

Abstract
Alcohol abuse is a major cause of pancreatitis in people, but the mechanism is unknown. It has been recently demonstrated that transient receptor potential vanilloid 1 (TRPV1) activation causes neurogenic inflammation and plays an important role in acute pancreatitis. Moreover, TRPV1 is activated by ethanol. We examined the direct effects of ethanol on acute pancreatitis. Acute inflammation of the pancreas was produced by injection of ethanol and palmitoleic acid (POA), a nonoxidative metabolite of ethanol, in wild-type C57BL/6J mice and Trpv1-knockout C57BL/6J mice. Inflammatory indexes were analyzed 24 h later. Injection of ethanol + POA produced acute pancreatitis indicated by significant increases in histopathological damage, serum amylase levels, and pancreatic MPO concentrations (P<0.05–0.001). All parameters of pancreatitis were blocked by pretreatment with the TRPV1 antagonist drug AMG9810. In addition, ethanol + POA administration to Trpv1knockout mice did not produce pancreatic inflammation. Treatment with vehicle, ethanol alone, or POA alone had no inflammatory effects. TRPV1 partially mediates inflammation induced by ethanol + POA in the mouse pancreas, consistent with the ability of ethanol to activate TRPV1. We propose that ethanol may contribute to alcohol-induced pancreatitis by a neurogenic mechanism.—Vigna, S. R., Shahid, R. A., and Liddle, R. A. Ethanol contributes to neurogenic pancreatitis by activation of TRPV1.
Keywords: acute pancreatitis, AMG9801, palmitoleic acid, TRP channel
Alcohol abuse is a major cause of acute pancreatitis in people, but the mechanisms by which ethanol damages the pancreas are not well understood. Recently, it was shown that nonoxidative metabolites of ethanol such as fatty acid ethyl esters (FAEEs) accumulate in higher concentrations in the pancreas than in other organs after ethanol consumption in people (1) and that administration of FAEEs to rats causes pancreatic damage in experimental models of pancreatitis (2). In addition, pretreatment of rats with inhibitors of oxidative metabolism of ethanol resulted in exacerbation of ethanol-induced pancreatic injury, providing further evidence for the role of nonoxidative ethanol metabolites such as FAEEs in acute alcoholic pancreatitis (3). It has been shown that the accumulation of FAEEs in the pancreas after ethanol consumption may damage the pancreas, at least in part, by causing toxic increases in cellular calcium concentrations in pancreatic acinar cells (4). Recently, a simple and reproducible method of ethanol/FAEE-induced pancreatic inflammation in the mouse has been described in which palmitoleic acid (POA; POA is released from palmitoleic acid ethyl ester by intracellular hydrolases) is administered together with ethanol (5).
In animal models of experimental pancreatitis, there is accumulating evidence for an important role for neurogenic mechanisms involving the transient receptor potential vanilloid 1 (TRPV1) receptor. For example, TRPV1 activation has been shown to mediate some of the inflammatory effects of secretagogue hyperstimulation with cholecystokinin-like peptides such as caerulein, ligation of the common pancreaticobiliary duct, and retrograde perfusion of the pancreatic duct with endoscopic retrograde cholangiopancreatography (ERCP) contrast medium (6, 7). TRPV1 is a ligand- and heat-gated cation channel expressed primarily by small-diameter primary sensory neurons (8). When activated, TRPV1 depolarizes primary sensory neurons, resulting in nociceptive neurotransmitter release centrally in the spinal cord and proinflammatory neurotransmitter release peripherally in organs such as the pancreas. TRPV1 is directly activated by heat, protons, some products of arachidonic acid metabolism, the active plant substances capsaicin and resiniferatoxin (RTX) (9–11), and ethanol (12). The effects of ethanol on TRPV1 are especially relevant in the present context because of the well-known relationship between alcoholism and pancreatitis in people. Notably, it has been demonstrated that ethanol sensitizes TRPV1 to capsaicin, protons, and heat and lowers the threshold for heat activation from ∼42 to ∼34°C (12). Thus, it is plausible that after ethanol ingestion, the inflammatory effects of TRPV1 activation in the pancreas may occur at normal body temperature and lead to pancreatitis. The goal of the present studies was to test this hypothesis using a mouse model of ethanol/FAEE-induced acute pancreatitis (5).
MATERIALS AND METHODS
These studies were approved by the Duke University Institutional Animal Care and Use Committee.
Animals and materials
Mice with a genetic deletion of the Trpv1 gene (Trpv1−/−) were obtained from Dr. D. Julius (University of California, San Francisco, CA, USA; ref. 13). Trpv1−/− mice and wild-type littermates were backcrossed onto a C57BL/6J background (The Jackson Laboratory, Bar Harbor, ME, USA). POA and AMG9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide] were purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in dimethyl sulfoxide (DMSO).
Induction of acute pancreatitis
Acute alcoholic pancreatitis was induced as described previously (5). Briefly, 8-wk-old male wild-type or Trpv1−/− C57BL/6J mice (20–30 g body weight) were injected intraperitoneally with either vehicle controls, 1.32 g/kg pure ethanol alone (1.673 μl/g body weight), 2 mg/kg POA alone (1.1175 μl/g body weight of POA diluted 1:500 in DMSO), or 1.32 g/kg ethanol followed 1 h later by 2 mg/kg POA. When the TRPV1 antagonist drug AMG9810 was used, it was given intraperitoneally at a dose of 30 mg/kg 30 min before ethanol administration (3 μl/g body weight of a 10 mg/ml solution in DMSO). The mice were killed by CO2 asphyxiation 24 h after the last injection and weighed, the pancreas was removed and weighed, and a portion of it was frozen at −80°C for later myeloperoxidase (MPO) assay; a separate portion was fixed overnight at 4°C in 10% formalin for histopathological analysis. Mixed arteriovenous blood was also collected by decapitation for serum amylase measurement.
Serum amylase activity
Mixed arteriovenous blood was centrifuged at room temperature for 10 min at 1500 g. The serum amylase concentration was measured as described previously (14) except that Phadebas amylase test tablets (Magle Life Sciences, Cambridge, MA, USA) were used as substrate instead of procion yellow starch. A standard curve was prepared using crude type VI-B α-amylase (Sigma-Aldrich), and serum amylase levels were expressed as units per milliliter and normalized to control values.
MPO activity
The harvested pancreata were immediately frozen on dry ice and then stored at −80°C until assay. We measured the tissue activity of MPO, an enzyme produced by neutrophils and used as a marker of inflammation associated with neutrophil infiltration, as described previously using the substrate tetramethylbenzidine (15, 16). Pancreatic MPO activity was expressed as units per milligram total protein and normalized to control values. Pancreatic total protein was measured using microBCA kits (Thermo Scientific, Rockford, IL, USA).
Histopathology
Portions of the pancreata were fixed overnight in phosphate-buffered 10% formalin. The tissue was then embedded in paraffin, sectioned at 5 μm, stained with hematoxylin and eosin, and coded for examination in a blinded procedure by two investigators unaware of the experimental design. The severity of pancreatitis was graded using modified scoring criteria previously described (17). The results were expressed in increments of 0.5 as a score of 0 to 3 for the histological parameters of edema and neutrophil infiltration and as a score of 0 to 7 for tissue necrosis and hemorrhage. The total histopathology score is the mean of the combined scores for edema, neutrophil infiltration, necrosis, and hemorrhage from both investigators.
Statistical analysis
Results are expressed as means ± sem. Mean differences between 2 groups were examined by the Student's t test and mean differences among several groups by 1-way ANOVA with the Dunnett's or Tukey-Kramer posttests, using GraphPad InStat 3.05 for Windows (GraphPad Software, San Diego, CA, USA). Values of P < 0.05 were considered significant.
RESULTS
To determine whether ethanol or POA affected the pancreas, we injected either ethanol (1.32 g/kg) or POA (2 mg/kg) intraperitoneally into mice. We observed that neither ethanol nor POA given alone affected pancreatic histology, serum amylase levels, pancreatic edema, or pancreatic MPO concentrations (Fig. 1). No damage to other abdominal organs was seen in response to ethanol or POA alone. In contrast, however, when given together, ethanol and POA caused marked changes in pancreatic histology, and significantly increased serum amylase levels and pancreatic MPO concentrations. Intense pancreatic edema, acinar cell necrosis, and neutrophil infiltration were observed in the pancreas of dual-treated mice (Fig. 1D). These findings are consistent with those seen in this and other experimental models of acute pancreatitis by us and others.
Figure 1.

Effects of ethanol (EtOH) alone, POA alone, and EtOH + POA on acute pancreatic inflammation in wild-type mice. A) Normal histology in control mice treated with vehicle. B) Lack of effects, other than mild edema, of treatment with EtOH alone on pancreatic histology. C) Lack of effects, other than mild edema, of treatment with POA alone on pancreatic histology. D) Extensive effects of treatment with EtOH + POA on pancreatic histology, including more intense edema, acinar cell necrosis, and neutrophil infiltration. E) Quantitation of the histopathology score in the various groups. F) Serum amylase levels, expressed as U/ml, in the various groups. G) Pancreatic edema, expressed as the ratio between pancreas wet weight and total body weight, in the various groups. H) MPO concentration in the pancreas, expressed as U/mg pancreatic protein, in the various groups. *P < 0.05, ***P < 0.001 vs. control.
Knowing that ethanol sensitizes TRPV1 and that TRPV1 activation causes pancreatitis, we hypothesized that ethanol may sensitize mice to the potential injurious effects of POA by activating TRPV1. We therefore tested the effects of pretreatment with the TRPV1 antagonist AMG9810 in animals treated with ethanol plus POA. AMG9810 was administered 30 min before ethanol and POA were given. As shown in Fig. 2, AMG9810 significantly inhibited some features of pancreatitis. Pancreatic histopathology improved by 63% (P<0.01), and serum amylase levels declined by 58% (P<0.05) There was a nonsignificant decline in pancreatic edema and MPO. On histological evaluation, AMG9810 markedly improved acinar cell necrosis and neutrophil infiltration (Fig. 2C).
Figure 2.
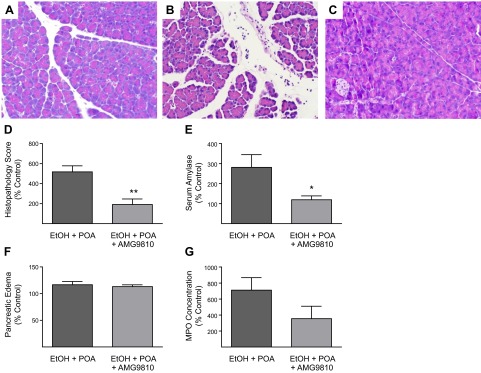
Effects of the TRPV1 antagonist AMG9810 on ethanol + POA-induced pancreatitis in wild-type mice. A) Normal histology in control mice treated with vehicle. B) Extensive effects of treatment with ethanol (EtOH) + POA on pancreatic histology, including more intense edema, acinar cell necrosis, and neutrophil infiltration. C) The protective effects of pretreating the mice with an intraperitoneal injection 30 mg/kg of AMG9810 30 min before EtOH + POA injection. D) Quantitation of the histopathology score, expressed as percentage of control, in the various groups. E) Serum amylase levels, expressed as percentage of control, in the various groups. F) Pancreatic edema, expressed as percentage of control, in the various groups. G) MPO concentration in the pancreas, expressed as percentage of control, in the various groups. Differences between groups in F and G are not statistically significant. *P < 0.05, **P < 0.01 vs. EtOH + POA.
We further investigated the possible role of TRPV1 in the pathogenesis of ethanol plus POA-induced pancreatitis by examining Trpv1−/− mice. These mice do not respond to selective noxious and deleterious stimuli that normally activate TRPV1, such as thermal pain or capsaicin administration (18). In a pattern similar to that seen after AMG9810 pretreatment, the histopathological score and serum amylase effects of ethanol plus POA on the pancreas were significantly reduced when these inflammatory insults were administered to mice with a genetic deletion of the Trpv1 gene (Fig. 3). On careful histological examination, the inflammatory changes normally induced by ethanol plus POA in wild-type animals were blocked by genetic deletion of Trpv1 (Fig. 3C). This was confirmed quantitatively by the histopathology score, which was reduced by 98% (P<0.01) in Trpv1−/− mice compared to wild-type mice receiving ethanol plus POA (Fig. 3D). Consistent with these histological findings, serum amylase levels were reduced by 76% (P<0.05) in the Trpv1−/− mice (Fig. 3E).
Figure 3.
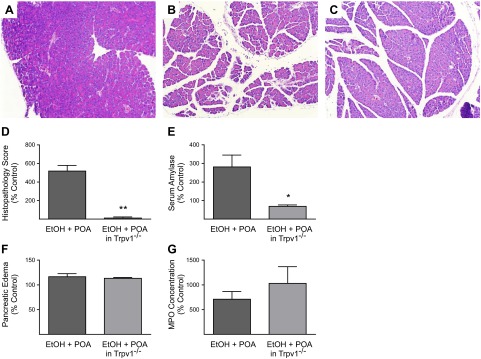
Effects of genetic deletion of the Trpv1 gene (Trpv1−/−) on ethanol (EtOH) + POA-induced pancreatitis in mice. A) Normal histology in control wild-type mice treated with vehicle. B) Extensive effects of treatment with EtOH + POA on pancreatic histology, including more intense edema, acinar cell necrosis, and neutrophil infiltration in wild-type mice. C) Protective effects of genetic deletion of the Trpv1 gene on EtOH + POA-induced acute pancreatitis in Trpv1−/− mice. D) Quantitation of the histopathology score, expressed as percentage of wild-type control, in Trpv1−/− vs. wild-type mice. E) Serum amylase levels, expressed as percentage of wild-type control, in Trpv1−/− vs. wild-type mice. F) Pancreatic edema, expressed as percentage of wild-type control, in Trpv1−/− vs. wild-type mice. G) MPO concentration in the pancreas, expressed as percentage of wild-type control, in Trpv1−/− vs. wild-type mice. Differences between groups in F and G are not statistically significant. *P < 0.05, **P < 0.01 vs. EtOH + POA.
DISCUSSION
We investigated the possibility that ethanol, through its ability to sensitize and activate TRPV1, contributes to development of acute pancreatitis. We used a model of acute pancreatitis that allowed us to study the effects of alcohol treatment independent of changes seen with chronic alcohol administration. We felt that this approach would allow us most closely to approximate the direct effects of alcohol on TRPV1 that had been observed in vitro. Notably, we observed that neither alcohol nor POA alone significantly damaged the pancreas. In contrast, however, together these agents produced a pattern of pancreatic damage that closely resembled other established models of acute pancreatitis and are consistent with those previously reported in this model (5). The mechanisms by which ethanol and POA cause acute pancreatitis in mice are illustrated in Fig. 4.
Figure 4.

Model depicting the mechanisms by which ethanol and POA cause acute pancreatitis in mice. Ethanol sensitizes TRPV1 expressed in pancreatic primary sensory neurons so that it is activated at body temperature, instead of at its normal temperature threshold of ∼42°C, resulting in the release of proinflammatory neurotransmitters, such as substance P, at peripheral and central nerve terminals. POA has been shown to act on pancreatic acinar cells to cause an unphysiological increase in cytosolic calcium levels, resulting in intracellular trypsin activation and acinar cell necrosis (4, 23). The combined effects of ethanol on TRPV1 and POA on acinar cells cause acute pancreatitis. Blocking TRPV1 activation either pharmacologically with AMG9810 or genetically by deletion of Trpv1 protects against ethanol plus POA-induced acute pancreatitis.
Neurogenic inflammation has been implicated in the pathogenesis of acute pancreatitis in a variety of experimental models including secretagogue-induced (caerulein) pancreatitis, bile acid injury, and a model of ERCP pancreatitis in mice and rats (6, 7). The central feature of neurogenic pancreatitis is the activation of TRPV1 on primary sensory nerves whose activation causes the release of inflammatory neuropeptides such as substance P both in the spinal cord (to signal pain) and in the pancreas leading to the features of local inflammation (e.g., neutrophil recruitment and necrosis). It is presumed that following pancreatic injury endogenous TRPV1 activators are produced locally in the pancreas that activate TRPV1 leading to subsequent neurogenic inflammation. We hypothesized that alcohol may sensitize TRPV1, lowering its activation threshold. If this hypothesis is correct, we would expect to see less severe pancreatitis in the setting of TRPV1 blockade. In the current study, we used both pharmacologic and genetic approaches to assess the role of TRPV1 in an alcohol model of acute pancreatitis. The drug AMG9810 is a pharmacological antagonist of TRPV1 activation and has been shown to be a potent and selective TRPV1 antagonist in rats (19, 20), dogs and monkeys (20), and mice (21). We found that pretreatment of mice with AMG9810 resulted in significant inhibition of both ethanol plus POA-induced histological damage of the pancreas and serum amylase levels consistent with protection against pancreatitis.
Similarly, when ethanol plus POA was given to Trpv1−/− mice, pancreatitis parameters were significantly reduced compared to wild-type mice. Taken together, the effects of AMG9810 and the results observed in Trpv1−/− mice indicate that ethanol plus POA administration results in activation of TRPV1 and subsequent neurogenic inflammation of the pancreas. Interestingly, neither pharmacological antagonism of TRPV1 nor deletion of the Trpv1 gene significantly reduced pancreatic MPO content, suggesting that the reductions in histopathology score and in serum amylase levels observed after these treatments may not be due to reduced neutrophil infiltration into the pancreas.
The pancreas has been shown to synthesize more FAEEs than any other human organ (1). Within cells, FAEEs are hydrolyzed, producing fatty acids such as POA, which is therefore likely to be produced preferentially in the pancreas. Infusion of FAEEs alone has been shown to cause acute inflammatory changes in the rat pancreas (2). When ethanol was infused by itself, similar damage was observed in the rat pancreas, and these changes were associated with the formation of FAEEs in the pancreas (3, 22). It has previously been demonstrated that FAEEs and POA (but not acetaldehyde, the main oxidative ethanol metabolite) cause sustained unphysiological increases in cytosolic calcium concentrations in mouse pancreatic acinar cells, leading to intracellular trypsin activation and cell death (4, 23). These observations together suggest that one of the mechanisms involved in alcohol-induced acute pancreatic damage involves direct effects of FAEEs on pancreatic acinar cells. In addition, it has been demonstrated that ethanol has little if any effect on cytosolic calcium concentrations in mouse pancreatic acinar cells, suggesting that if ethanol directly participates in the etiology of pancreatitis it is not via disruption of acinar cell calcium levels (4). Our results indicated that POA when given alone (at the same dose that caused acute damage when combined with ethanol) had no effect on the pancreas, suggesting that ethanol has an inflammatory effect independent of its metabolism to FAEEs. It is possible that this additional effect involves a neurogenic mechanism via activation of TRPV1 in primary sensory neurons; such a mechanism has been shown to be involved in multiple experimental models of acute pancreatitis (7).
The sensitizing effect of ethanol on TRPV1 in primary sensory nerves was observed previously to occur at ethanol concentrations of 0.1–3% (17–510 mM; ref. 12). This is clearly within the range of blood ethanol concentrations that might be found in people consuming alcohol. The dose of ethanol used in the present study resulted in a peak serum ethanol concentration of 35 mM (5), which is within this range, making it likely that human ethanol consumption results in TRPV1 sensitization at normal body temperature. This sensitization may be sufficient in susceptible individuals to result in reaching the threshold for initiation of an acute neurogenic inflammatory response in the pancreas and suggests a potential novel approach to treating alcohol-induced acute pancreatitis in humans.
Acknowledgments
The authors thank Dr. D. Julius (University of California, San Francisco, CA, USA) for the generous gift of Trpv1−/− mice.
This work was supported by U.S. National Institutes of Health grant R01 DK-064213.
Footnotes
- AMG9810
- (E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide
- DMSO
- dimethyl sulfoxide
- ERCP
- endoscopic retrograde cholangiopancreatography
- FAEE
- fatty acid ethyl ester
- MPO
- myeloperoxidase
- POA
- palmitoleic acid
- TRPV1
- transient receptor potential vanilloid 1
REFERENCES
- 1. Laposata E. A., Lange L. G. (1986) Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science 231, 497–499 [DOI] [PubMed] [Google Scholar]
- 2. Werner J., Laposata M., Fernandez-del Castillo C., Saghir M., Iozzo R. V., Lewandrowski K. B., Warshaw A. L. (1997) Pancreatic injury in rats induced by fatty acid ethyl ester, a nonoxidative metabolite of alcohol. Gastroenterology 113, 286–294 [DOI] [PubMed] [Google Scholar]
- 3. Werner J., Saghir M., Warshaw A. L., Lewandrowski K. B., Laposata M., Iozzo R. V., Carter E. A., Schatz R. J., Fernandez-Del Castillo C. (2002) Alcoholic pancreatitis in rats: injury from nonoxidative metabolites of ethanol. Am. J. Physiol. 283, G65–G73 [DOI] [PubMed] [Google Scholar]
- 4. Criddle D. N., Raraty M. G., Neoptolemos J. P., Tepikin A. V., Petersen O. H., Sutton R. (2004) Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc. Natl. Acad. Sci. U. S. A. 101, 10738–10743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang W., Mukherjee R., Elliott V., Booth D. M., Tepikin A., Criddle D. N., Sutton R. (2010) A novel model of acute alcoholic pancreatitis by concomitant administration of ethanol and fatty acid. Pancreas 39, 1324 [Google Scholar]
- 6. Liddle R. A., Nathan J. D. (2004) Neurogenic inflammation and pancreatitis. Pancreatology 4, 551–559, discussion 559–560 [DOI] [PubMed] [Google Scholar]
- 7. Liddle R. A. (2007) The role of transient receptor potential vanilloid 1 (TRPV1) channels in pancreatitis. Biochim. Biophys. Acta 1772, 869–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Caterina M. J., Schumacher M. A., Tominaga M., Rosen T. A., Levine J. D., Julius D. (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816. [DOI] [PubMed] [Google Scholar]
- 9. Szallasi A., DiMarzo V. (2000) New perspectives on enigmatic vanilloid receptors. Trends Neurosci. 23, 491. [DOI] [PubMed] [Google Scholar]
- 10. Caterina M. J., Julius D. (2001) The vanilloid receptor: A molecular gateway to the pain pathway. Ann. Rev. Neurosci. 24, 487–517 [DOI] [PubMed] [Google Scholar]
- 11. Hwang S. W., Cho H., Kwak J., Lee S. Y., Kang C. J., Jung J., Cho S., Min K. H., Suh Y. G., Kim D., Oh U. (2000) Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. U. S. A. 97, 6155–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trevisani M., Smart D., Gunthorpe M. J., Tognetto M., Barbieri M., Campi B., Amadesi S., Gray J., Jerman J. C., Brough S. J., Owen D., Smith G. D., Randall A. D., Harrison S., Bianchi A., Davis J. B., Geppetti P. (2002) Ethanol elicits and potentiates nociceptor responses via the vanilloid receptor-1. Nat. Neurosci. 5, 546–551 [DOI] [PubMed] [Google Scholar]
- 13. Romac J. M., McCall S. J., Humphrey J. E., Heo J., Liddle R. A. (2008) Pharmacologic disruption of TRPV1-expressing primary sensory neurons but not genetic deletion of TRPV1 protects mice against pancreatitis. Pancreas 36, 394–401 [DOI] [PubMed] [Google Scholar]
- 14. Nathan J. D., Romac J., Peng R. Y., Peyton M., Macdonald R. J., Liddle R. A. (2005) Transgenic expression of pancreatic secretory trypsin inhibitor-I ameliorates secretagogue-induced pancreatitis in mice. Gastroenterology 128, 717–727 [DOI] [PubMed] [Google Scholar]
- 15. Bradley P. P., Priebat D. A., Christensen R. D., Rothstein G. (1982) Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J. Invest. Dermatol. 78, 206–209 [DOI] [PubMed] [Google Scholar]
- 16. Dawra R., Ku Y. S., Sharif R., Dhaulakhandi D., Phillips P., Dudeja V., Saluja A. K. (2008) An improved method for extracting myeloperoxidase and determining its activity in the pancreas and lungs during pancreatitis. Pancreas 37, 62–68 [DOI] [PubMed] [Google Scholar]
- 17. Spormann H., Sokolowski A., Letko G. (1989) Effect of temporary ischemia upon development and histological patterns of acute pancreatitis in the rat. Pathol. Res. Pract. 184, 507–513 [DOI] [PubMed] [Google Scholar]
- 18. Caterina M. J., Leffler A., Malmberg A. B., Martin W. J., Trafton J., Petersen-Zeitz K. R., Koltzenburg M., Basbaum A. I., Julius D. (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313 [DOI] [PubMed] [Google Scholar]
- 19. Gavva N. R., Tamir R., Qu Y., Klionsky L., Zhang T. J., Immke D., Wang J., Zhu D., Vanderah T. W., Porreca F., Doherty E. M., Norman M. H., Wild K. D., Bannon A. W., Louis J. C., Treanor J. J. (2005) AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J. Pharmacol. Exp. Ther. 313, 474–484 [DOI] [PubMed] [Google Scholar]
- 20. Gavva N. R., Bannon A. W., Surapaneni S., Hovland D. N., Jr., Lehto S. G., Gore A., Juan T., Deng H., Han B., Klionsky L., Kuang R., Le A., Tamir R., Wang J., Youngblood B., Zhu D., Norman M. H., Magal E., Treanor J. J., Louis J. C. (2007) The vanilloid receptor TRPV1 is tonically activated in vivo and involved in body temperature regulation. J. Neurosci. 27, 3366–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Long D. J., Devantier H. R., Brennan F. X., Bryant R. W., Salemme F. R., Palmer R. K. Pharmacologic antagonism of the oral aversive taste-directed response to capsaicin in a mouse brief access taste aversion assay. J. Pharmacol. Exp. Ther. 332, 525–530 [DOI] [PubMed] [Google Scholar]
- 22. Werner J., Saghir M., Fernandez-del Castillo C., Warshaw A. L., Laposata M. (2001) Linkage of oxidative and nonoxidative ethanol metabolism in the pancreas and toxicity of nonoxidative ethanol metabolites for pancreatic acinar cells. Surgery 129, 736–744 [DOI] [PubMed] [Google Scholar]
- 23. Gerasimenko J. V., Lur G., Sherwood M. W., Ebisui E., Tepikin A. V., Mikoshiba K., Gerasimenko O. V., Petersen O. H. (2009) Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc. Natl. Acad. Sci. U. S. A. 106, 10758–10763 [DOI] [PMC free article] [PubMed] [Google Scholar]