

Abstract
Children who are exposed to environmental respiratory insults often develop asthma that persists into adulthood. In this study, we used a neonatal mouse model of ovalbumin (OVA)-induced allergic airway inflammation to understand the long-term effects of early childhood insults on airway structure and function. We showed that OVA sensitization and challenge in early life led to a 2-fold increase in airway smooth muscle (ASM) innervation (P<0.05) and persistent airway hyperreactivity (AHR). In contrast, OVA exposure in adult life elicited short-term AHR without affecting innervation levels. We found that postnatal ASM innervation required neurotrophin (NT)-4 signaling through the TrkB receptor and that early-life OVA exposure significantly elevated NT4 levels and TrkB signaling by 5- and 2-fold, respectively, to increase innervation. Notably, blockade of NT4/TrkB signaling in OVA-exposed pups prevented both acute and persistent AHR without affecting baseline airway function or inflammation. Furthermore, biophysical assays using lung slices and isolated cells demonstrated that NT4 was necessary for hyperreactivity of ASM induced by early-life OVA exposure. Together, our findings show that the NT4/TrkB-dependent increase in innervation plays a critical role in the alteration of the ASM phenotype during postnatal growth, thereby linking early-life allergen exposure to persistent airway dysfunction.—Aven, L., Paez-Cortez, J., Achey, R., Krishnan, R., Ram-Mohan, S., Cruikshank, W. W., Fine, A., Ai X. An NT4/TrkB-dependent increase in innervation links early-life allergen exposure to persistent airway hyperreactivity.
Keywords: smooth muscle, ASM, childhood asthma, innervation, contractility, lung slice
Asthma is a chronic inflammatory airway disease with increasing prevalence despite improved treatment. It is one of the leading causes of missed days from work and school. Patients with asthma present common symptoms of airflow obstruction, wheezing, and coughing. In asthma, the airway smooth muscle (ASM) undergoes profound phenotypic changes. These include hypertrophy, hyperplasia, and, notably, airway hyperreactivity (AHR) in response to nonspecific and specific agonists, such as methacholine (1).
Asthma typically presents early in childhood and often continues into adulthood (2–4). Clinical data indicate that 80–90% of adults with chronic asthma have disease onset before age 5, and there is an association between the severity of symptoms in childhood and persistence of asthma into adult life (4). In this context, a major risk factor for asthma is early-life exposure to environmental insults, including aeroallergens, ozone, cigarette smoke, and respiratory viral infection (1, 3). As the lung continues to develop after birth, early-life insults could have profound, long-term effects on airway structure, function, and disease susceptibility. In support of this hypothesis, recent studies in rodents show that peri- and neonatal exposure to tobacco smoke and respiratory syncytial virus (RSV) lead to AHR later in life, whereas late, postnatal exposure has no such effect (5, 6). These findings indicate that the immature lung is more prone to long-term impairment by environmental insults than is the mature, adult lung. However, little is known about the molecular nature and mechanisms of such impairment.
A growing body of evidence indicates a link between alterations in airway innervation and asthma. Previous studies of peri- and neonatal primate and rodent models showed that environmental insults lead to changes in innervation by neuropeptide Y- and substance P-containing axons and that these changes are associated with increased expression of nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF), respectively (7, 8). NGF and BDNF, together with neurotrophin (NT)-3 and NT-4, belong to the NT family, which has established roles in controlling neural innervation of target tissues (9). In addition, changes in airway innervation have been reported in infant rhesus monkeys that were exposed to ozone and house dust mite allergen (10). Furthermore, human infants with acute RSV infection, a risk factor for asthma later in life (1, 6), have high levels of NGF and BDNF in their bronchoalveolar lavage (BAL) fluid (11). Notably, high serum levels of NT4 correlate positively with asthma severity in children (12). These observations suggest that changes in NT expression and innervation caused by early-life environmental insults are associated with airway dysfunction.
The mammalian airway is innervated by 2 distinct groups of neurons: the intrinsic neurons, whose cell bodies are located in the main bronchi, and the extrinsic neurons, whose cell bodies are located outside of the lung and extend their axons into the airways (13–15). We have shown that the intrinsic neurons are dependent on glial cell–derived neurotrophic factor for ASM innervation in the main bronchi (16). Only a few intrinsic neurons are located in the secondary bronchi, with little to none in the more distal part of the mouse lung (16). In contrast, the extrinsic neurons require ASM-derived BDNF to innervate ASM within the whole lung during embryogenesis (17). In support of our findings, mice deficient in TrkB, the tyrosine kinase receptor for both BDNF and NT4, exhibit reduced ASM innervation (18). However, the mechanisms controlling postnatal airway innervation have not been established.
The crosstalk between the neuron and its target plays a critical role in shaping the phenotypes of both the neuron and the target (19–24). In relation to the smooth muscle phenotype, we have shown in the esophagus that reduction in innervation in the embryonic stage causes profound impairment of smooth muscle contractility in adult life (21). Building on this well-established paradigm and our findings regarding NTs in ASM innervation, we speculated that early-life environmental insults alter airway innervation, which in turn alters the ASM phenotype, leading to persistent AHR.
In this study, we used a mouse model of allergic inflammation to investigate how allergen exposure during early postnatal life affects airway function later in life. We found that an increase in NT4/TrkB signaling functionally links early-life allergen exposure to persistent AHR by regulating the ASM phenotype.
MATERIALS AND METHODS
Mice
Wild-type (WT), NT4−/− (stock no. 002497), and NG2-dsRed (008241) mice were purchased from The Jackson Laboratories (Bar Harbor, ME, USA). The TrkBF616A/F616A (TrkBFA/FA) line was kindly provided by Dr. David Ginty (Johns Hopkins University, Baltimore, MD, USA; ref. 25). The αSMA-GFP mice had a green fluorescent protein (GFP) gene expressed under the control of the α-smooth muscle actin (αSMA) promoter (26). All mice used were on a C57BL/6J background. All animal studies were approved by the Boston University School of Medicine Institutional Animal Care and Use Committee.
Neonatal allergic asthma model
Pups were sensitized by intraperitoneal injections of 10 μg ovalbumin (OVA, A5503; Sigma-Aldrich, St. Louis, MO, USA) in Imject alum (77161; ThermoScientific, Waltham, MA, USA) on postnatal d 5 (P5) and P10, followed by three 10-min challenges with 3% aerosolized OVA solution on P18, P19, and P20. Control pups were challenged with PBS. The mice were subjected to physiological assessment of airway function with the FlexiVent apparatus (SciReq, Montreal, QC, Canada) on P21. To block TrkB signaling in the TrkBFA/FA pups, 10 μl 1NMPP1 (25 μM) in 2% ethanol was delivered by intraperitoneal injection, whereas control animals were injected with 2% ethanol alone (25). For studies on TrkB-dependent postnatal lung innervation, the pups received a daily 1NMPP1 injection up to P14. To block overactivation of TrkB in the OVA-exposed pups without affecting the normal basal level of TrkB signaling, we injected TrkBFA/FA pups with 1NMPP1 daily between P11 and P20. A subset of OVA-exposed WT and NT4−/− pups was allowed to grow into adulthood, followed by physiological and histological assays at 8 wk of age.
Adult acute allergic asthma model
Adult mice at 3 mo of age were sensitized by intraperitoneal injections of OVA protein (50 μg) in alum on d 0 and 7 (27). The mice were challenged with aerosolized 2% OVA solution for 20 min on d 14, 15, 16, 17, and 18 (27). The control mice were sensitized with OVA, followed by a challenge with aerosolized PBS. The mice were analyzed on d 19. For the persistence group, mice were analyzed 2.5 wk (18 d) after the last challenge.
ELISA
Blood and BAL fluid were collected from pups at P21. OVA-specific IgE levels in serum were measured with ELISA kits (M036005; MD BioProducts, Saint Paul, MN, USA). IL-13 levels in BAL fluid were assayed with another ELISA kit (KMC2221; Life Technologies, Carlsbad, CA, USA).
Histology, αSMA staining, and quantification
Serial, sagittal, paraffin-embedded sections (5 μm) in the middle of the left lobe of the lung were collected before rehydration and PBS washes. Mucin staining was performed with periodic acid-Schiff (PAS; kit 395B-1KTZ; Sigma-Aldrich). ASM was immune labeled with an antibody against αSMA (1:100, MS-113; ThermoScientific) and visualized with DAB. Medium-sized airways were imaged with ×40 bright-field microscopy (Axioskop 40; Zeiss, Thornwood, NY, USA). The αSMA density was calculated by dividing the αSMA+ area by the length of the basement membrane, measured with Image J (U.S. National Institutes of Health, Bethesda, MD, USA). A minimum of 12 airways from 3 mice, 4 airways/mouse, were quantified and averaged for each treatment group.
Physiological measurement of airway reactivity
Measurement of airway resistance was performed with the FlexiVent (27). Baseline airway resistance was measured after airway delivery of nebulized vehicle, and similar measurements were performed at increasing concentrations of nebulized methacholine. Data are presented as normalized to baseline level. After final measurements, the mouse was disconnected from the ventilator for organ and body fluid harvest.
BAL fluid counts
Mouse lungs were flushed for 30 s with 0.5 ml cold PBS for P21 pups or 1 ml cold PBS for adults before BAL fluid was collected. Total cell counts in BAL fluid were determined with a hemocytometer. BAL fluid cells were pelleted, resuspended in 200 μl cold PBS, and spun onto a histology slide by using Cytospin (ThermoScientific). The slides were fixed and stained with Hema3 Stain (Fisher Scientific, Pittsburgh, PA, USA). For differential cell counts, 200 cells were counted for each sample, to determine relative abundance of different immune cell types.
Fluorescent labeling and confocal microscopy
The right apical lobe of the lung was collected and fixed in 4% paraformaldehyde in PBS at 4°C overnight. After cryoprotection in 30% sucrose in PBS, 50 μm sections were collected along the rostral–caudal axis of the lobe. Four sections, 1 mm apart, were immunostained (17) and mounted with Fluoromount-G (eBioscience, San Diego, CA, USA). Primary antibodies included biotinylated mouse antineural class III β-tubulin (TuJ1, 1:200, BAM1195; R&D Systems, Minneapolis, MN, USA) and mouse anti-CGRP (calcitonin gene-related peptide; 1:1000, C8198; Sigma-Aldrich). Fluorescently stained sections were imaged with an Axiovert 100M LSM 510 microscope (Zeiss). Compressed z-stack confocal images were presented and quantified. To determine the innervation density, we quantified the TuJ1 or CGRP immune-reactive area and divided the result by the area of each airway using Image J. Ten medium-sized airways were imaged and quantified per slide for a total of 40 airways/mouse. These numbers were then averaged per mouse and for each treatment.
Western blot analysis
Protein samples were collected from right basal lung lobes after homogenization in RIPA buffer containing proteinase inhibitor (Roche Diagnostics, Indianapolis, IN, USA). Protein concentration of whole lung lobe homogenate was determined with the BCA protein assay (ThermoScientific). Western blot analysis for protein samples (20 μg) was performed (17). Primary antibodies included rabbit anti-NF(H&M) (1:1000, MAB1592; Millipore, Billerica, MA, USA), rabbit anti-NF(H) (1:2000, AB1991; Millipore), rabbit anti-BDNF (1:100, 546 N-20; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-NT4 (1:100, 545 N-20; Santa Cruz), mouse anti-GAPDH (1:100,000, ab8245l Abcam, Cambridge, MA, USA), and mouse anti-phosphorylated TrkB (1:500, ab52191; Abcam). Antigen-antibody complexes were detected with SuperSignal West Pico Chemiluminescent Substrate (ThermoScientific) and exposed on film. Densitometry units of individual protein bands were measured by Image J and were normalized to GAPDH levels.
Lung slice preparation and measurement of airway contraction
Precision-cut lung slices (200 μm in thickness) were prepared (28) with a VF-300 tissue slicer (Precisionary Instruments, San Jose, CA, USA). The slices were cultured in 1:1 DMEM/F-12 supplemented antibiotics (Invitrogen-Life Technologies). The next day, the slices were secured within a standard 12-well culture plate with a ring of nylon mesh together with a metal washer. The multiwell plate was imaged with an inverted microscope (DMI6000B; Leica Microsystems, Buffalo Grove, IL, USA). The lung slices were pretreated with isoproterenol (30 μM; Sigma-Aldrich), to induce relaxation, followed by stimulations with 10−7 M, 10−6 M, 10−5 M, and 10−4 M methacholine (Sigma-Aldrich). Midsized airways with a baseline luminal area between 14,000 and 20,000 μm2 were assayed. Each airway was imaged every minute for a total duration of 35 min. From the acquired images, we quantified the airway luminal area (Image J) and normalized its magnitude to the pretreatment baseline value.
Cell contractility–traction force microscopy
ASM cells were isolated from PBS-treated and OVA-exposed αSMA-GFP;NG2-dsRed and NT4−/−;αSMA-GFP;NG2-dsRed pups (29). GFP+ ASM cells were cultured sparsely on collagen-1-coated polyacrylamide gel substrates (stiffness, 4 kPa). Embedded in the substrate were fiducial markers consisting of fluorescent nanobeads. By tracking deformation of the beads and solving the inverse problem of the forces necessary to induce those displacements, we used MATLAB (The MathWorks, Natick, MA, USA) to quantify the contractile forces (called tractions) that cells exert on their substrate (30, 31). From the traction map, we extracted a single measure of overall cell contraction, called the contractile moment (30).
Statistics
All data are represented as means ± sem from a minimum of 3 separate experiments. For comparisons between 2 genotypes or 2 conditions, such as in Western blot analyses, immunohistochemistry quantification, and differential BAL fluid cell counts, statistical analysis was performed with the 2-tailed Student's t test. Airway resistance data and contraction assays of lung slices were compared between genotypes, treatments, and methacholine doses with 2-way, repeated-measures ANOVA. Values of P < 0.10 were considered significant.
RESULTS
OVA exposure in early life, but not in adulthood, leads to persistent AHR
To evaluate the effect of early-life allergen exposure on the airways, we created a neonatal mouse model by sensitizing C57BL/6 pups to OVA allergen at P5 and P10, followed by 3 aerosolized OVA challenges between P18 and P20 (Fig. 1A). The mice were analyzed at P21, when the airway begins to assume a mature configuration (32). Compared to saline (PBS)-challenged pups, OVA-challenged pups displayed key features of asthma: overproduction of mucin (Fig. 1B), elevated serum levels of OVA-specific IgE (Fig. 1C), AHR in response to methacholine (Fig. 1D), ASM thickening (Fig. 1E, F), increased levels of the Th2 cytokine IL-13 (Fig. 1J), and elevated infiltration of eosinophils and neutrophils (Fig. 1K). Notably, OVA-exposed pups had persistent AHR after they reached adulthood at 8 wk of age (P21→adult; Fig. 1G), despite the lack of sustained ASM thickening (Fig. 1H, I) and diminished inflammation (Fig. 1J, K).
Figure 1.

OVA exposure in early postnatal life led to persistent AHR. A) Experimental protocol for inducing allergic airway inflammation by OVA in a neonatal mouse model. Controls received saline (PBS) challenges. B) Mucin staining in the airways of PBS- and OVA-exposed pups at P21. Arrow indicates mucin+ cells. C) Serum levels of OVA-specific IgE in PBS- and OVA-exposed pups at P21, measured by ELISA. Each circle represents a sample. Red line indicates mean of the OVA-challenged group. D) Airway reactivity of control (n=14) and OVA-exposed (n=13) pups was assessed at P21 with the FlexiVent apparatus with increasing concentrations of methacholine. E) Representative images of αSMA immunostaining in the airways of PBS- and OVA-exposed pups at P21. Arrows indicate αSMA+ ASM; asterisks indicate αSMA blood vessels. F) Quantification of αSMA density in the airways of PBS- and OVA-exposed pups at P21. G) Control (n=9) and OVA-exposed (n=13) pups were allowed to grow for 5 wk into adulthood (P21→adult). Airway resistance was measured with the FlexiVent apparatus at 8 wk of age. H) Representative images of αSMA immunostaining in the airways of 8-wk-old adult mice (P21→adult) exposed to PBS and OVA as neonates. Arrows indicate αSMA+ ASM; asterisks indicate αSMA blood vessels. I) Quantification of αSMA density. J) Serum levels of IL-13 in PBS- and OVA-exposed pups at P21 and P21→adult mice, measured by ELISA. K) Differential cell count in BAL fluid of PBS- and OVA-exposed pups at P21 and P21→adult mice. Percentages of macrophages (Mac), eosinophils (Eos), lymphocytes (Lymph), and neutrophils (Neut) are shown. Scale bars = 50 μm. n.s., not significant. *P < 0.10; **P < 0.05; ***P < 0.01.
To test whether OVA exposure in adult life also leads to persistent AHR, we sensitized and challenged 3-mo-old adult mice with OVA (Fig. 2A and ref. 27) and then examined them 24 h after the last challenge. Compared to the PBS-challenged controls, the OVA-challenged adult mice exhibited features of asthma that included AHR (Fig. 2C), mucous hyperplasia (Fig. 2B), ASM thickening (Fig. 2D), and increased serum levels of OVA-specific IgE and IL-13 (Fig. 2E, G). However, AHR was found to be diminished completely 2.5 wk after the last challenge (adult→2.5 wk recovery), along with resolution of the inflammation (Fig. 2F, G and ref. 33). These observations confirm that the developing lung is more prone to long-term functional impairment by insults than is the mature, adult lung (5, 6).
Figure 2.

OVA exposure in adulthood did not lead to persistent AHR. A) Experimental protocol for inducing acute asthma in adult mice. Control mice received PBS challenges. B) Mucin staining in the airways of PBS- and OVA-exposed adult mice. Arrows indicate mucin+ cells. C) Airway reactivity of control (n=8) and OVA-challenged (n=5) adult mice was assessed at d 19 by measuring airway resistance with the FlexiVent apparatus. D) Quantification of αSMA density in the airways of PBS- and OVA-exposed adult mice. E) Serum levels of OVA-specific IgE in PBS- and OVA-exposed adult mice, measured by ELISA. Each circle represents a sample. Red line indicates mean of the OVA-challenged group. F) Airway reactivity of control and OVA-exposed mice was assessed 2.5 wk after the last challenge. G) Serum levels of IL-13 in PBS- and OVA-exposed adult mice at d 19 and after a 2.5 wk recovery, measured by ELISA. Scale bar = 50 μm. *P < 0.10; **P < 0.05; ***P < 0.01.
OVA exposure in early life, but not in adulthood, leads to an increase in ASM innervation
We used OVA-exposed pups as a model to investigate how early-life allergen exposure affects long-term airway structure and function. In these mice, persistent AHR occurred without sustained ASM thickening (Fig. 1G–I). This finding suggests persistent changes in the ASM phenotype. Since ASM is innervated, we hypothesized that early-life allergen exposure affects ASM innervation, ultimately leading to a change in the ASM phenotype. To test this hypothesis, we measured ASM innervation levels in OVA-exposed pups at P21 and after they grew into adulthood. Western blot analysis showed that early-life OVA exposure significantly (by 2-fold) increased the level of neurofilament (NF; Fig. 3A, B). Because ASM is the major target of innervation in the mouse lung, we concluded that early-life OVA exposure leads to elevated ASM innervation. Consistently, immunostaining for neural-specific tubulin with a TuJ1 antibody showed that the OVA-exposed lungs had more overall axons and higher ASM innervation density than did the controls at P21 (Fig. 3C, E). The abundance of sensory afferents marked by CGRP was unchanged after OVA exposure at P21 (Fig. 3D, F), suggesting that the increase in ASM innervation most likely involves efferent nerves. Similar increases in ASM innervation were observed in pups exposed to cockroach allergen (Supplemental Fig. S1). However, the overall airway innervation in the adult mice that were exposed to OVA as neonates (8 wk of age; P21→adult) was no longer increased over that in the control mice (Fig. 3C, E), whereas CGRP+ sensory afferents in the adult mice were more abundant than in the control mice (Fig. 3D, F). These findings indicate that early-life OVA exposure leads to dynamic remodeling of ASM innervation.
Figure 3.
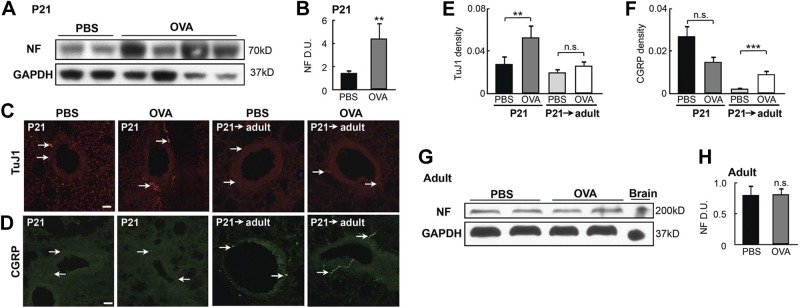
Early-life OVA exposure increased ASM innervation. A) Western blot analysis of NF in lung homogenates collected at P21 from controls and OVA-exposed pups. Each lane represents 1 mouse. B) Densitometry analysis of NF signal in the Western blot assay in (A). Data were normalized to the total amount of loaded protein. C) TuJ1 immunolabeling in airways of PBS- and OVA-exposed pups at P21 and in 8-wk-old mice exposed to PBS or OVA as neonates (P21→adult). Arrows indicate TuJ1-labeled axons. D) Immunostaining for CGRP in the airways of PBS- and OVA-exposed pups at P21 and in 8-wk-old adult mice exposed to PBS or OVA as neonates (P21adult). Arrows indicate CGRP-containing sensory afferents. E) Innervation density in control and OVA-exposed P21 pups and P21→adult mice. TuJ1 immunoreactivity was normalized to the size of each airway to calculate innervation density. Forty airways from 10 pups of each group were analyzed. F) Quantification of CGRP staining in the airways of P21 pups and P21→adult mice by normalizing CGRP immunoreactivity to the size of the airway. Forty airways from 10 pups of each group were analyzed. G) Western blot analysis of NF in lung homogenates of adult mice that were challenged with PBS or OVA. Lungs were harvested at d 19. H) NF levels were quantified by normalization to GAPDH levels. Data represent the average of 4 separate experiments with ≥3 mice/group for each experiment. Scale bars = 50 μm. n.s., not significant; **P < 0.05; ***P < 0.01.
In contrast, OVA exposure to the adult lung had no effect on innervation levels, as assayed by Western blot analysis (Fig. 3G, H), and CGRP production increased transiently within 48 h after the last challenge (34). These data show that allergen exposure elicits a distinct neurogenic response in the developing postnatal lung.
NT4-activated TrkB signaling is necessary for ASM innervation in the postnatal lung
To understand how early-life OVA exposure increased ASM innervation, we first characterized how the lung normally becomes innervated in postnatal life. Using TuJ1 immunostaining and Western blot analysis for NF, we showed that ASM innervation increased postnatally and peaked around P14 (Fig. 4A, B, D). Next, we sought to identify the key signals for postnatal ASM innervation. We focused on the NGF family because we had shown that ASM expresses BDNF and NT4, 2 NTs that both signal through the TrkB receptor (9), and that extrinsic neurons require BDNF-activated TrkB signaling for axon extension, to innervate the ASM during embryogenesis (17, 18). Western blot analysis showed that BDNF levels were greatly reduced after birth, whereas NT4 expression increased and peaked at P14 (Fig. 4C, D), coinciding with the maturation of postnatal lung innervation (Fig. 4B, D).
Figure 4.

NT4-activated TrkB signaling was necessary for ASM postnatal innervation. A) Representative images of TuJ1-labeled lung sections from WT mice before birth (E18.5) and at P7, P14, and P21. TuJ1 staining of lung sections from NT4−/− and 1NMPP1-treated TrkBFA/FA pups at P21 is also shown. Arrows indicate TuJ1-labeled axons in the airway. B) Western blot analysis of NF levels in lungs of pups at P7, P14, and P21. C) Western blot analysis of BDNF and NT4 levels in the lungs at E18.5, P7, P14, and P21. D) Densitometry analysis of NF, BDNF, and NT4 levels at E18.5 and during postnatal development, as shown in B and C. Signals were normalized to GAPDH levels. E) Western blot analysis of NF (high and medium molecular weight) levels in E18.5 whole-lung homogenates of WT and NT4−/− pups. F) NF signals, quantified by densitometry and normalized to GADPH levels. G) Western blot analysis of NF levels in P21 whole-lung homogenates of WT and NT4−/− pups. H) NF signals quantified and normalized. I) Western blot analysis of TuJ1 levels in P14 lungs of TrkBFA/FA pups and TrkBFA/+ littermates treated with 1NMPP1. J) TuJ1 signals, quantified and normalized to GADPH levels. Data represent results from 3 separate experiments with ≥3 mice/group for each experiment. Scale bar = 50 μm. n.s., not significant; **P < 0.05.
To investigate the role of NT4 in postnatal lung innervation, we compared NF (of both high and medium molecular weight) levels between WT and NT4−/− mice at E18.5 and P21. NT4−/− mice have minor neural defects but are otherwise viable and fertile (35) and show normal airway structure (Supplemental Fig. S3A). We found no change in airway innervation levels in NT4−/− mice at E18.5 (Fig. 4E, F), further supporting BDNF as the key neurogenic signal in the embryonic lung (17). However, the NT4−/− pups showed a 2- to 3-fold reduction in NF levels and decreased TuJ1 immunoreactivity compared with that of the WT pups at P21 (Fig. 4A, G, H), indicating that NT4 is necessary for postnatal ASM innervation. To validate this observation, we blocked signaling from the NT4 receptor, TrkB, by treating TrkBF616A/F616A (TrkBFA/FA) pups with 1NMPP1and assessed changes in ASM innervation. TrkBFA/FA mice harbor a point mutation in the TrkB kinase domain that renders the receptor susceptible to inhibition by 1NMPP1, a specific inhibitor of mutant kinase (25). This inhibition is reversible in vivo, and without 1NMPP1 treatment, TrkBFA/FA mice are normal (25). We found that daily intranasal 1NMPP1 delivery for 2 wk after birth resulted in ∼70% reduction in lung innervation of P14 TrkBFA/FA pups compared with that in similarly treated TrkBFA/+ littermates (Fig. 4I).
To test whether reduced airway innervation in the NT4−/− pups and 1NMPP1-treated TrkBFA/FA pups was due to a defect in ASM development, we examined the expression of αSMA in the lungs of control and mutant pups at P21 and found no difference (Supplemental Fig. S3A, G). We also detected no TrkB mRNA expression in the whole lung or in isolated ASM (Supplemental Fig. S2 and refs. 36, 37). In contrast, TrkB mRNA was readily detectable in the cell bodies of extrinsic neurons within peripheral sensory ganglia (Supplemental Fig. S2A and refs. 13, 15, 37, 38). We reasoned that the amount of TrkB mRNA in ASM-innervating axons is likely to be too low for detection (39). A lack of NT4 receptor expression by ASM indicates that NT4 does not directly signal in ASM. Collectively, these findings support that NT4-activated TrkB signaling is necessary for postnatal lung innervation but not for ASM formation.
Early-life OVA exposure elevates NT4/TrkB signaling to increase ASM innervation
To test whether early-life OVA exposure alters NT4/TrkB signaling to increase ASM innervation, we compared the NT4 level and TrkB signaling in the lungs of PBS controls and OVA-exposed pups by Western blot analysis. We found that the OVA-exposed pups had a 5-fold increase in NT4 protein expression (Fig. 5A) and a 2-fold increase in TrkB phosphorylation, indicative of receptor activation (Fig. 5B). We then assessed the effect of elevated NT4/TrkB signaling on ASM innervation in the OVA-exposed pups. For this, we sensitized and challenged NT4−/− pups (described in Fig. 1A) and analyzed lung innervation at P21. OVA exposure had no effect on the low basal levels of ASM innervation in the NT4−/− pups (Fig. 5C, D), in contrast to the increased innervation in the OVA-exposed WT pups (Figs. 3A, B). Intranasal 1NMPP1 treatment of TrkBFA/FA pups between P11 and P20 also prevented the OVA-induced increase in ASM innervation, without affecting normal levels of innervation density, as assayed by TuJ1 immunohistochemistry (Fig. 5E). These findings demonstrate that early-life OVA exposure increased ASM innervation by elevating NT4-activated TrkB signaling.
Figure 5.

NT4-activated TrkB signaling was necessary for an early-life OVA-induced increase in lung innervation and AHR. A) NT4 levels in the lungs of controls and OVA-exposed pups at P21, analyzed by Western blot analysis and normalized to GAPDH levels. B) Western blot analysis of activated TrkB by phosphorylation in control and OVA-exposed lungs. C) Western blot analysis of NF at P21in WT and NT4−/− pups that were exposed to PBS and OVA. D) NF signals, quantified by densitometry and normalized to GAPDH levels. E) WT and TrkBFA/FA pups were exposed to PBS or OVA. TrkBFA/FA pups also received short-term 1NMPP1 treatment, to prevent OVA-induced overactivation of TrkB signaling. Innervation density in the lungs of each group was analyzed by TuJ1 immunolabeling. Graphs represent an average of 40 airways from 10 pups/group. F) Airway resistance of P21 WT and NT4−/− mice that were exposed to PBS or OVA (n=11/group). G) Airway resistance of WT and 1NMPP1-treated TrkBFA/FA pups that were exposed to PBS or OVA (WT-PBS, n=12; WT-Ova, n=9; TrkBFA/FA-PBS, n=9; and TrkBFA/FA-OVA, n=12). H) Airway resistance of adult WT and NT4−/− mice that had been exposed to PBS and OVA as pups (WT-PBS, n=9; NT4−/−-PBS; n=9; WT-OVA, n=13; and NT4−/−-OVA, n=16). Data represent the average of each group from 3 separate experiments. Data from the WT mice analyzed in F and G partially overlapped; results of WT mice at 8 wk of age in Fig. 1C were replotted in H, to allow a direct comparison between WT and NT4−/− mice. **P < 0.05; ***P < 0.01.
NT4/TrkB signaling is necessary for OVA-induced AHR
The increased ASM innervation may link early-life OVA exposure to persistent AHR into adulthood. To establish this link, we assessed the airway function and inflammation after OVA exposure in NT4−/− and 1NMPP1-treated TrkBFA/FA pups. These pups, which were exposed to PBS, had normal airway reactivity (Fig. 5F–H). OVA exposure also induced similar levels of inflammation in the pups, as compared to that in the WT pups, assessed by measurements of IL-13 and OVA-specific IgE levels, BAL fluid cell counts, and ASM thickening (Supplemental Fig. S3). However, in contrast to the WT mice, the OVA-exposed NT4−/− pups exhibited no AHR at P21 or after they reached adulthood (Fig. 5F, H). In addition, 1NMPP1 treatment also diminished AHR in the OVA-exposed TrkBFA/FA pups at P21 (Fig. 5G). These observations suggest a functional link between increased innervation after early-life allergen exposure and altered ASM phenotype.
NT4 is necessary for the OVA-induced hyperreactive phenotype of ASM
To assess the role of NT4 signaling in regulating ASM contractility, we compared ASM contraction in lung slices from PBS- and OVA-exposed WT and NT4−/− pups at P21 (28). We chose to assay medium-sized airways in which no intrinsic neurons are located (16). In this assay, ASM contraction was induced by methacholine free of neural input. All lung slices were pretreated with 30 μM isoproterenol, a β2-adrenergic receptor agonist, to completely relax the ASM and eliminate potential baseline differences in contraction. Following methacholine treatment, all airways exhibited a dose-dependent reduction of the luminal area, indicative of ASM contraction (Fig. 6A). Control slices from WT pups exhibited little contraction at 10−7 M methacholine and contracted by up to 50% area at higher doses (Fig. 6A). In comparison, lung slices from the OVA-exposed WT pups significantly reduced the luminal area (25%) at 10−7 M and maximally reduced the area (80%) at 10−5 M methacholine (Fig. 6A). These findings indicate that early-life OVA exposure significantly increased the sensitivity and the magnitude of contraction of ASM in response to methacholine in WT mice. Notably, although NT4 deficiency had no effect on baseline contraction, NT4−/− lung slices from both the PBS- and the OVA-exposed pups exhibited a greatly reduced methacholine response. The maximum contraction of NT4−/− lung slices from PBS-exposed pups led to only a 20% reduction in the airway luminal area, and lung slices from the OVA-exposed NT4−/− pups exhibited a dose curve of methacholine similar to that of PBS-exposed WT pups (Fig. 6A). These data further support a role for NT4-dependent innervation in controlling the contractile phenotype of ASM.
Figure 6.

NT4 is required for OVA-induced increase in ASM contractile phenotypes. A) Measurement of methacholine-induced ASM contraction using lung slices. All lung slices were pretreated with isoproterenol to diminish differences in baseline contraction. A minimum of 26 airways from 3 mice were analyzed for each condition. There was a significant difference in lung slice contraction between OVA-exposed WT and NT4−/− mice. B) Differential GFP and dsRed expression in αSMA-GFP;NG2-dsRed pups at P21. Arrowheads indicate GFP+ ASM; asterisks indicate GFP+ and dsRed+ blood vessels. C) Contractile moment measurement of individual ASM cells based on their contraction map. ASM cells were isolated from αSMA-GFP;NG2-dsRed and NT4−/−;αSMA-GFP;NG2-dsRed pups after PBS and OVA exposure at P21. Each symbol represents the measurement of a single ASM cell; horizontal line represents the average within the group. D) Representative contraction maps of a single ASM cell isolated from lungs of OVA-exposed WT and NT4−/− pups. Insets: imaged GFP+ ASM cells. Scale bars = 50 μm. n.s., not significant. ***P < 0.01.
We then compared biophysical properties of individual ASM cells isolated from WT and NT4−/− lungs by traction–force microscopy. To allow purification of ASM cells by cell sorting, we generated an αSMA-GFP;NG2-dsRed double-fluorescent mouse. In the lungs of this mouse, GFP was expressed by smooth muscle cells in both the airway and vasculature (26), and dsRed was found to be expressed in pericytes, some immune cells, and vascular smooth muscle cells, but not in ASM (Fig. 6B and ref. 40). As a result, ASM cells can be separated from vascular smooth muscle cells by single GFP positivity (Fig. 6B and ref. 29). Dissociated lung cells in suspension were stained for additional markers to allow separation of dead cells (efluor+), endothelial cells (CD31+), and immune cells (CD45+). A stringent sorting algorithm was designed to further exclude possible myofibroblasts with low GFP expression (Supplemental Fig. S4 and ref. 29). We bred the NT4−/− mouse into this double-fluorescent background so that a pure population of ASM cells (efluor−CD45−CD31−hrGFP+dsRed−) could be isolated from the lungs of both genotypes. Isolated ASM cells were plated on collagen 1-coated plates that were impregnated with red fluorescent nanobeads. After 72 h in culture to allow cell attachment, individual ASM cells and nanobeads were live imaged on the basis of their fluorescence. The contractile moment was calculated by quantifying the extent of the bead displacement before and after cell removal by trypsin (30, 31). We found that NT4 deficiency had no effect on the basal contractile moment measurement (Fig. 6C). In addition, the ASM cells isolated from the OVA-exposed WT pups at P21 had greater contractile moment than did the WT PBS control cells (Fig. 6C), consistent with an OVA-induced increase in basal tone of ASM cells. In contrast, OVA-exposure had no effect on contractile moment of the NT4−/− ASM cells (Fig. 6C, D). Collectively, our results indicate that NT4 is necessary for the hyper-reactive phenotype of ASM after OVA-exposure.
DISCUSSION
In this study, we compared mouse models of allergic inflammation in neonates and in adults. We found that allergen exposure in early life, but not in adult life, leads to the elevated NT4 levels and increased ASM innervation that are associated with persistent AHR in adulthood. These findings provide a potential explanation for the positive correlation between NT4 levels and asthma disease severity in children (12). Our study also suggests that early-life events that promote persistent airway dysfunction may involve alterations along the ASM-nerve axis (1, 3).
By using mice with genetic disruption or pharmacologic blockade of NT4/TrkB signaling, we showed that this pathway was necessary for the development of acute and sustained AHR in vivo. Notably, this pathway was not involved in normal lung development, function, or inflammation. Assays of single, isolated ASM cells also showed that elevated cellular tone after early-life allergen exposure was dependent on NT4. On the basis of these findings, we propose that increased innervation after early-life allergen exposure is functionally linked to persistent alterations in the ASM phenotype. In our model (Fig. 7), NT4 acts as a target-derived signal for ASM innervation by TrkB-expressing neurons during postnatal development. In pathological states such as early-life allergen exposure, up-regulated NT4/TrkB signaling leads to increased ASM innervation. Elevated innervation alters the ASM phenotype, ultimately resulting in persistent AHR (Fig. 7A). Deficiency in NT4/TrkB signaling blocks the allergen-induced increase in ASM innervation, thereby preventing aberrant changes in ASM contractility (Fig. 7B).
Figure 7.

Model of how increased ASM innervation after early-life allergen exposure leads to persistent AHR. A) Postnatal lung innervation and early-life, allergen-induced AHR. NT4 is an ASM-derived neurotrophic factor for postnatal ASM innervation by TrkB-expressing extrinsic neurons. Early-life allergen exposure leads to ASM thickening and elevates NT4-activated TrkB signaling to increase ASM innervation. Increased innervation changes the ASM phenotype, ultimately resulting in persistent AHR without ASM thickening. B) Without NT4 signaling, ASM innervation in postnatal lung is reduced. In addition, although NT4 deficiency has no effect on ASM thickening after early-life allergen exposure, it blocks the increases in ASM innervation, which in turn prevents changes in ASM reactivity.
The assay of ex vivo lung slices supported our conclusion that NT4 signaling is necessary for allergen-induced AHR. The results showed that only the OVA-exposed WT airways exhibited hyper-reactivity to methacholine, whereas the OVA-exposed NT4−/− airways exhibited contraction similar to that of the control WT airways at all tested concentrations of methacholine. Compared to physiological assays in vivo, in which no difference in airway reactivity between PBS- and OVA-exposed NT4−/− pups was found, lung slice assays showed an effect of OVA exposure on methacholine responsiveness. This discrepancy may be caused by a difference in methacholine doses between these two assays and may also reflect a lack of neural input and humoral and structural components in lung slices. However, it does raise the possibility that NT4 regulates ASM phenotypes, such as reactivity to methacholine in the normal lung. Considering that NT4 is needed for ASM innervation, this possibility is consistent with our model in which NT4-dependent neural innervation regulates the ASM phenotype in asthma.
The current study in the lung, along with our previous study in the esophagus (21), highlights the functional link between innervation and smooth muscle phenotypes. Our data suggest that the deregulated release of neurotransmitters and neuropeptides from innervating nerves during the neonatal period contribute to long-term changes in the ASM phenotype; candidate neurotransmitters in this process include acetylcholine and substance P, two known activators of ASM reactivity (41–43). Future studies will detail changes in specific types of ASM innervation, such as cholinergic efferents induced by early-life allergen exposure. In addition, the molecular nature of hyper-reactive ASM induced by nerve-derived signals requires further characterization. We found no difference in muscarinic receptor mRNA expression when we used isolated ASM from WT and NT4−/− mice. This finding suggests that neural regulation of the ASM contractile phenotype is mediated through a posttranscriptional mechanism or other signaling mechanisms that may have a broad effect on ASM response to a variety of agonists.
We identified an overall increase in airway innervation after early-life allergen exposure. However, previous studies have shown that early-life exposure to tobacco smoke, ozone, and house dust mite allergen elicits specific changes in the type of innervation (7, 8). One possibility is that changes in NT expression and airway innervation are dependent on the type of provocative insult and are also developmentally stage specific. Supporting this possibility, the ozone-induced increase in substance P-containing axons is dependent on the postnatal time window of exposure. In addition, other studies in primates after ozone and allergen exposure focused on neural changes in the airway epithelium (10), a structure that is not abundantly innervated in mice. Of note, several of these studies have suggested that increased substance P levels contribute to long-term AHR (5, 7), which is consistent with our hypothesis.
In summary, this study provided evidence that neural innervation plays a key role in controlling the ASM contractile phenotype. The results further support a paradigm wherein insults to the developing lung can lead to irreversible changes in lung cell phenotype, thereby resulting in adult disease. Based on our findings, it may be possible to develop treatment strategies that prevent the progression from childhood to adult asthma.
Supplementary Material
Acknowledgments
The authors thank Dr. David Ginty (Johns Hopkins University, Baltimore, MD, USA) for the TrkBF616A/F616A mouse line; Dr. Janice Weinberg for statistical analysis; Kavon Kaboli for airway resistance assay by Flexivent; and Kelsi Radzikinas, Kruti Patel, and Kavitha Rajendran for technical assistance.
This work was supported by grants to X.A. and A.F. (HL112619) and W.C. (CA122737) and a T32 training grant to L.A. (HL007035) from the U.S. National Institutes of Health; a grant from The Parker Francis Foundation to R.K.; and a grant to X.A. from the American Asthma Foundation (12–0086).
The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- αSMA
- α-smooth muscle actin
- AHR
- airway hyperreactivity
- ASM
- airway smooth muscle
- BAL
- bronchoalveolar lavage
- BDNF
- brain-derived neurotrophic factor
- CGRP
- calcitonin gene-related peptide
- GFP
- green fluorescent protein
- NF
- neurofilament
- NGF
- nerve growth factor
- NT
- neurotrophin
- OVA
- ovalbumin
- P
- postnatal day
- PAS
- periodic acid-Schiff
- RSV
- respiratory syncytial virus
- WT
- wild type
REFERENCES
- 1. Maddox L., Schwartz D. A. (2002) The pathophysiology of asthma. Annu. Rev. Med. 53, 477–498 [DOI] [PubMed] [Google Scholar]
- 2. Stern D. A., Morgan W. J., Halonen M., Wright A. L., Martinez F. D. (2008) Wheezing and bronchial hyper-responsiveness in early childhood as predictors of newly diagnosed asthma in early adulthood: a longitudinal birth-cohort study. Lancet 372, 1058–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martinez F. D. (2009) The connection between early life wheezing and subsequent asthma: the viral march. Allergol. Immunopathol. (Madr.) 37, 249–251 [DOI] [PubMed] [Google Scholar]
- 4. Bisgaard H., Bønnelykke K. (2010) Long-term studies of the natural history of asthma in childhood. J. Allergy Clin. Immunol. 126, 187–197 [DOI] [PubMed] [Google Scholar]
- 5. Wu Z. X., Hunter D. D., Kish V. L., Benders K. M., Batchelor T. P., Dey R. D. (2009) Prenatal and early, but not late, postnatal exposure of mice to sidestream tobacco smoke increases airway hyperresponsiveness later in life. Environ. Health Perspect. 117, 1434–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gelfand E. W. (2012) Development of asthma is determined by the age-dependent host response to respiratory virus infection: therapeutic implications. Curr. Opin. Immunol. 24, 713–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu M., Zheng X., Peake J., Joad J. P., Pinkerton K. E. (2008) Perinatal environmental tobacco smoke exposure alters the immune response and airway innervation in infant primates. J. Allergy Clin. Immunol. 122, 640–647 [DOI] [PubMed] [Google Scholar]
- 8. Wu Z. X., Benders K. B., Hunter D. D., Dey R. D. (2012) Early postnatal exposure of mice to side-steam tobacco smoke increases neuropeptide Y in lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 302, L152–L159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang E. J., Reichardt L. F. (2003) Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642 [DOI] [PubMed] [Google Scholar]
- 10. Larson S. D., Schelegle E. S., Walby W. F., Gershwin L. J., Fanuccihi M. V., Evans M. J., Joad J. P., Tarkington B. K., Hyde D. M., Plopper C. G. (2004) Postnatal remodeling of the neural components of the epithelial-mesenchymal trophic unit in the proximal airways of infant rhesus monkeys exposed to ozone and allergen. Toxicol. Appl. Pharmacol. 194, 211–220 [DOI] [PubMed] [Google Scholar]
- 11. Tortorolo L., Langer A., Polidori G., Vento G., Stampachiacchere B., Aloe L., Piedimonte G. (2005) Neurotrophin overexpression in lower airways of infants with respiratory syncytial virus infection. Am. J. Respir. Crit. Care Med. 172, 233–237 [DOI] [PubMed] [Google Scholar]
- 12. Szczepankiewicz A., Rachel M., Sobkowiak P., Kycler Z., Wojsyk-Banaszak I., Schöneich N., Skibinska M., Brêborowicz A. A. (2012) Serum neurotrophin-3 and neurotrophin-4 levels are associated with asthma severity in children. Eur. Respir. J. 39, 1035–1037 [DOI] [PubMed] [Google Scholar]
- 13. Belvisi M. G. (2002) Overview of the innervation of the lung. Curr. Opin. Pharmacol. 2, 211–215 [DOI] [PubMed] [Google Scholar]
- 14. Kc P., Martin R. J. (2010) Role of central neurotransmission and chemoreception on airway control. Respir. Physiol. Neurobiol. 173, 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aven L., X., Ai. (2013) Mechanisms of respiratory innervation during embryonic development. Organogensis 9, 194–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Langsdorf A., Radzikinas K., Kroten A., Jain S., Ai X. (2011) Neural crest cell origin and signals for intrinsic neurogenesis in the mammalian respiratory tract. Am. J. Respir. Cell Mol. Biol. 44, 293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Radzikinas K., Aven L., Jiang Z., Tran T., Paez-Cortez J., Boppidi K., Lu J., Fine A., Ai X. (2011) A Shh/miR-206/BDNF cascade coordinates innervation and formation of airway smooth muscle. J. Neurosci. 31, 15407–15415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. García-Suárez O., Pérez-Pinera P., Laurà R., Germana A., Esteban I., Cabo R., Silos-Santiago I., Cobo J. L., Vega J. A. (2009) TrkB is necessary for the normal development of the lung. Respir. Physiol. Neurobiol. 167, 281–291 [DOI] [PubMed] [Google Scholar]
- 19. Shuler M. G., Krimm R. F., Hill D. L. (2004) Neuron/target plasticity in the peripheral gustatory system. J. Comp. Neurol. 472, 183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chevrel G., Hohlfeld R., Sendtner M. (2006) The role of neurotrophins in muscle under physiological and pathological conditions. Muscle Nerve 33, 462–476 [DOI] [PubMed] [Google Scholar]
- 21. Ai X., Kitazawa T., Do A. T., Kusche-Gullberg M., Labosky P. A., Emerson C. P., Jr. (2007) SULF1 and SULF2 regulate heparan sulfate-mediated GDNF signaling for esophageal innervation. Development 134, 3327–3338 [DOI] [PubMed] [Google Scholar]
- 22. Nekrep N., Wang J., Miyatsuka T., German M. S. (2008) Signals from the neural crest regulate beta-cell mass in the pancreas. Development 135, 2151–2160 [DOI] [PubMed] [Google Scholar]
- 23. Luther J. A., Birren S. J. (2009) Neurotrophins and target interactions in the development and regulation of sympathetic neuron electrical and synaptic properties. Auton. Neurosci. 151, 46–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Knox S. M., Lombaert I. M., Reed X., Vitale-Cross L., Gutkind J. S., Hoffman M. P. (2010) Parasympathetic innervation maintains epithelial progenitor cells during salivary organogenesis. Science 329, 1645–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen X., Ye H., Kuruvilla R., Ramanan N., Scangos K. W., Zhang C., Johnson N. M., England P. M., Shokat K. M., Ginty D. D. (2005) A chemical-genetic approach to studying neurotrophin signaling. Neuron 46, 13–21 [DOI] [PubMed] [Google Scholar]
- 26. Ghosh S., Paez-Cortez J., Boppidi K., Vasconcelos M., Roy M., Cardoso W., Ai X., Fine A. (2011) Activation dynamics and signaling properties of Notch3 receptor in the developing pulmonary artery. J. Biol. Chem. 286, 22678–22687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morgan R. K., McAllister B., Cross L., Green D. S., Kornfeld H., Center D. M., Cruikshank W. W. (2007) Histamine 4 receptor activation induces recruitment of FoxP3+ T cells and inhibits allergic asthma in a murine model. J. Immunol. 178, 8081–8089 [DOI] [PubMed] [Google Scholar]
- 28. Bai Y., Sanderson M. J. (2006) Modulation of the Ca2+ sensitivity of airway smooth muscle cells in murine lung slices. Am. J. Respir. Cell. Mol. Physiol. 291, L208–L221 [DOI] [PubMed] [Google Scholar]
- 29. Paez-Cortez J., Krishnan R., Arno A., Aven L., Ram-Mohan S., Patel K. R., Lu J., King O. D., Ai X., Fine A. (2013) A new approach for the study of lung smooth muscle phenotypes and its application in a murine model of allergic airway inflammation. PLoS ONE 8, e74469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Butler J. P., Tolic-Norrelykke I. M., Fabry B., Fredberg J. J. (2002) Traction fields, moments, and strain energy that cells exert on their surroundings. Am. J. Physiol. Cell Physiol. 282, C595–C605 [DOI] [PubMed] [Google Scholar]
- 31. Krishnan R., et al. (2009) Reinforcement versus fluidization in cytoskeletal mechanoresponsiveness. PLoS ONE 4, e5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ten Have-Opbroek A. A. (1991) Lung development in the mouse embryo. Exp. Lung Res. 17, 111–130 [DOI] [PubMed] [Google Scholar]
- 33. Rogerio A. P., Haworth O., Croze R., Oh S. F., Uddin M., Carlo T., Pfeffer M. A., Priluck R., Serhan C. N., Levy B. D. (2012) Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J. Immunol. 189, 1983–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fischer A., McGregor G. P., Saria A., Philippin B., Kummer W. (1996) Induction of tachykinin gene and peptide expression in guinea pig nodose primary afferent neurons by allergic airway inflammation. J. Clin. Invest. 98, 2284–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu X., Ernfors P., Wu H., Jaenisch R. (1995) Sensory but not motor neuron deficits in mice lacking NT4 and BDNF. Nature 375, 238–241 [DOI] [PubMed] [Google Scholar]
- 36. Kerzel S., Päth G., Nockher W. A., Quarcoo D., Raap U., Groneberg D. A., Dinh D. T., Fischer A., Braun A., Renz H. (2003) Pan-neurotrophin receptor p75 contributes to neuronal hyperreactivity and airway inflammation in a murine model of experimental asthma. Am. J. Respir. Cell Mol. Biol. 28, 170–178 [DOI] [PubMed] [Google Scholar]
- 37. Nassenstein C., Möhring U. H., Luttmann W., Virchow J. C., Jr., Braun A. (2006) Pulmonary distribution, regulation, and functional role of Trk receptors in a murine model of asthma. J. Allergy Clin. Immunol. 118, 597–605 [DOI] [PubMed] [Google Scholar]
- 38. Springall D. R., Cadieux A., Oliveira H., Su H., Royston D., Polak J. M. (1987) Retrograde tracing shows that CGRP immunoreactive nerves of rat trachea and lung originate from vagal and dorsal root ganglia. J. Auton. Nerv. Syst. 20, 155–166 [DOI] [PubMed] [Google Scholar]
- 39. Giuditta A., Kaplan B. B., van Minnen J., Alvarez J., Koenig E. (2002) Axonal and presynaptic protein synthesis: new insights into the biology of the neuron. Trends Neurosci. 25, 400–404 [DOI] [PubMed] [Google Scholar]
- 40. Zhu X., Bergles D. E., Nishiyama A. (2008) NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development 135, 145–157 [DOI] [PubMed] [Google Scholar]
- 41. De Vries A., Engels F., Henricks P. A., Leusink-Muis T., McGregor G. P., Braun A., Groneberg D. A., Dessing M. C., Nijkamp F. P., Fischer A. (2006) Airway hyper-responsiveness in allergic asthma in guinea-pigs is mediated by nerve growth factor via the induction of substance P: a potential role for trkA. Clin. Exp. Allergy 36, 1192–1200 [DOI] [PubMed] [Google Scholar]
- 42. Caceres A. I., Brackmann M., Elia M. D., Bessac B. F., del Camino D., D'Amours M., Witek J. S., Fanger C. M., Chong J. A., Hayward N. J., Homer R. J., Cohn L., Huang X., Moran M. M., Jordt S. E. (2009) A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc. Natl. Acad. Sci. U. S. A. 106, 9099–9104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grainge C. L., Lau L. C., Ward J. A., Dulay V., Lahiff G., Wilson S., Holgate S., Davies D. E., Howarth P. H. (2011) Effect of bronchoconstriction on airway remodeling in asthma. N. Engl. J. Med. 364, 2006–2015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.