

Abstract
Cystic fibrosis (CF) is caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) that impair its expression and/or chloride channel function. Here, we provide evidence that type 4 cyclic nucleotide phosphodiesterases (PDE4s) are critical regulators of the cAMP/PKA-dependent activation of CFTR in primary human bronchial epithelial cells. In non-CF cells, PDE4 inhibition increased CFTR activity under basal conditions (ΔISC 7.1 μA/cm2) and after isoproterenol stimulation (increased ΔISC from 13.9 to 21.0 μA/cm2) and slowed the return of stimulated CFTR activity to basal levels by >3-fold. In cells homozygous for ΔF508-CFTR, the most common mutation found in CF, PDE4 inhibition alone produced minimal channel activation. However, PDE4 inhibition strongly amplified the effects of CFTR correctors, drugs that increase expression and membrane localization of CFTR, and/or CFTR potentiators, drugs that increase channel gating, to reach ∼25% of the chloride conductance observed in non-CF cells. Biochemical studies indicate that PDE4s are anchored to CFTR and mediate a local regulation of channel function. Taken together, our results implicate PDE4 as an important determinant of CFTR activity in airway epithelia, and support the use of PDE4 inhibitors to potentiate the therapeutic benefits of CFTR correctors and potentiators.—Blanchard, E., Zlock, L., Lao, A., Mika, D., Namkung, W., Xie, M., Scheitrum, C., Gruenert, D.C., Verkman, A.S., Finkbeiner, W.E., Conti, M., Richter, W. Anchored PDE4 regulates chloride conductance in wild type and ΔF508-CFTR human airway epithelia.
Keywords: cystic fibrosis, cyclic nucleotide phosphodiesterase, cAMP, corrector
The cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-stimulated anion channel that is critical for the regulation of ion and water transport across multiple epithelia. CFTR dysfunction plays a central role in several diseases. CFTR hyperfunction is a characteristic of secretory diarrhea and polycystic kidney disease (PKD; ref. 1). Conversely, CFTR hypofunction is the underlying cause of cystic fibrosis (CF), a lethal genetic disorder caused by mutations in the CFTR gene that impair expression and/or function of the channel (2, 3). Deletion of phenylalanine 508 (ΔF508) is the most common mutation found in patients with CF and triggers misfolding and premature degradation of the translated protein. As a result, the number of channels inserted into epithelial cell membranes is insufficient to support normal ion transport.
Physiologically, phosphorylation by the cAMP-stimulated protein kinase (PKA) is the main mechanism of activation of CFTR. This finding prompted first efforts to modulate the cAMP signaling pathway as a therapeutic approach for CF nearly 20 yr ago (4–6). Early work showed that, in principal, ΔF508-CFTR channels can still be activated via the cAMP/PKA pathway, such as in overexpression systems (6). However, treatment with adenylyl cyclase activators, such as forskolin (FSK), and/or broad-spectrum inhibitors of cyclic nucleotide phosphodiesterases (PDEs), such as 3-isobutyl-1-methylxanthine (IBMX), was ineffective in restoring CFTR-dependent ion transport in cells expressing endogenous ΔF508-CFTR (6, 7), likely due to insufficient amounts of ΔF508-CFTR at the apical membrane. This lack of efficacy discouraged further development of drugs targeting the cAMP/PKA pathway. Instead, current therapeutic approaches for CF aim to restore CFTR protein levels through gene therapy or treatment with so-called small-molecule correctors, drugs that prevent degradation and enhance membrane localization of CFTR (2, 3, 8–10). Alternatively, so-called potentiators are designed to augment the function of mutant CFTR that has reached its normal location in the membrane.
PDEs, the enzymes that hydrolyze and inactivate cAMP, comprise a group of 21 genes that are divided into 11 PDE families based on kinetic, pharmacologic, and regulatory properties (11). Many PDE genes are expressed as multiple variants, giving rise to as many as 100 individual PDE proteins. Each PDE plays unique and nonoverlapping physiological and pathophysiological roles in the body by tightly controlling cAMP levels in specific subcellular compartments (11, 12). This provides the opportunity to selectively modulate microdomains of cAMP/PKA signaling by targeting individual PDEs. In the present study, we wished to determine whether selective inactivation of specific PDE subtypes could serve to augment wild-type and/or ΔF508-CFTR function in primary human airway epithelial cells, experimental models highly relevant to airway disease.
MATERIALS AND METHODS
Materials
CFTR antibodies (A1-660, A3-217 and A4-596) were kindly provided by Dr. J. R. Riordan (University of North Carolina at Chapel Hill, Chapel Hill, NC, USA) via the CFTR Antibody Distribution Program of the Cystic Fibrosis Foundation, and CFTR correctors VRT325 and VRT640, as well as the potentiator VRT532, were kindly provided by Dr. Robert Bridges (Rosalind Franklin University, North Chicago, IL, USA) via the CF Compound Distribution Program of Cystic Fibrosis Foundation Therapeutics. The PAN-PDE4 antibody K116, the PDE4D antibody M3S1, the adenovirus encoding the EPAC2 cAMP sensor, and the PDE4 expression constructs have been described previously (13–16). The GFP-CFTR expression vector and VX809 (Selleckchem, Houston, TX, USA) were kind gifts from Dr. Peter Haggie (University of California San Francisco). The phospho-Ser/Thr-PKA substrate antibody was from Cell Signaling Technology (Danvers, MA, USA) and the CFTR antibody M3A7 from Millipore (Billerica, MA, USA).
Cell culture
Primary human bronchial epithelial (pHBE) cells were isolated from tracheas and mainstem bronchi obtained from autopsies of patients without CF, and primary CF bronchial epithelial (pCFBE) cells from patients homozygous for ΔF508-CFTR (ΔF508-pCFBE cells). Cells were cultured as described previously (17). Use of human tissues for these studies was approved by the Institutional Review Board of the University of California San Francisco. 16HBE14o− (18) and CFBE41o−(+WT) cells (19) were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 30 μg/ml penicillin, 100 μg/ml streptomycin and 300 μg/ml hygromycin B [CFBE41o−(+WT) cells only] on plastic ware precoated with human fibronectin (1 mg/ml; Becton Dickinson, Franklin Lakes, NJ, USA), bovine collagen I (3 mg/ml; Becton Dickinson) and bovine serum albumin (0.1%). Calu3 and HEK293 cells were cultured in DMEM supplemented with 10% FBS, 30 μg/ml penicillin, and 100 μg/ml streptomycin. Cells were transfected with plasmids encoding PDE4s or CFTR using Effectene (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol or infected with adenoviruses at an MOI of 1000. All cells were cultured at 37°C and under a 5% CO2 atmosphere. For temperature correction, cultures were incubated for 48 h at 27°C prior to measurement of short-circuit currents, which were performed at 37°C.
Short-circuit current (Isc) measurements
Isc was measured as described previously (20). In short, epithelial cell lines or primary cells were grown on 1.12 cm2 Snapwell inserts. The filters were mounted into Ussing chambers, and a chloride gradient was applied by bathing the cells in high-chloride basolateral buffer (140 mM NaCl, 5 mM KCl, 0.36 mM K2HPO4, 0.44 mM KH2PO4, 1.3 mM CaCl2, 0.5 mM MgCl2, 4.2 mM NaHCO3, 10 mM HEPES, and 10 mM glucose, pH 7.4) and low-chloride apical buffer (133.3 mM Na-gluconate, 5 mM K-gluconate, 2.5 mM NaCl, 0.36 mM K2HPO4, 0.44 mM KH2PO4, 5.7 mM CaCl2, 0.5 mM MgCl2, 4.2 mM NaHCO3, 10 mM Hepes, and 10 mM mannitol, pH 7.4). The buffers were aerated with a mix of 95% O2 and 5% CO2 and temperature was kept at 37°C throughout the experiment. The cultures were voltage-clamped at 0 mV using an EVC4000 MultiChannel V/I Clamp (World Precision Instruments, Sarasota, FL, USA). After a 30-min stabilization period, drugs were added at specified times, while Isc was continuously recorded.
Immunocytochemistry
Cells grown on Snapwell inserts were fixed in 4% paraformaldehyde for 30 min at room temperature, followed by a 5-min incubation with 0.5% Triton X-100 in phosphate-buffered saline (PBS). Filters were subsequently blocked for 60 min at room temperature with PBS containing 10% normal goat serum, 1% bovine serum albumin, and 0.1% Triton X-100. They were then incubated for 2 h at room temperature with PAN-PDE4 antiserum (K116) or normal rabbit serum as a control and anti-CFTR antibody A1-660 or normal mouse IgG as a control, all diluted 1:500 in blocking buffer. After 3 washes with blocking buffer, the filters were incubated for 1 h with fluorescein isothiocyanate (FITC)-labeled anti-rabbit-IgG and cyanine dye 3 (Cy3)-labeled anti-mouse-IgG (Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA). Filters were then washed 3 times with blocking buffer and 1 time with PBS, dehydrated and mounted upside down in VectaShield mounting medium (Vector Laboratories, Burlingame, CA, USA) on glass slides. Images were acquired using a laser-scanning confocal microscope (Nikon, Tokyo, Japan).
Immunoprecipitation (IP)
Cells were harvested in buffer containing 20 mM HEPES (pH 7.4), 1 mM EDTA, 0.2 mM EGTA, 150 mM NaCl, 20% sucrose, 1 μM microcystin-LR, complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA), and 1% Triton X-100. After a 30-min rotation at 4°C, cell debris was pelleted with a 10-min centrifugation at 20,000 g, and soluble extracts were subjected to IP using 30 μl Protein G Sepharose and the respective antibody. After incubation for 3 h at 4°C, the resin was washed 3 times and proteins recovered in the IP pellets were detected by Western blotting or PDE activity assay.
PDE activity assay
Cyclic AMP-PDE activity was measured as described previously (13). PDE3 and PDE4 activity were defined as the fractions of total cAMP-PDE activity inhibited by 1 μM cilostamide (CIL) or 10 μM rolipram (ROL), respectively.
Data analysis
GraphPad Prism software (GraphPad Inc., San Diego, CA, USA) was used for all statistical analysis. Statistical significance was determined using unpaired Student's t test, and values of P < 0.05 were considered significant.
RESULTS
PDE4 controls basal CFTR activity in pHBE cells
As the first step to investigate the role of PDEs in the regulation of CFTR activity in human airway epithelia, we measured CFTR-dependent ISC in pHBE cells from non-CF subjects. Average basal ISC was 26.2 ± 1.0 μA/cm2 prior to and 22.1 ± 0.9 μA/cm2 after application of the epithelial sodium channel (ENaC) inhibitor amiloride (10 μM; apical; Fig. 1A). Maximal cAMP/PKA-dependent activation of CFTR by treatment with the adenylyl cyclase activator FSK (10 μM) increased basal ISC by 24.5 μA/cm2. Subsequent application of the CFTR inhibitor CFTRinh-172 (20 μM; apical) not only reversed the effect of FSK treatment but lowered ISC below baseline (Fig. 1A), which suggests that FSK-induced currents are CFTR dependent and that CFTR is already active under baseline conditions and contributes to basal/nonstimulated ISC.
Figure 1.
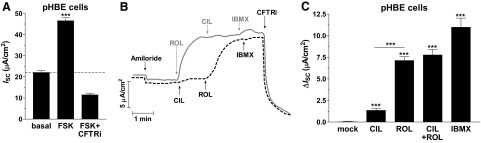
PDE4 controls basal CFTR activity in non-CF pHBE cells. A) Characterization of CFTR-dependent ISC in pHBE cells. The graph shows the average current measured after treatment with amiloride (10 μM; apical) and subsequent application of FSK (10 μM) and CFTRinh-172 (CFTRi; 20 μM) (n≥90). B) Representative ISC traces in response to treatment with the PDE3 inhibitor CIL (1 μM), the PDE4 inhibitor ROL (10 μM), the pan-selective PDE inhibitor IBMX (200 μM) and CFTRi. C) Average changes in ISC in response to treatment with PDE inhibitors (n≥14). Data in bar graphs represent means ± sem. ***P< 0.001.
Of the 11 PDE families, PDE3 and PDE4 have been shown to regulate CFTR in immortalized epithelial cell lines (21–23). Thus, we tested the role of these PDE subtypes in pHBE cells. Treatment with the PDE4-selective inhibitor ROL (10 μM) produced a substantial increase in basal CFTR-dependent ISC, whereas treatment with the PDE3-selective inhibitor CIL (1 μM), either by itself or after ROL application, produced only a minor effect (Fig. 1B,C). Inhibition of PDE4 per se induces a large portion of the CFTR-dependent ISC induced by inhibition of all cellular PDE activity using the pan-selective PDE inhibitor IBMX (200 μM), which suggests that PDE4 is the predominant PDE subtype regulating basal CFTR activity in pHBE cells. (Fig. 1C).
PDE4 inhibition augments and sustains the β-adrenergic stimulation of CFTR activity in non-CF pHBE cells
Next, we wished to probe the role of PDEs in the regulation of hormone-stimulated ISC. Given that long-acting β-adrenergic receptor (β-AR) agonists are widely used in CF therapy, we tested whether subtype-selective PDE inhibition modulates the β-AR-dependent stimulation of CFTR. In mock-treated cells, application of the β-AR agonist isoproterenol (ISO; 10 nM, basolateral) induced a submaximal stimulation of CFTR-dependent ISC, which rapidly returned to basal levels on blockade of β-AR signaling with the β-AR antagonist propranolol (PROP; 1 μM, basolateral) (Fig. 2A). Inhibition of PDE4 with ROL augmented CFTR-dependent ISC induced by ISO to the maximal activation obtained with FSK treatment (Fig. 2A, B). In addition, PDE4 inhibition delayed the return of ISC to basal levels on termination of β-AR signaling by PROP (Fig. 2B, C). Conversely, inhibition of PDE3 with CIL did not affect the amplitude or decay of ISO-induced CFTR currents in pHBE cells. (Fig. 2B, C).
Figure 2.
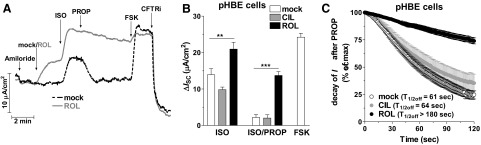
PDE4 inhibition augments and prolongs the β-AR stimulation of CFTR in non-CF pHBE cells. Cultures were stimulated with the β-AR agonist ISO (10 nM, basolateral) in the presence or absence of the PDE3 inhibitor CIL (1 μM) or the PDE4 inhibitor ROL (10 μM). When ISO-induced currents peaked, β-AR stimulation was blocked by adding the β-AR antagonist PROP (1 μM, basolateral) followed by treatment with FSK (10 μM) and CFTRinh-172 (CFTRi; 20 μM). A) Representative traces of ISC in the absence or presence of ROL. B) Average changes in ISC in response to ISO or the ISO-induced current 3 min after addition of PROP in the absence (n≥26) or presence of CIL (n≥10) or ROL (n≥33). Data represent means ± sem. C) Decay of ISC on application of PROP in the presence or absence of CIL or ROL. Time points represent means ± sem. **P< 0.01, ***P< 0.001.
PDE4 controls the activity of ΔF508-CFTR
Given the predominant role of PDE4 in regulating CFTR in non-CF cells, we wished to determine whether PDE4 also controls the activity of ΔF508-CFTR (Fig. 3). If analyzed under identical conditions as non-CF cells, ΔF508-pCFBE cells showed only a nonsignificant trend toward increased CFTR currents in response to PDE4 inhibitor and/or FSK treatment (Fig. 3A, B, D). The simplest explanation for this observation is a lack of sufficient amounts of ΔF508-CFTR at the apical membrane to produce measurable currents. Incubation at low temperature (27°C for 48 h) has been shown to increase protein expression and membrane localization of ΔF508-CFTR (24, 25). Indeed, on temperature correction, we observed consistent responses and higher amplitudes of CFTR-dependent currents in response to PDE4 inhibitor and/or FSK treatment (Fig. 3C, D). That PDE4 inhibition induced CFTR-dependent currents in temperature-corrected cells suggests that PDE4 also controls the activity of ΔF508-CFTR in CF epithelia. As low-temperature correction is not a therapeutic option, we next tested whether PDE4 regulates ΔF508-CFTR in cells that had been CFTR corrected pharmacologically with 3 structurally distinct compounds: VRT640 (Fig. 4A, B; ref. 10), VRT325 (Fig. 4C; refs. 10, 26, 27) and VX809 (Fig. 4D; ref. 28). The effect of PDE4 inhibition in small-molecule-corrected ΔF508-pCFBE cells mirrored the responses in non-CF cells (Fig. 4) except for their amplitude (Supplemental Fig. S1). PDE4 inhibition increased basal ISC, potentiated ISO-stimulated ISC, and delayed the return of ISC to basal levels on termination of β-AR stimulation (Fig. 4). CFTR function in patients homozygous for ΔF508 is ≤2% compared to that of non-CF subjects. As an increase of ΔF508-CFTR function to as little as 5% of normal levels may ameliorate the symptoms of CF (29, 30), the effects of PDE4 inhibition on ISC in ΔF508-pCFBE cells may be therapeutically relevant.
Figure 3.
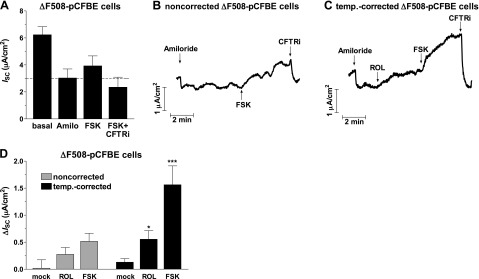
CFTR activity in noncorrected and temperature-corrected primary bronchial epithelial cells homozygous for ΔF508-CFTR (ΔF508-pCFBE cells). A) Mean ISC in noncorrected pCFBE cells under basal conditions and after subsequent addition of amiloride (Amilo; 10 μM), FSK (10 μM) and CFTRinh-172 (CFTRi; 20 μM) (n≥13). B, C) Representative traces of ISC in noncorrected (B) and temperature-corrected (C) ΔF508-pCFBE cells in response to treatment with the PDE4 inhibitor ROL (10 μM) and/or FSK. D) Average changes in ISC in response to ROL or FSK treatment in noncorrected and temperature-corrected ΔF508-pCFBE cells. Data represent means ± sem of ≥6 experiments. *P < 0.05, ***P < 0.001.
Figure 4.
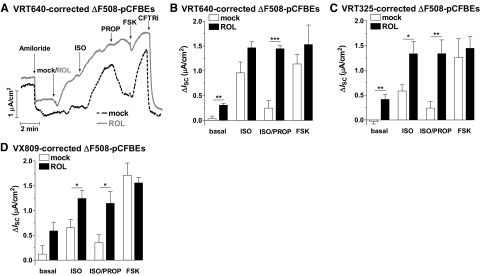
PDE4 controls CFTR activity in small-molecule-corrected ΔF508-pCFBE cell cultures. Cells were pretreated with CFTR correctors VRT640 (20 μM), VRT325 (20 μM), or VX809 (10 μM) for 48 h, and CFTR-dependent ISC was then recorded in response to subsequent treatment with the PDE4 inhibitor ROL (10 μM), ISO (10 nM, basolateral), PROP (1 μM, basolateral), FSK (10 μM), and CFTRinh-172 (CFTRi; 20 μM). A) Representative ISC traces in VRT640-corrected ΔF508-pCFBE cells in the presence or absence of ROL. B–D) Average changes in ISC in response to ROL, ISO, PROP and FSK in VRT640-corrected (n≥4; B), VRT325-corrected (n≥8; C) and VX809-corrected (n≥5; D) ΔF508-pCFBE cells. Data represent means ± sem. *P< 0.05, **P< 0.01, ***P< 0.001.
PDE4 inhibitors synergize with CFTR correctors and potentiators in rescuing ΔF508-CFTR function
To further maximize ΔF508-CFTR function, we tested the effect of PDE4 inhibition in combination with CFTR correctors, as well as a CFTR potentiator, VRT532 (20 μM; ref. 26). Potentiators, such as VRT532, augment channel gating beyond the physiological range but require prior PKA phosphorylation of CFTR for efficacy. As a result, treatment with VRT532 or maximizing PKA phosphorylation of CFTR with FSK by itself induces only minor currents, whereas combining both treatments reveals substantial synergy (Supplemental Fig. S2). This synergy in augmenting CFTR function is apparent in noncorrected cells (Supplemental Fig. S2A, C) but is further enhanced by small-molecule CFTR correction with VX809 (Supplemental Fig. S2B, C). Next, we probed the effect of PDE3 and/or PDE4 inhibition in VX809-corrected ΔF508-pCFBE cells in the presence of the potentiator VRT532. Similar to its effects in non-CF cells (Figs. 1 and 2), PDE4 inhibition with ROL increased basal CFTR-dependent ISC (Fig. 5A, B), potentiated ISO-stimulated ISC (Fig. 5A, B), and delayed the return of ISC to basal levels on termination of β-AR stimulation (Fig. 5). Conversely, inhibition of PDE3 with CIL had no effect on CFTR function (Fig. 5B, C).
Figure 5.
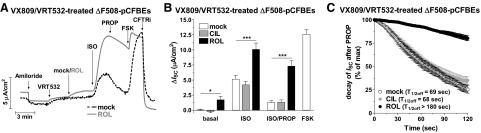
PDE4 inhibition synergizes with CFTR correctors and potentiators to stimulate CFTR in ΔF508-pCFBE cells. Cultures were treated with CFTR corrector VX809 (10 μM) for 48 h, and CFTR-dependent ISC was then recorded in response to subsequent treatment with the CFTR potentiator VRT532 (20 μM), followed by the PDE3 inhibitor CIL (1 μM), the PDE4 inhibitor ROL (10 μM) or solvent (mock treatment) and subsequent treatment with ISO (10 nM, basolateral), PROP (1 μM, basolateral), FSK (10 μM) and CFTRinh-172 (CFTRi; 20 μM). A) Representative ISC traces in VX809/VRT532-treated ΔF508-pCFBE cells in the presence or absence of ROL. B) Average change in ISC in response to CIL, ROL, ISO, PROP and FSK in VX809/VRT532-treated ΔF508-pCFBE cells. Data represent means ± sem. C) Decay of ISC on application of PROP in the absence (mock; n=17) or presence of CIL (n=15) or ROL (n=22). Time points represent means ± sem. *P< 0.05, ***P< 0.001.
Expression and subcellular localization of PDE4 in airway epithelial cells
Recent reports indicate that CFTR deficiency in CF may trigger adaptive changes in the cAMP signaling machinery in airway epithelia (31, 32). To probe whether changes in PDE expression might contribute to this effect, we measured total and subtype-selective PDE activity in airway epithelial cells from control subjects and patients with CF. We found that total PDE activity, as well as PDE3 and PDE4 activity, are expressed at comparable levels in both non-CF and CF cells (Fig. 6) implying that the function of these PDEs might be unaffected by CFTR deficiency in CF cells.
Figure 6.

Pattern of PDE expression in pHBE cells. Detergent extracts prepared from non-CF pHBE and ΔF508-pCFBE cells were subjected to PDE activity assays. Total cAMP-PDE activity is defined as the activity measured in the absence of PDE inhibitors (A), whereas PDE3 and PDE4 activities (B) are defined as the fraction of activity inhibited by CIL (1 μM) and ROL (10 μM), respectively. Data represent means ± sem of ≥3 experiments.
CFTR function is controlled by macromolecular signaling complexes that sequester many components of the cAMP signaling machinery in the vicinity of the CFTR (33), and these might also recruit PDEs to locally regulate cAMP levels (22, 34). Although PDE4 contributes a major portion of total cAMP-PDE activity in primary airway epithelial cells (Fig. 6B), several findings suggest that PDE4s are recruited to CFTR signaling complexes and control CFTR function through a local regulation of cAMP/PKA activity in the vicinity of the CFTR. First, PDE4s are not evenly distributed throughout the cell but are highly enriched at the apical membrane of primary airway epithelia as shown by immunocytochemistry (Fig. 7A) providing a first indication of PDE4 sequestration into the vicinity of the CFTR. To probe for a physical interaction between CFTR and PDE4s we next performed coimmunoprecipitation (co-IP) experiments. The amount of cells/protein that can be obtained from primary cultures of airway epithelial cells is not sufficient for these kinds of biochemical approaches. However, we can detect CFTR/PDE4 complexes by co-IP of the endogenous CFTR from airway epithelial cell lines, such as Calu3 (Fig. 7B). In addition, we observe interaction between exogenous PDE4 and CFTR in the HEK293 overexpression system (Fig. 7C).
Figure 7.

Subcellular compartmentalization of PDE4 with CFTR. A) Subcellular localization of CFTR and PDE4 in pHBE cells, as detected by immunocytochemistry with antibodies against CFTR (A1-660; red) and PDE4 (K116; green). Top panels; confocal x-y images from just below the apical membrane. Bottom panels: x-z images generated by 3D reconstruction. B) Detergent extracts prepared from Calu3 airway epithelial cells were subjected to IP with the CFTR antibody M3A7. The graph shows the cAMP-PDE activity recovered in IP pellets measured in the presence or absence of the PDE4 inhibitor ROL (10 μM). C) Detergent extracts prepared from HEK293 cells expressing GFP-tagged CFTR either by itself or coexpressing GFP-CFTR and exogenous PDE4D5 were subjected to IP of PDE4D. Images show the amount of GFP-CFTR recovered in PDE4D IP pellets, as detected by immunoblotting with CFTR antibody A4-596.
Compartmentalized PDE4 regulates CFTR in airway epithelial cells
To further confirm our hypothesis that PDE4 regulates CFTR function by controlling its PKA phosphorylation and does so in a compartmentalized fashion by regulating local PKA activity in the vicinity of the channel, we probed the role of PDE4 in two human airway epithelial cell lines, 16HBE14o− and CFBE41o−(+WT) cells (Fig. 8), which express higher levels of CFTR and are less resistant to adenovirus infection compared to primary cell cultures. In both cell lines, PDE4, but not PDE3, is the main regulator of CFTR function, as determined by measurements of CFTR-dependent ISC (Fig. 8B, C, E). In addition, inhibition of PDE4, but not inhibition of PDE3, strongly augments PKA-phosphorylation of CFTR, as measured by immunoblots with phospho-PKA substrate antibodies (Fig. 8D, F). This latter finding supports the idea that PDE4 regulates CFTR-dependent chloride transport by controlling the PKA-dependent activation of this channel. PDE4 plays a critical role in regulating CFTR phosphorylation and CFTR-dependent currents in CFBE41o−(+WT) cells despite the fact that PDE4s represent a minor fraction of total PDE activity (∼25%) in these cells (Fig. 8G), indicating a local role of PDE4 in regulating CFTR. In addition, expression of a catalytically active PDE4 (PDE4-WT) reduced PKA-phosphorylation of CFTR in CFBE41o−(+WT) cells, whereas expression of a catalytically inactive, dominant-negative PDE4 (PDE4-DN), which functions by displacing endogenous PDE4s from macromolecular signaling complexes (35), promotes PKA phosphorylation of CFTR (Fig. 8H). The latter findings further support the idea of a compartmentalized function of PDE4 in controlling CFTR via regulation of local cAMP/PKA levels.
Figure 8.
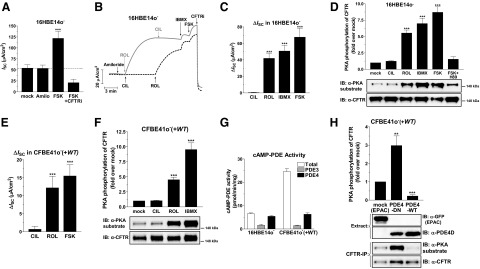
PDE4 controls PKA phosphorylation of CFTR and CFTR-dependent ISC in 16HBE14o− and CFBE41o−(+WT) airway epithelial cell lines. Panels show CFTR-dependent ISC and measurements of PKA phosphorylation of CFTR in 16HBE14o− (A–D) and CFBE41o−(+WT) (E, F, H) airway epithelial cell lines, respectively. A) Average current measured at baseline (mock) and after consecutive application of amiloride (Amilo; 10 μM), FSK (10 μM) and CFTRinh-172 (CFTRi; 20 μM). B) Representative ISC traces in response to subsequent treatment of 16HBE14o− cells with the PDE3 inhibitor CIL (1 μM), the PDE4 inhibitor ROL (10 μM), the pan-selective PDE inhibitor IBMX (200 μM), FSK, and CFTRi. C) Average increase in ISC in response to PDE inhibitors and FSK in 16HBE14o− cells. D) 16HBE14o− cells, pretreated for 1 h in the presence or absence of the PKA inhibitor H89 (20 μM), were treated for 5 min with PDE inhibitors or FSK prior to cell lysis and IP of CFTR using antibody A3-217. Panel shows representative immunoblots for CFTR protein (antibody A4-596) and PKA-phosphorylation levels of CFTR recovered in IP pellets. E) Average ISC induced by PDE inhibitor treatment or FSK in CFBE41o−(+WT) cells. F) Quantification of PKA phosphorylation of CFTR induced by treatment of CFBE41o−(+WT) cells with PDE inhibitors. G) Comparison of total and subtype-specific cAMP-PDE activity in detergent extracts of 16HBE14o− and CFBE41o−(+WT) cells. H) CFBE41o−(+WT) airway epithelial cells were infected with adenovirus encoding catalytically active PDE4D5 (PDE4-WT), catalytically inactive PDE4D5 (PDE4-DN) or the EPAC2 cAMP sensor (EPAC/mock) as a control. Cells were lysed 48 h after infection, and the resulting lysates were subjected to IP of CFTR using antibody A3-217. PKA-phosphorylation and protein levels of CFTR recovered in IP pellets was detected by immunoblotting with PKA substrate antibodies and CFTR antibody A4-596, respectively. Graphs show means ± sem of ≥3 experiments. **P< 0.01, ***P< 0.001.
DISCUSSION
Targeting PDE4 to correct CFTR hypofunction in CF
In the present study, we have identified PDE4 as a critical regulator of CFTR activity in pHBE cells from control subjects and patients with CF. Previous reports indicated that treatment with adenylyl cyclase activators and/or broad-spectrum PDE inhibitors was ineffective in restoring CFTR-dependent ion transport in cells expressing endogenous ΔF508-CFTR (6, 7), and this lack of efficacy discouraged development of drugs targeting the cAMP/PKA pathway. Our current findings challenge this conclusion. In ΔF508-CF cells, though PDE4 inhibition alone produced minimal channel activation (Fig. 3), PDE4 inhibition strongly amplified the effects of CFTR correctors and/or CFTR potentiators (Figs. 4 and 5). Thus, augmentation of cAMP/PKA signaling via PDE4 inhibition can serve to maximize the therapeutic benefits derived from CFTR correctors and/or potentiators that are currently in development and vice versa. Different correctors augment CFTR expression and membrane localization via distinct mechanism of action, including effects on protein folding, degradation and/or trafficking (36, 37). Our observation that PDE4 inhibition is effective in ΔF508-CF epithelia treated with 3 structurally distinct CFTR correctors (Fig. 4), suggests that combination treatment with a PDE4 inhibitor might potentiate the effects of a range of mechanistically diverse drugs. The same might apply to CFTR potentiators. Indeed, similar to the CFTR potentiator VRT532 used in the present study, the efficacy of the most clinically advanced CFTR potentiator, VX770/Ivacaftor, also depends on prior cAMP/PKA activation of CFTR (2, 38, 39).
CFTR correctors and potentiators restore little CFTR conductance in the absence of cAMP/PKA stimuli (Fig. 5A and Supplemental Fig. S2). Thus, given the transient nature of second messenger signaling, cellular cAMP/PKA activity and, hence, PKA phosphorylation of CFTR and the efficacy of correctors and potentiators to augment CFTR function are likely never saturated in vivo. This provides an opportunity for the application of cAMP activators, such as PDE4 inhibitors, to enhance PKA phosphorylation and function of CFTR. Individuals heterozygous for the ΔF508-CFTR mutation do not develop CF, which suggests that ≤50% of normal CFTR function would be sufficient to cure the disease (also see ref. 40). PDE4 inhibition augments hormone-stimulated CFTR currents in VX809/VRT532-corrected cells (Fig. 5A, B) to ∼25% of the maximal CFTR current measured in non-CF cells (Fig. 1A), which suggests that cAMP augmentation via PDE4 inhibition contributes to restoring therapeutically significant levels of CFTR function. Notably, given that a first PDE4 inhibitor, roflumilast (41, 42), has recently been approved for use in humans, augmentation of cAMP/PKA signaling via PDE4 inhibition represents a viable therapeutic approach, whereas treatment with saturating concentrations of FSK or Gs-protein-coupled receptor (GsPCR) agonists, which are commonly applied in in vitro assays measuring CFTR function, does not.
Previous reports implied a role for PDE3 in the regulation of CFTR in airway epithelia as the PDE3 inhibitor milrinone (100 μM) was shown to stimulate CFTR function (4, 43). Conversely, we observe only minimal effects of PDE3 inhibition in our hands. A possible explanation for this discrepancy is the fact that milrinone at a concentration of 100 μM is rather nonselective (IC50 for PDE3 and PDE4 is 0.5 and 15 μM, respectively) and may also produce off-target effects unrelated to PDE inhibition (44).
Local role for PDE4 in regulating CFTR
To increase CFTR signaling specificity and efficiency, large macromolecular signaling complexes localize the channel at the epithelial apical membrane and sequester key players involved in its cAMP-dependent regulation (45). These include GsPCRs, such as β-ARs, the cAMP effector PKA, and protein phosphatases that counter the action of PKA by dephosphorylating CFTR (22, 33, 34, 46, 47). Our finding that PDE4 is sequestered to the apical membrane of pHBE cells (Fig. 7A) and physically interacts with CFTR in airway epithelial cells, as well as in overexpression systems (Fig. 7B, C), supports the hypothesis that these complexes also sequester a pool of PDE that controls CFTR function via regulation of local cAMP/PKA levels (22, 23, 34). Inactivation of this pool of PDE should be sufficient to augment CFTR function without the need to increase global cellular cAMP levels. This idea is confirmed by the observation that PDE4 inhibition exerts a predominant effect on CFTR phosphorylation and function in CFBE41o−(+WT) cells (Fig. 8E, F), despite representing a minor fraction of total cellular PDE activity (Fig. 8G), and that disruption of PDE4 complexes via overexpression of dominant-negative PDE4 constructs augments PKA phosphorylation of the channel (Fig. 8H).
The PDE4 family consists of 4 genes, PDE4A to PDE4D. Which particular PDE4 subtype or subtypes controls CFTR function in primary human epithelia remains to be determined. Besides the variant PDE4D5 shown in Fig. 7C, other PDE4 variants, such as PDE4A4, PDE4B3, and PDE4D9, also coimmunoprecipitate with CFTR in the HEK293 overexpression system (data not shown) suggesting that either multiple PDE4s can independently interact with the channel, or at least one domain conserved among PDE4 subtypes [such as the upstream conserved regions (UCRs) or the catalytic domain; ref. 48] mediates binding to the CFTR, a pattern previously observed for other PDE4 interacting proteins, such as receptor for activated C kinase 1 (RACK1) or β-arrestin (49).
PDE4 as a therapeutic target in CF and chronic obstructive pulmonary disease (COPD)
Pan-selective PDE4 inhibitors produce a range of physiological responses; most notably anti-inflammatory (12, 50) as well as potent memory- and cognition-enhancing effects (51). A first PDE4 inhibitor, roflumilast, has recently been approved for treatment of COPD (41, 42). The therapeutic benefits of the drug are thought to be due to its anti-inflammatory properties. However, exposure to cigarette smoke, a major underlying cause of COPD, has been shown to impair CFTR function (52) by inducing CFTR internalization (53) and/or reducing CFTR expression (54), and smoking-induced CFTR hypofunction has been causally linked to mucus stasis and COPD pathogenesis (53–55). Thus, in light of our finding that PDE4 inhibitors augment CFTR function in non-CF airway epithelia, it is possible that the therapeutic benefits of PDE4 inhibitors in COPD may not only result from their anti-inflammatory properties but be derived in part from correcting CFTR hypofunction, thereby restoring airway surface liquid and facilitating mucus clearance.
That CF and severe COPD share a number of clinical manifestations (e.g., CFTR hypofunction, mucus hypersecretion, airway infection, and hyperinflammation) also implies a therapeutic potential for PDE4 inhibitors in CF therapy. By restoring CFTR function and by attenuating inflammatory responses, PDE4 inhibitors may exert two independent therapeutic benefits in CF; the former ameliorating the underlying cause of the disease, the latter serving to break the vicious cycle of airway obstruction, chronic infection, and hyperinflammation, which is ultimately responsible for tissue destruction, loss of respiratory function, and mortality in CF (56–58).
Development of pan-selective PDE4 inhibitors for inflammatory diseases (59–61) has been hampered by their significant side effects (e.g., emesis, nausea, diarrhea, and weight loss), often limiting the doses that could be administered below therapeutically effective levels. Although still associated with these side effects (41, 42), roflumilast partially overcomes this hurdle through its unique pharmacokinetic properties (62, 63). Given that individual PDE4 subtypes and variants serve unique and nonoverlapping physiological functions, development of subtype-selective PDE4 inhibitors has been proposed as a strategy to separate the therapeutically beneficial from the side effects of nonselective PDE4 inhibitors (51, 61, 64, 65). Our finding that PDE4s are sequestered via physical interactions to the CFTR (Fig. 7) and that displacement of PDE4 from the CFTR signaling complex can serve to modulate CFTR activity (Fig. 8) provides impetus for the development of small-molecule disruptors of CFTR/PDE4 complexes as CF therapeutics with reduced side effects compared to active site-directed inhibitors of PDE activity.
Supplementary Material
Acknowledgments
This work was supported by grants from the Cystic Fibrosis Foundation (pilot grant to W.R.; RDP R613-CR11 to A.S.V. and W.E.F.) and grants from the U.S. National Institutes of Health (HL107960 to W.R.; HL0927088 to M.C.; DK072517 to A.S.V and W.E.F.). E.B. is supported by an Elizabeth Nash Postdoctoral Fellowship from Cystic Fibrosis Research Inc. (CFRI), and D.C.G. was supported, in part, by funds from Pennsylvania Cystic Fibrosis, Inc.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- β-AR
- β-adrenergic receptor
- CF
- cystic fibrosis
- CFTR
- cystic fibrosis transmembrane conductance regulator
- CIL
- cilostamide
- co-IP
- coimmunoprecipitation
- COPD
- chronic obstructive pulmonary disease
- FSK
- forskolin
- IBMX
- 3-isobutyl-1-methylxanthine
- IP
- immunoprecipitation
- ISC
- short-circuit current
- ISO
- isoproterenol
- PDE
- cyclic nucleotide phosphodiesterase
- pCFBE
- primary cystic fibrosis bronchial epithelial
- pHBE
- primary human bronchial epithelial
- PKA
- protein kinase A
- PROP
- propranolol
- ROL
- rolipram
REFERENCES
- 1. Verkman A. S., Galietta L. J. (2009) Chloride channels as drug targets. Nat. Rev. Drug Discov. 8, 153–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim Chiaw P., Eckford P. D., Bear C. E. (2011) Insights into the mechanisms underlying CFTR channel activity, the molecular basis for cystic fibrosis and strategies for therapy. Essays Biochem. 50, 233–248 [DOI] [PubMed] [Google Scholar]
- 3. Lukacs G. L., Verkman A. S. (2012) CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol. Med. 18, 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kelley T. J., Al-Nakkash L., Cotton C. U., Drumm M. L. (1996) Activation of endogenous deltaF508 cystic fibrosis transmembrane conductance regulator by phosphodiesterase inhibition. J. Clin. Invest. 98, 513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Powell K., Zeitlin P. L. (2002) Therapeutic approaches to repair defects in deltaF508 CFTR folding and cellular targeting. Adv. Drug Deliv. Rev. 54, 1395–1408 [DOI] [PubMed] [Google Scholar]
- 6. Schultz B. D., Frizzell R. A., Bridges R. J. (1999) Rescue of dysfunctional deltaF508-CFTR chloride channel activity by IBMX. J. Membr. Biol. 170, 51–66 [DOI] [PubMed] [Google Scholar]
- 7. Grubb B., Lazarowski E., Knowles M., Boucher R. (1993) Isobutylmethylxanthine fails to stimulate chloride secretion in cystic fibrosis airway epithelia. Am. J. Respir. Cell Mol. Biol. 8, 454–460 [DOI] [PubMed] [Google Scholar]
- 8. Amaral M. D. (2011) Targeting CFTR: how to treat cystic fibrosis by CFTR-repairing therapies. Curr. Drug Targets 12, 683–693 [DOI] [PubMed] [Google Scholar]
- 9. Pettit R. S. (2012) Cystic fibrosis transmembrane conductance regulator-modifying medications: the future of cystic fibrosis treatment. Ann. Pharmacother. 46, 1065–1075 [DOI] [PubMed] [Google Scholar]
- 10. Rowe S. M., Pyle L. C., Jurkevante A., Varga K., Collawn J., Sloane P. A., Woodworth B., Mazur M., Fulton J., Fan L., Li Y., Fortenberry J., Sorscher E. J., Clancy J. P. (2010) DeltaF508 CFTR processing correction and activity in polarized airway and non-airway cell monolayers. Pulm. Pharmacol. Ther. 23, 268–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conti M., Beavo J. (2007) Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 76, 481–511 [DOI] [PubMed] [Google Scholar]
- 12. Jin S. L., Ding S. L., Lin S. C. (2012) Phosphodiesterase 4 and its inhibitors in inflammatory diseases. Chang Gung Med. J. 35, 197–210 [DOI] [PubMed] [Google Scholar]
- 13. Richter W., Conti M. (2002) Dimerization of the type 4 cAMP-specific phosphodiesterases is mediated by the upstream conserved regions (UCRs). J. Biol. Chem. 277, 40212–40221 [DOI] [PubMed] [Google Scholar]
- 14. Richter W., Conti M. (2004) The oligomerization state determines regulatory properties and inhibitor sensitivity of type 4 cAMP-specific phosphodiesterases. J. Biol. Chem. 279, 30338–30348 [DOI] [PubMed] [Google Scholar]
- 15. Richter W., Day P., Agrawal R., Bruss M. D., Granier S., Wang Y. L., Rasmussen S. G., Horner K., Wang P., Lei T., Patterson A. J., Kobilka B., Conti M. (2008) Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 27, 384–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richter W., Jin S. L., Conti M. (2005) Splice variants of the cyclic nucleotide phosphodiesterase PDE4D are differentially expressed and regulated in rat tissue. Biochem. J. 388, 803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L., Gallup M., Zlock L., Finkbeiner W. E., McNamara N. A. (2013) Rac1 and Cdc42 differentially modulate cigarette smoke-induced airway cell migration through p120-catenin-dependent and -independent pathways. Am. J. Pathol. 182, 1986–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cozens A. L., Yezzi M. J., Kunzelmann K., Ohrui T., Chin L., Eng K., Finkbeiner W. E., Widdicombe J. H., Gruenert D. C. (1994) CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 10, 38–47 [DOI] [PubMed] [Google Scholar]
- 19. Illek B., Maurisse R., Wahler L., Kunzelmann K., Fischer H., Gruenert D. C. (2008) Cl transport in complemented CF bronchial epithelial cells correlates with CFTR mRNA expression levels. Cell. Physiol. Biochem. 22, 57–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie M., Rich T. C., Scheitrum C., Conti M., Richter W. (2011) Inactivation of multidrug resistance proteins disrupts both cellular extrusion and intracellular degradation of cAMP. Mol. Pharmacol. 80, 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu S., Veilleux A., Zhang L., Young A., Kwok E., Laliberte F., Chung C., Tota M. R., Dube D., Friesen R. W., Huang Z. (2005) Dynamic activation of cystic fibrosis transmembrane conductance regulator by type 3 and type 4D phosphodiesterase inhibitors. J. Pharmacol. Exp. Ther. 314, 846–854 [DOI] [PubMed] [Google Scholar]
- 22. Penmatsa H., Zhang W., Yarlagadda S., Li C., Conoley V. G., Yue J., Bahouth S. W., Buddington R. K., Zhang G., Nelson D. J., Sonecha M. D., Manganiello V., Wine J. J., Naren A. P. (2010) Compartmentalized cyclic adenosine 3′,5′-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol. Biol. Cell 21, 1097–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barnes A. P., Livera G., Huang P., Sun C., O'Neal W. K., Conti M., Stutts M. J., Milgram S. L. (2005) Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J. Biol. Chem. 280, 7997–8003 [DOI] [PubMed] [Google Scholar]
- 24. LeSimple P., Liao J., Robert R., Gruenert D. C., Hanrahan J. W. (2010) Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J. Physiol. 588, 1195–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng J., Cebotaru V., Cebotaru L., Guggino W. B. (2010) Syntaxin 6 and CAL mediate the degradation of the cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 21, 1178–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Van Goor F., Straley K. S., Cao D., Gonzalez J., Hadida S., Hazlewood A., Joubran J., Knapp T., Makings L. R., Miller M., Neuberger T., Olson E., Panchenko V., Rader J., Singh A., Stack J. H., Tung R., Grootenhuis P. D., Negulescu P. (2006) Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L1117–L1130 [DOI] [PubMed] [Google Scholar]
- 27. Loo T. W., Bartlett M. C., Wang Y., Clarke D. M. (2006) The chemical chaperone CFcor-325 repairs folding defects in the transmembrane domains of CFTR-processing mutants. Biochem. J. 395, 537–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Stack J. H., Straley K. S., Decker C. J., Miller M., McCartney J., Olson E. R., Wine J. J., Frizzell R. A., Ashlock M., Negulescu P. A. (2011) Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U. S. A. 108, 18843–18848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amaral M. D. (2005) Processing of CFTR: traversing the cellular maze–how much CFTR needs to go through to avoid cystic fibrosis? Pediatr. Pulmonol. 39, 479–491 [DOI] [PubMed] [Google Scholar]
- 30. Ramalho A. S., Beck S., Meyer M., Penque D., Cutting G. R., Amaral M. D. (2002) Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 27, 619–627 [DOI] [PubMed] [Google Scholar]
- 31. Monterisi S., Favia M., Guerra L., Cardone R. A., Marzulli D., Reshkin S. J., Casavola V., Zaccolo M. (2012) CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J. Cell Sci. 125, 1106–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Watson M. J., Worthington E. N., Clunes L. A., Rasmussen J. E., Jones L., Tarran R. (2011) Defective adenosine-stimulated cAMP production in cystic fibrosis airway epithelia: a novel role for CFTR in cell signaling. FASEB J. 25, 2996–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li C., Naren A. P. (2005) Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol. Ther. 108, 208–223 [DOI] [PubMed] [Google Scholar]
- 34. Lee J. H., Richter W., Namkung W., Kim K. H., Kim E., Conti M., Lee M. G. (2007) Dynamic regulation of cystic fibrosis transmembrane conductance regulator by competitive interactions of molecular adaptors. J. Biol. Chem. 282, 10414–10422 [DOI] [PubMed] [Google Scholar]
- 35. Richter W., Mika D., Blanchard E., Day P., Conti M. (2013) beta1-adrenergic receptor antagonists signal via PDE4 translocation. EMBO Rep. 14, 276–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Farinha C. M., King-Underwood J., Sousa M., Correia A. R., Henriques B. J., Roxo-Rosa M., Da Paula A. C., Williams J., Hirst S., Gomes C. M., Amaral M. D. (2013) Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR Rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 20, 943–955 [DOI] [PubMed] [Google Scholar]
- 37. Okiyoneda T., Veit G., Dekkers J. F., Bagdany M., Soya N., Xu H., Roldan A., Verkman A. S., Kurth M., Simon A., Hegedus T., Beekman J. M., Lukacs G. L. (2013) Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 9, 444–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Cao D., Neuberger T., Turnbull A., Singh A., Joubran J., Hazlewood A., Zhou J., McCartney J., Arumugam V., Decker C., Yang J., Young C., Olson E. R., Wine J. J., Frizzell R. A., Ashlock M., Negulescu P. (2009) Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U. S. A. 106, 18825–18830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McPhail G. L., Clancy J. P. (2013) Ivacaftor: the first therapy acting on the primary cause of cystic fibrosis. Drugs Today 49, 253–260 [DOI] [PubMed] [Google Scholar]
- 40. Stoltz D. A., Rokhlina T., Ernst S. E., Pezzulo A. A., Ostedgaard L. S., Karp P. H., Samuel M. S., Reznikov L. R., Rector M. V., Gansemer N. D., Bouzek D. C., Alaiwa M. H., Hoegger M. J., Ludwig P. S., Taft P. J., Wallen T. J., Wohlford-Lenane C., McMenimen J. D., Chen J. H., Bogan K. L., Adam R. J., Hornick E. E., Nelson G. A. t., Hoffman E. A., Chang E. H., Zabner J., McCray P. B., Jr., Prather R. S., Meyerholz D. K., Welsh M. J. (2013) Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J. Clin. Invest. 123, 2685–2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Calverley P. M., Rabe K. F., Goehring U. M., Kristiansen S., Fabbri L. M., Martinez F. J. (2009) Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 374, 685–694 [DOI] [PubMed] [Google Scholar]
- 42. Fabbri L. M., Calverley P. M., Izquierdo-Alonso J. L., Bundschuh D. S., Brose M., Martinez F. J., Rabe K. F. (2009) Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with long acting bronchodilators: two randomised clinical trials. Lancet 374, 695–703 [DOI] [PubMed] [Google Scholar]
- 43. Kelley T. J., al-Nakkash L., Drumm M. L. (1995) CFTR-mediated chloride permeability is regulated by type III phosphodiesterases in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 13, 657–664 [DOI] [PubMed] [Google Scholar]
- 44. Shakur Y., Fong M., Hensley J., Cone J., Movsesian M. A., Kambayashi J., Yoshitake M., Liu Y. (2002) Comparison of the effects of cilostazol and milrinone on cAMP-PDE activity, intracellular cAMP and calcium in the heart. Cardiovasc. Drugs Ther. 16, 417–427 [DOI] [PubMed] [Google Scholar]
- 45. Guggino W. B., Stanton B. A. (2006) New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat. Rev. Mol. Cell Biol. 7, 426–436 [DOI] [PubMed] [Google Scholar]
- 46. Huang P., Trotter K., Boucher R. C., Milgram S. L., Stutts M. J. (2000) PKA holoenzyme is functionally coupled to CFTR by AKAPs. Am. J. Physiol. Cell Physiol. 278, C417–422 [DOI] [PubMed] [Google Scholar]
- 47. Thelin W. R., Kesimer M., Tarran R., Kreda S. M., Grubb B. R., Sheehan J. K., Stutts M. J., Milgram S. L. (2005) The cystic fibrosis transmembrane conductance regulator is regulated by a direct interaction with the protein phosphatase 2A. J. Biol. Chem. 280, 41512–41520 [DOI] [PubMed] [Google Scholar]
- 48. Conti M., Richter W., Mehats C., Livera G., Park J. Y., Jin C. (2003) Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J. Biol. Chem. 278, 5493–5496 [DOI] [PubMed] [Google Scholar]
- 49. Bolger G. B., Baillie G. S., Li X., Lynch M. J., Herzyk P., Mohamed A., Mitchell L. H., McCahill A., Hundsrucker C., Klussmann E., Adams D. R., Houslay M. D. (2006) Scanning peptide array analyses identify overlapping binding sites for the signalling scaffold proteins, beta-arrestin and RACK1, in cAMP-specific phosphodiesterase PDE4D5. Biochem. J. 398, 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Page C. P., Spina D. (2011) Phosphodiesterase inhibitors in the treatment of inflammatory diseases. Handbook Exp. Pharmacol. 204, 391–414 [DOI] [PubMed] [Google Scholar]
- 51. Richter W., Menniti F. S., Zhang H. T., Conti M. (2013) PDE4 as a target for cognition enhancement. Expert Opin. Ther. Targets, 17 1011–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cantin A. M., Hanrahan J. W., Bilodeau G., Ellis L., Dupuis A., Liao J., Zielenski J., Durie P. (2006) Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am. J. Respir. Crit. Care. Med. 173, 1139–1144 [DOI] [PubMed] [Google Scholar]
- 53. Clunes L. A., Davies C. M., Coakley R. D., Aleksandrov A. A., Henderson A. G., Zeman K. L., Worthington E. N., Gentzsch M., Kreda S. M., Cholon D., Bennett W. D., Riordan J. R., Boucher R. C., Tarran R. (2012) Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 26, 533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sloane P. A., Shastry S., Wilhelm A., Courville C., Tang L. P., Backer K., Levin E., Raju S. V., Li Y., Mazur M., Byan-Parker S., Grizzle W., Sorscher E. J., Dransfield M. T., Rowe S. M. (2012) A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS One 7, e39809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dransfield M. T., Wilhelm A. M., Flanagan B., Courville C., Tidwell S. L., Raju S. V., Gaggar A., Steele C., Tang L. P., Liu B., Rowe S. M. (2013) Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 144, 498–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Elizur A., Cannon C. L., Ferkol T. W. (2008) Airway inflammation in cystic fibrosis. Chest 133, 489–495 [DOI] [PubMed] [Google Scholar]
- 57. Nichols D., Chmiel J., Berger M. (2008) Chronic inflammation in the cystic fibrosis lung: alterations in inter- and intracellular signaling. Clin. Rev. Allergy Immunol. 34, 146–162 [DOI] [PubMed] [Google Scholar]
- 58. Nichols D. P., Konstan M. W., Chmiel J. F. (2008) Anti-inflammatory therapies for cystic fibrosis-related lung disease. Clin. Rev. Allergy Immunol. 35, 135–153 [DOI] [PubMed] [Google Scholar]
- 59. Francis S. H., Houslay M. D., Conti M. (2011) Phosphodiesterase inhibitors: factors that influence potency, selectivity, and action. Handbook Exp. Pharmacol. 204, 47–84 [DOI] [PubMed] [Google Scholar]
- 60. Houslay M. D., Schafer P., Zhang K. Y. (2005) Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov. Today 10, 1503–1519 [DOI] [PubMed] [Google Scholar]
- 61. Zhang K. Y., Ibrahim P. N., Gillette S., Bollag G. (2005) Phosphodiesterase-4 as a potential drug target. Expert Opin. Ther. Targets 9, 1283–1305 [DOI] [PubMed] [Google Scholar]
- 62. Giembycz M. A., Field S. K. (2010) Roflumilast: first phosphodiesterase 4 inhibitor approved for treatment of COPD. Drug Des. Devel. Ther. 4, 147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hatzelmann A., Morcillo E. J., Lungarella G., Adnot S., Sanjar S., Beume R., Schudt C., Tenor H. (2010) The preclinical pharmacology of roflumilast—a selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 23, 235–256 [DOI] [PubMed] [Google Scholar]
- 64. Giembycz M. A. (2008) Can the anti-inflammatory potential of PDE4 inhibitors be realized: guarded optimism or wishful thinking? Br. J. Pharmacol. 155, 288–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Srivani P., Usharani D., Jemmis E. D., Sastry G. N. (2008) Subtype selectivity in phosphodiesterase 4 (PDE4): a bottleneck in rational drug design. Curr. Pharm. Des. 14, 3854–3872 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.