 production are mediated by events requiring very high and persistent cAMP levels and do not reflect signaling by adenosine receptors or, likely, other Gs-linked receptors, in fibroblasts.
A2AR differentially regulates activation of MAP kinases and AKT
In dermal fibroblasts, the PI3 kinase/AKT pathway mediates an increase in Col1 (34), and cAMP inhibits ERK via PKA to prevent MMP-1 expression (35), which is negatively correlated with Col1 synthesis (36). We therefore sought to analyze the role of MAPKs and AKT in adenosine receptor-stimulated collagen synthesis. A2AR activation by CGS21680 for 5 min inhibited ERK and p38 MAPK activation (phosphorylation) in a dose-dependent fashion but increased AKT phosphorylation at low, but not higher, concentrations of CGS21680 (Fig. 2). In contrast, when examined 12 h after stimulation, there was a clear, dose-dependent increase in ERK, p38 MAPK, and AKT activation.
Figure 2.
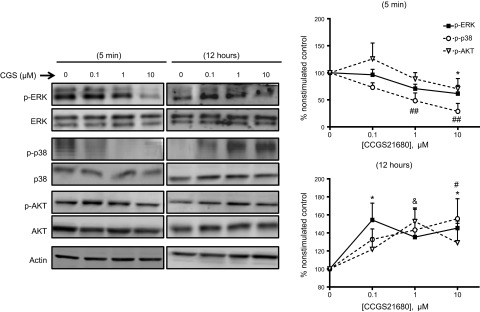
A2AR stimulation differentially regulates ERK, p38, and AKT. NHDFs were serum starved for 4 h and stimulated with increasing concentrations of the A2AR agonist CGS21680 for 5 min or 12 h at the indicated concentrations. Data represent means ± se of 4 independent experiments. Statistical analysis was performed by 1-way ANOVA with post hoc Dunnett's test. *P < 0.05 for p-ERK vs. nonstimulated control; #P < 0.05, ##P < 0.01 for p-p38 vs. nonstimulated control; &P < 0.05 for p-AKT vs. nonstimulated control.
Inhibition of ERK, p38, and AKT differentially regulate Col1 and Col3 expression after A2AR activation
As shown above, A2AR activation with the specific agonist CGS21680 stimulated an increase in Col1 expression at nanomolar and micromolar concentrations, whereas Col3 was increased only at micromolar concentrations (Fig. 3; control). Interestingly, ERK inhibition with U0126 did not significantly affect CGS21680-stimulated changes in Col1 expression but permitted increased Col3 expression after stimulation with even nanomolar CGS21680. p38 MAPK blockade by SB203580 only partially prevented Col1 increases but completely abrogated the CGS21680-stimulated increase in Col3 expression, as previously reported for hepatic stellate cells (13). AKT inhibition with either LY294002 or wortmannin prevented both Col1 and Col3 expression at all tested CGS21680 concentrations (Fig. 3). Further evidence for the ERK1/2 and AKT role in Col1 and Col3 regulation is provided by specific knockdowns (Fig. 4).
Figure 3.

Effects of ERK, p38, and AKT blockade on Col1 and Col3 expression. A) Representative Western blots showing Col1 and Col3 expression after CGS21680 stimulation on ERK1/2 and p38 blockade with U0126 (1 μM) and SB203580 (1 μM), respectively, and AKT blockade with either LY294002 (10 μM) or wortmannin (0.1 μM). B) Bands were quantified; data represent means ± sem of ≥3 independent experiments. Statistical analysis was performed by 2-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Figure 4.

Effect of ERK1/2 and AKT1/2/3 knockdown on collagen expression. A) Double siRNA ERK1/2 and triple siRNA AKT1/2/3 significantly reduce ERK1/2 and AKT expression. B) Effect of ERK1/2 and AKT1/2/3 knockdowns on Col1 and Col3 expression after CGS21680 incubation. Bands were quantified, and data represent means ± se of 4 independent experiments. ***P < 0.001 vs. control siRNA, Student's t test; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. control siRNA, 2-way ANOVA.
As noted above and previously reported, Col1, but not Col3, is increased on stimulation by CGS21680 at a concentration of 0.1 μM, but Col3 is increased at 0.1 μM CGS21680only when PKA is inhibited (14). In a parallel experiment, 0.1 μM CGS21680 significantly increased Col3 only on ERK inhibition (Table 1), signaling molecules that are activated at low A2AR signal strength. Further corroboration that ERK1/2 negatively regulates Col3 after A2AR activation is provided by double knockdown of ERK1 and ERK2 in cultured fibroblasts (Fig. 4). These findings strongly suggest that at different signal strengths, A2AR stimulation differentially activates downstream signaling steps that lead to differential regulation of Col1 and Col3 expression.
Table 1.
Col3 expression after incubation with CGS21680 (0.1 μM)
| Treatment | Expression (% of control) | n | t test (P) |
|---|---|---|---|
| Control | 99.9 ± 6.8 | 16 | – |
| SB203580 | 93.0 ± 5.9 | 4 | 0.6304 |
| U0126 | 157.0 ± 3.8 | 3 | 0.0024** |
| LY294002 | 51.3 ± 11.9 | 5 | 0.0023** |
| Wortmannin | 119.6 ± 18.8 | 4 | 0.2398 |
Values are means ± sem.
P < 0.01.
A2AR increases Col1 and Col3 via a Smad-independent pathway
It is well known that TGF-β promotes collagen production via the canonical Smad pathway, as well as via Smad-independent signaling (21–23). Although the A2AR has been repeatedly shown to promote collagen increase in vivo and in vitro (8, 11, 13, 37, 38), the intracellular pathways involved in dermal fibroblasts have not been fully elucidated. We first confirmed that in our system, TGF-β1 activates both Col1 and Col3 (Fig. 5A) and that TGF-β1 is a potent stimulator of both Smad2 and Smad3 phosphorylation (Fig. 5B). Interestingly, we found that A2AR activation does not stimulate Smad2 or Smad3 phosphorylation. Moreover, A2AR activation does not regulate expression of Smad 6/7 (Fig. 5C), providing further evidence that A2AR stimulation does not regulate the canonical Smad pathway. Knockdown experiments demonstrated that the Col1 and Col3 increase after A2AR activation by 1 μM CGS21680 is independent of Smad2/3 (Fig. 6). Interestingly, Col1 up-regulation by TGF-β1 was independent of both Smad2 and Smad3, suggesting activation of the TGF-βRII, as previously shown in dermal fibroblasts (22), while Smad3 was needed for the TGF-β1-increased Col3.
Figure 5.
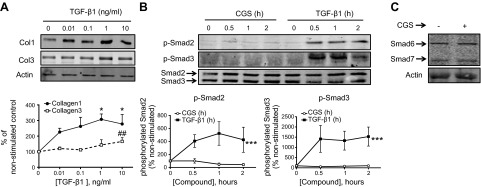
A2AR does not activate the Smad2/3 or Smad6/7 pathway A) TGF-β1 increases both Col1 and Col3 in a dose-response fashion: Western blots for Col1 and Col3 were performed after stimulation with increasing concentration of TGF-β1. Statistical analysis was performed by 1-way ANOVA with post hoc Dunnett's test. *P < 0.05 for Col1 vs. nonstimulated control; ##P < 0.01 for Col3 vs. nonstimulated control. B) NHDFs were serum starved for 4 h and stimulated at the indicated time points with the A2AR agonist CGS21680 (1 μM) or TGF-β1 (5 ng/ml). Statistical analysis was performed by 2-way ANOVA ***P < 0.001 for TGF-β1 vs. CGS21680. Data represent means ± se of 4 independent experiments. C) Stimulation with CGS21680 (1 μM) for 24 h does not alter the expression of Smad6 or Smad7.
Figure 6.

Effect of Smad2, Smad3, or double Smad2/3 knockdown on Col1 and Col3 expression after CGS21680 (1 μM) or TGF-β1 (5 ng/ml). A) siRNAs specifically reduce protein expression of Smad2, Smad3, or Smad2/3. Bands were quantified, and data represent means ± se of 8 independent experiments. Statistics were performed by Student's t test. *P < 0.05, ***P < 0.001 vs. control siRNA. B) NHDFs were stimulated for 24 h with the A2AR agonist CGS21680 (1 μM) or TGF-β1 (5 ng/ml). Statistics were performed by Student's t test. #P < 0.05, ##P < 0.01 vs. nonstimulated control. Data represent means ± se of 4 independent experiments.
DISCUSSION
In recent studies, we have reported that ligation of adenosine A2AR stimulates increased collagen production by stimulated primary human fibroblasts (14) and hepatic stellate cells (13). Here, we report that A2AR stimulation results in collagen production through multiple signaling pathways downstream of AC. These pathways differ with respect to which type of collagen is synthesized (Col1 vs. Col3) and the signal strength required to activate them. At low levels of A2AR stimulation (0.1 μM CGS21680), Col1 expression is increased in a manner dependent on AKT phosphorylation and activation. In contrast, higher levels of receptor activation (1–10 μM CGS21680) are required to stimulate Col3 expression by an AKT/p38 MAPK-dependent mechanism. Moreover, because stimulation of Col3 is negatively regulated by ERK1 and ERK2 activation, greater positive stimulation (and thus greater receptor activation) is required to stimulate Col3 expression. Increasing the complexity of collagen regulation by the A2AR, we previously reported that Fli1 and CTGF are important signals for the synthesis of Col1 by A2AR (40). However, we found that, unlike the effect on direct activation of collagen production, increased CTGF levels resulting from A2AR stimulation were not reduced by AKT knockdown (Supplemental Fig. S2). Thus, our findings indicate that these are independent pathways for collagen regulation.
The most well-known effect of adenosine A2AR receptor stimulation is an increase in intracellular cAMP (26, 30, 31) and the A2AR has been shown not to couple to the Gq/PLC/PKC pathway (26, 27). In fact, direct activation of PKC by PMA dramatically represses both Col1 and Col3 in NHDFs (14), which strongly suggests that the Col1 and Col3 increase by A2AR activation occurs via cAMP. However, there is some controversy in this regard, since the two main effectors of cAMP, Epac and PKA, have both been reported to decrease collagen synthesis in human fibroblasts (28). Moreover, the cAMP-stimulating agents forskolin and isoproterenol have been shown to inhibit the TGF-β-induced increase of collagen (28, 39–41). In the same line, it has been suggested that, although cAMP integrates profibrotic and antifibrotic signals (28), increasing cAMP levels or activating Epac could be used to inhibit fibrosis (29). Paradoxically, Park et al. (35) reported that in human skin fibroblasts cAMP inhibits ERK via PKA, thereby reducing MMP-1 expression, which is characteristic of systemic sclerosis (SSc) fibroblasts and may contribute to excessive collagen deposition seen in patients with SSc (24). Similarly, collagen is increased by activation of AC-coupled receptors, such as the angiotensin AT-1R (42) and the adenosine A2AR (11, 13), the latter via PKA and Epac (13, 14).
Interestingly, cAMP systems have been previously found to work as an all-or-none switch, where a narrow range of cAMP cytosolic concentrations elicits diametrically different behaviors (16, 43). For example, the reduction of intracellular levels of cAMP in a narrow range of 10–50 nM switches the turning response from attraction to repulsion of the axon growth cone (16) and altering the cytosolic cAMP switches between attractant and repellent cone growth responses to netrin 1 (17). Similarly, a slight intracellular cAMP increase activates fibroblast migration, which is, nonetheless, inhibited by micromolar cAMP levels (28). To our knowledge, the present work is the first showing that cAMP also works as an all-or-none switch for fibroblast proliferation and collagen production (Fig. 1), so that slight cAMP physiological increases on A2AR activation by CGS21680 leads to increases in proliferation and Col1, but supraphysiological intracellular cAMP levels by direct AC activation inhibit both proliferation and Col1 (Fig. 1). In agreement, we found that nanomolar concentrations of two different cAMP-analogs increase Col1 expression, but it is decreased on higher concentrations (Supplemental Fig. S3). These findings help to understand why previous studies using high concentrations of cAMP analogs (≥50 μM; refs. 28, 39, 44–46) or cAMP-increasing agents (≥10 μM; refs. 39, 46–49) report inhibition of collagen and proliferation (reviewed in ref. 29). However, we and others have found that collagen is increased by more pharmacologically relevant concentrations of cAMP-increasing agents (≤10 μM; refs. 11, 13, 14, 42, and present work), strongly suggesting that cAMP integrates profibrotic and antifibrotic signals, as previously suggested (28). In support of previous findings that higher cAMP levels are needed to increase Col3 than Col1 (14), in the present work, we show that Col3 is up-regulated on micromolar CGS21680 and at all the tested forskolin concentrations (Fig. 1C).
MMP-1 expression in human skin fibroblasts is reduced by cAMP via ERK inhibition by PKA (35). In agreement, we recently showed that nanomolar CGS21680 increases Col3 only when PKA is inhibited (14). Similarly, in the present work we show that A2AR activation promotes ERK inhibition at early A2AR activation (Fig. 2) and that pharmacological blockade or knockdown of ERK permits Col3 to increase after nanomolar CGS21680 (Fig. 3), which suggests that CGS21680 activates Col3 at high concentrations via ERK inhibition by PKA. Moreover, and unlike Col1, Col3 is increased at maximal cAMP (Fig. 1C). Taken together, these results help to understand the dramatic decrease in the Col1:Col3 ratio in hypertrophic and immature scars (15), where adenosine levels are increased, and which is prevented by A2AR blockade (8), suggesting that PKA inhibition of ERK on Gs-protein-coupled receptor activation perpetuates skin fibrosis progression. However, it has been suggested that there must be other mediators, since cAMP still suppresses MMP-1 expression even when ERK activity is unaffected by it (35). In this respect, AKT has been previously found to up-regulate collagen and down-regulate MMP1 in human dermal fibroblasts (24). Moreover, Bujor et al. (24) reported that AKT is not involved in the TGF-β-induced up-regulation of collagen and that its inhibition does not affect Smad2/3 activation. In fact, it was suggested that although inhibiting the components of the PI3K/AKT network ultimately leads to decreased collagen deposition by human dermal fibroblasts, a distinct cellular mechanism other than TGF-β may be involved in mediating this effect (24). Similarly, it has been previously reported that ANG II activation of the AT-1 receptor increases collagen production via cAMP and AKT (42). In agreement, in the present work, we show that AKT blockade or knockdown completely prevents the A2AR-mediated Col1 and Col3 increase (Figs. 3, 4), which is independent of Smad2/3 (Figs. 5 and 6), strongly suggesting that cAMP-increasing receptors promote collagen synthesis via AKT independently of Smad2/3. Interestingly, we have previously found that, in a murine model of dermal fibrosis, A2AR activation increases TGF-β1 (37), which suggests that in vivo A2AR may indirectly activate Smad2/3.
In summary, our findings suggest that although TGF-β and adenosine promote fibrosis, both follow distinct signaling pathways. As depicted in Fig. 7, TGF-β and adenosine tissue levels increase on fibrotic insults. While both agents promote collagen synthesis, TGF-β signals via Smad3 and adenosine activation of A2AR promotes increased cAMP intracellular levels within a physiological range that promotes collagen synthesis. We also show that A2AR regulates ERK1/2, p38 MAPK, and AKT for a differential Col1 and Col3 regulation. Moreover, the present work indicates that intracellular cAMP works as an all-or-none switch for collagen type I synthesis and fibroblast proliferation, providing an attractive explanation for the paradox of how A2AR induces fibrosis via cAMP, and suggests that buffering cAMP within a specific range holds promise as a new pharmacologic strategy to prevent tissue fibrosis.
Figure 7.

Diagram depicting the parallel TGF-β and adenosine pathways for collagen synthesis.
Supplementary Material
Acknowledgments
This work was supported by grants from the U.S. National Institutes of Health (NIH; AR56672 and AR54897), the New York University (NYU)–Health and Hospitals Corporation Clinical and Translational Science Institute (UL1RR029893), the NYU Cancer Institute (Center Support grant), and the NIH/National Cancer Institute (5 P30CA16087-31).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- A2AR
- adenosine A2A receptor
- AC
- adenylyl cyclase
- Col1
- collagen type I
- Col3
- collagen type III
- CTGF
- connective tissue growth factor
- Epac
- exchange protein directly activated by cAMP
- GPCR
- G-protein-coupled receptor
- NHDF
- normal human dermal fibroblast
- TGF-β1
- transforming growth factor β1
REFERENCES
- 1. Hasko G., Cronstein B. N. (2004) Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 25, 33–39 [DOI] [PubMed] [Google Scholar]
- 2. Ohta A., Sitkovsky M. (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414, 916–920 [DOI] [PubMed] [Google Scholar]
- 3. Lazarova T., Brewin K. A., Stoeber K., Robinson C. R. (2004) Characterization of peptides corresponding to the seven transmembrane domains of human adenosine A2A receptor. Biochemistry 43, 12945–12954 [DOI] [PubMed] [Google Scholar]
- 4. Thevenin D., Roberts M. F., Lazarova T., Robinson C. R. (2005) Identifying interactions between transmembrane helices from the adenosine A2A receptor. Biochemistry 44, 16239–16245 [DOI] [PubMed] [Google Scholar]
- 5. Victor-Vega C., Desai A., Montesinos M. C., Cronstein B. N. (2002) Adenosine A2A receptor agonists promote more rapid wound healing than recombinant human platelet-derived growth factor (Becaplermin gel). Inflammation 26, 19–24 [DOI] [PubMed] [Google Scholar]
- 6. Katebi M., Fernandez P., Chan E. S., Cronstein B. N. (2008) Adenosine A2A receptor blockade or deletion diminishes fibrocyte accumulation in the skin in a murine model of scleroderma, bleomycin-induced fibrosis. Inflammation 31, 299–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lazzerini P. E., Natale M., Gianchecchi E., Capecchi P. L., Montilli C., Zimbone S., Castrichini M., Balistreri E., Ricci G., Selvi E., Garcia-Gonzalez E., Galeazzi M., Laghi-Pasini F. (2012) Adenosine A2A receptor activation stimulates collagen production in sclerodermic dermal fibroblasts either directly and through a cross-talk with the cannabinoid system. J. Mol. Med. (Berl.) 90, 331–342 [DOI] [PubMed] [Google Scholar]
- 8. Perez-Aso M., Chiriboga L., Cronstein B. N. (2012) Pharmacological blockade of adenosine A2A receptors diminishes scarring. FASEB J. 26, 4254–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan E. S., Montesinos M. C., Fernandez P., Desai A., Delano D. L., Yee H., Reiss A. B., Pillinger M. H., Chen J. F., Schwarzschild M. A., Friedman S. L., Cronstein B. N. (2006) Adenosine A2A receptors play a role in the pathogenesis of hepatic cirrhosis. Br. J. Pharmacol. 148, 1144–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Montesinos M. C., Gadangi P., Longaker M., Sung J., Levine J., Nilsen D., Reibman J., Li M., Jiang C. K., Hirschhorn R., Recht P. A., Ostad E., Levin R. I., Cronstein B. N. (1997) Wound healing is accelerated by agonists of adenosine A2 (G alpha s-linked) receptors. J. Exp. Med. 186, 1615–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan E. S., Fernandez P., Merchant A. A., Montesinos M. C., Trzaska S., Desai A., Tung C. F., Khoa D. N., Pillinger M. H., Reiss A. B., Tomic-Canic M., Chen J. F., Schwarzschild M. A., Cronstein B. N. (2006) Adenosine A2A receptors in diffuse dermal fibrosis: pathogenic role in human dermal fibroblasts and in a murine model of scleroderma. Arthritis Rheum. 54, 2632–2642 [DOI] [PubMed] [Google Scholar]
- 12. Peng Z., Fernandez P., Wilder T., Yee H., Chiriboga L., Chan E. S., Cronstein B. N. (2008) Ecto-5′-nucleotidase (CD73) -mediated extracellular adenosine production plays a critical role in hepatic fibrosis. FASEB J. 22, 2263–2272 [DOI] [PubMed] [Google Scholar]
- 13. Che J., Chan E. S., Cronstein B. N. (2007) Adenosine A2A receptor occupancy stimulates collagen expression by hepatic stellate cells via pathways involving protein kinase A, Src, and extracellular signal-regulated kinases 1/2 signaling cascade or p38 mitogen-activated protein kinase signaling pathway. Mol. Pharmacol. 72, 1626–1636 [DOI] [PubMed] [Google Scholar]
- 14. Perez-Aso M., Mediero A., Cronstein B. N. (2013) Adenosine A2A receptor is a fine-tune regulator of the collagen1:collagen3 balance. [E-pub ahead of print] Purinergic Signal. 10.1007/s11302-013-9368-1 [DOI] [PMC free article] [PubMed]
- 15. Stadelmann W. K., Digenis A. G., Tobin G. R. (1998) Physiology and healing dynamics of chronic cutaneous wounds. Am. J. Surg. 176, 26S–38S [DOI] [PubMed] [Google Scholar]
- 16. Ming G. L., Song H. J., Berninger B., Holt C. E., Tessier-Lavigne M., Poo M. M. (1997) cAMP-dependent growth cone guidance by netrin-1. Neuron 19, 1225–1235 [DOI] [PubMed] [Google Scholar]
- 17. Moore S. W., Kennedy T. E. (2006) Protein kinase A regulates the sensitivity of spinal commissural axon turning to netrin-1 but does not switch between chemoattraction and chemorepulsion. J. Neurosci. 26, 2419–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen S. J., Yuan W., Lo S., Trojanowska M., Varga J. (2000) Interaction of smad3 with a proximal smad-binding element of the human alpha2(I) procollagen gene promoter required for transcriptional activation by TGF-β. J. Cell. Physiol. 183, 381–392 [DOI] [PubMed] [Google Scholar]
- 19. Chen S. J., Yuan W., Mori Y., Levenson A., Trojanowska M., Varga J. (1999) Stimulation of type I collagen transcription in human skin fibroblasts by TGF-β: involvement of Smad 3. J. Invest. Dermatol. 112, 49–57 [DOI] [PubMed] [Google Scholar]
- 20. Flanders K. C. (2004) Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 85, 47–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cho H. J., Yoo J. (2007) Rho activation is required for transforming growth factor-β-induced epithelial-mesenchymal transition in lens epithelial cells. Cell Biol. Int. 31, 1225–1230 [DOI] [PubMed] [Google Scholar]
- 22. Pannu J., Nakerakanti S., Smith E., ten Dijke P., Trojanowska M. (2007) Transforming growth factor-β receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J. Biol. Chem. 282, 10405–10413 [DOI] [PubMed] [Google Scholar]
- 23. Wang S., Wilkes M. C., Leof E. B., Hirschberg R. (2005) Imatinib mesylate blocks a non-Smad TGF-β pathway and reduces renal fibrogenesis in vivo. FASEB J. 19, 1–11 [DOI] [PubMed] [Google Scholar]
- 24. Bujor A. M., Pannu J., Bu S., Smith E. A., Muise-Helmericks R. C., Trojanowska M. (2008) Akt blockade downregulates collagen and upregulates MMP1 in human dermal fibroblasts. J. Invest. Dermatol. 128, 1906–1914 [DOI] [PubMed] [Google Scholar]
- 25. Jun J. B., Kuechle M., Min J., Shim S. C., Kim G., Montenegro V., Korn J. H., Elkon K. B. (2005) Scleroderma fibroblasts demonstrate enhanced activation of Akt (protein kinase B) in situ. J. Invest. Dermatol. 124, 298–303 [DOI] [PubMed] [Google Scholar]
- 26. Fredholm B. B., AP I. J., Jacobson K. A., Klotz K. N., Linden J. (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 53, 527–552 [PMC free article] [PubMed] [Google Scholar]
- 27. Fredholm B. B., Arslan G., Halldner L., Kull B., Schulte G., Wasserman W. (2000) Structure and function of adenosine receptors and their genes. Naunyn. Schmiedebergs Arch. Pharmacol. 362, 364–374 [DOI] [PubMed] [Google Scholar]
- 28. Yokoyama U., Patel H. H., Lai N. C., Aroonsakool N., Roth D. M., Insel P. A. (2008) The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc. Natl. Acad. Sci. U. S. A. 105, 6386–6391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Insel P. A., Murray F., Yokoyama U., Romano S., Yun H. R., Brown L., Snead A., Lu D. V., Aroonsakool N. (2012) cAMP and Epac in the regulation of tissue fibrosis. Br. J. Pharmacol. 166, 447–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Milne G. R., Palmer T. M. (2011) Anti-inflammatory and immunosuppressive effects of the A2A adenosine receptor. ScientificWorldJournal. 11, 320–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohta A., Madasu M., Kini R., Subramanian M., Goel N., Sitkovsky M. (2009) A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J. Immunol. 183, 5487–5493 [DOI] [PubMed] [Google Scholar]
- 32. Butkowski R. J. (1982) Estimation of the size of collagenous proteins by electrophoresis and gel chromatography. Methods Enzymol. 82, 410–423 [Google Scholar]
- 33. Rentz T. J., Poobalarahi F., Bornstein P., Sage E. H., Bradshaw A. D. (2007) SPARC regulates processing of procollagen I and collagen fibrillogenesis in dermal fibroblasts. J. Biol. Chem. 282, 22062–22071 [DOI] [PubMed] [Google Scholar]
- 34. Shi G. X., Rehmann H., Andres D. A. (2006) A novel cyclic AMP-dependent Epac-Rit signaling pathway contributes to PACAP38-mediated neuronal differentiation. Mol. Cell. Biol. 26, 9136–9147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Park C. H., Moon Y., Shin C. M., Chung J. H. (2010) Cyclic AMP suppresses matrix metalloproteinase-1 expression through inhibition of MAPK and GSK-3β. J. Invest. Dermatol. 130, 2049–2056 [DOI] [PubMed] [Google Scholar]
- 36. Goffin L., Seguin-Estevez Q., Alvarez M., Reith W., Chizzolini C. (2010) Transcriptional regulation of matrix metalloproteinase-1 and collagen 1A2 explains the anti-fibrotic effect exerted by proteasome inhibition in human dermal fibroblasts. Arthritis Res. Ther. 12, R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fernandez P., Trzaska S., Wilder T., Chiriboga L., Blackburn M. R., Cronstein B. N., Chan E. S. (2008) Pharmacological blockade of A2A receptors prevents dermal fibrosis in a model of elevated tissue adenosine. Am. J. Pathol. 172, 1675–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chan E. S., Liu H., Fernandez P., Luna A., Perez-Aso M., Bujor A. M., Trojanowska M., Cronstein B. N. (2013) Adenosine A2A receptors promote collagen production by a Fli1- and CTGF-mediated mechanism. Arthritis Res. Ther. 15, R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schiller M., Dennler S., Anderegg U., Kokot A., Simon J. C., Luger T. A., Mauviel A., Bohm M. (2010) Increased cAMP levels modulate transforming growth factor-beta/Smad-induced expression of extracellular matrix components and other key fibroblast effector functions. J. Biol. Chem. 285, 409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu X., Sun S. Q., Hassid A., Ostrom R. S. (2006) cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol. Pharmacol. 70, 1992–2003 [DOI] [PubMed] [Google Scholar]
- 41. Duncan M. R., Frazier K. S., Abramson S., Williams S., Klapper H., Huang X., Grotendorst G. R. (1999) Connective tissue growth factor mediates transforming growth factor β-induced collagen synthesis: down-regulation by cAMP. FASEB J. 13, 1774–1786 [PubMed] [Google Scholar]
- 42. Yano N., Suzuki D., Endoh M., Zhao T. C., Padbury J. F., Tseng Y. T. (2007) A novel phosphoinositide 3-kinase-dependent pathway for angiotensin II/AT-1 receptor-mediated induction of collagen synthesis in MES-13 mesangial cells. J. Biol. Chem. 282, 18819–18830 [DOI] [PubMed] [Google Scholar]
- 43. Lipshtat A., Jayaraman G., He J. C., Iyengar R. (2010) Design of versatile biochemical switches that respond to amplitude, duration, and spatial cues. Proc. Natl. Acad. Sci. U. S. A. 107, 1247–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Okunishi K., Sisson T. H., Huang S. K., Hogaboam C. M., Simon R. H., Peters-Golden M. (2011) Plasmin overcomes resistance to prostaglandin E2 in fibrotic lung fibroblasts by reorganizing protein kinase A signaling. J. Biol. Chem. 286, 32231–32243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang S. K., Wettlaufer S. H., Chung J., Peters-Golden M. (2008) Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. Am. J. Respir. Cell Mol. Biol. 39, 482–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Villarreal F., Epperson S. A., Ramirez-Sanchez I., Yamazaki K. G., Brunton L. L. (2009) Regulation of cardiac fibroblast collagen synthesis by adenosine: roles for Epac and PI3K. Am. J. Physiol. Cell Physiol. 296, C1178–C1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. D'Souza K. M., Malhotra R., Philip J. L., Staron M. L., Theccanat T., Jeevanandam V., Akhter S. A. (2011) G protein-coupled receptor kinase-2 is a novel regulator of collagen synthesis in adult human cardiac fibroblasts. J. Biol. Chem. 286, 15507–15516 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Liu X., Ostrom R. S., Insel P. A. (2004) cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am. J. Physiol. Cell Physiol. 286, C1089–C1099 [DOI] [PubMed] [Google Scholar]
- 49. Hayashi T., Nishihira J., Koyama Y., Sasaki S., Yamamoto Y. (2006) Decreased prostaglandin E2 production by inflammatory cytokine and lower expression of EP2 receptor result in increased collagen synthesis in keloid fibroblasts. J. Invest. Dermatol. 126, 990–997 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.