

Abstract
Chronic inflammation is associated with cachexia-induced skeletal muscle mass loss in cancer. Levels of IL-6 cytokine family members are increased during cancer-related cachexia and induce intracellular signaling through glycoprotein130 (gp130). Although muscle STAT3 and circulating IL-6 are implicated in cancer-induced muscle wasting, there is limited understanding of muscle gp130's role in this process. Therefore, we investigated the role of skeletal muscle gp130 (skm-gp130) in cancer-induced alterations in the regulation of muscle protein turnover. Lewis lung carcinoma (LLC) cells were injected into 8-wk-old skm-gp130-knockout (KO) mice or wild-type mice. Skeletal muscle loss was attenuated by 16% in gp130-KO mice, which coincided with attenuated LLC-induced phosphorylation of muscle STAT3, p38, and FOXO3. gp130 KO did not restore mTOR inhibition or alter AMP-activated protein kinase (AMPK) expression. The induction of atrogin expression and p38 phosphorylation in C2C12 myotubes exposed to LLC-treated medium was attenuated by gp130 inhibition, but mTOR inhibition was not restored. STAT signaling inhibition in LLC-treated myotubes did not attenuate the induction of p38 or AMPK phosphorylation. We concluded that, during LLC-induced cachexia, skm-gp130 regulates muscle mass signaling through STAT3 and p38 for the activation of FOXO3 and atrogin, but does not directly regulate the suppression of mTOR.—Puppa, M. J., Gao, S., Narsale, A. A., Carson, J. A. Skeletal muscle glycoprotein 130's role in Lewis lung carcinoma–induced cachexia.
Keywords: inflammation, cancer, STAT3, protein turnover, C2C12 myotube, IL-6
Cachexia, the unintentional loss of body weight, including muscle and fat mass, is associated with many cancer types (1–3). It occurs in ∼20% of all patients with cancer and is responsible for 40% of colon cancer–related deaths (4, 5). Chronic inflammation is associated with diseases that induce muscle wasting, including cancer (6). Several cytokines are up-regulated during cachexia in both human cancer and animal models, such as the ApcMin/+ mouse, C26 adenocarcinoma, and Lewis lung carcinoma (LLC), including IL-6, TNF-α, IL-1β, LIF, CNTF, IFN-γ, and IL-10 (7–10). It is clear that cytokines play a vital role in the development of muscle atrophy in cachexia. Anticytokine therapy has been widely used to combat muscle wasting during cancer-induced cachexia (11–13). However, further research is needed to understand whether inflammatory cytokines exert direct or indirect effects on skeletal muscle, in altering protein turnover and inducing wasting.
Circulating levels of the IL-6 family of cytokines are associated with cachexia in cancer (6, 10, 14–17). IL-6 can signal via the classic pathway, through the membrane receptor, or through the trans pathway, utilizing soluble IL-6 receptor (IL-6r) to induce signaling; however, glycoprotein 130 (gp130) is necessary for this signaling (18). The IL-6 family of cytokines signals through gp130 by forming either a heterodimer or homodimer with the cytokine, its receptor, and gp130. Gp130 dimerization leads to activation of several intracellular signaling pathways, including JAK/STAT, p38/MAPK, and PI3K/Akt (19, 20). In skeletal muscle, these signaling pathways have been associated with the regulation of growth and atrophy (21–23). Although researchers have examined the role of STAT3 and IL-6 specifically, the role of gp130 in skeletal muscle wasting during cachexia has not been investigated.
Skeletal muscle mass is regulated by a balance of protein synthesis and protein degradation, termed protein turnover. Altered protein turnover is an established regulatory point of both skeletal muscle mass loss and muscle growth. During cancer-related cachexia, there is an increase in skeletal muscle protein degradation and suppression of muscle protein synthesis (MPS; ref. 6). Muscle STAT3 signaling is sufficient to induce skeletal muscle atrophy both in vitro and in vivo, and STAT3 inhibition can attenuate cancer-induced muscle atrophy (10, 21). P38/MAPK and C/EBPβ also mediate cancer-induced muscle atrophy through the inhibition of FOXO1/3 phosphorylation and the activation of atrogin-1 (24, 25). Although there is strong evidence that gp130-mediated signaling regulates cancer-induced protein degradation, its role in the suppression of protein synthesis during cachexia remains poorly defined.
We and others have defined important roles for IL-6, muscle STAT3, and p38/MAPK signaling in cancer-induced muscle wasting (21, 24, 26), yet significant gaps remain in our understanding of the relationship of these signaling pathways to the IL-6 family of cytokines and their regulation of the suppression of MPS in cachexia. Gp130 is a common regulatory point for the IL-6 family of cytokines and for STAT3 and p38 signaling; however, STAT3 and p38 can be activated by other signaling cascades. In addition, gp130 regulates pathways other than STAT3 and p38/MAPK, such as PI3K/Akt (19). The purpose of this study was to examine the role of skeletal muscle–specific gp130 (skm-gp130) in the regulation of muscle protein turnover during LLC-induced cachexia. We hypothesized that skm-gp130 is necessary for muscle STAT3-mediated inhibition of mTOR signaling and FOXO3a activation during LLC-induced cachexia. We examined a muscle-specific knockout (KO) of gp130 during LLC-induced cachexia. The role of systemic IL-6 and STAT signaling were investigated with the administration of pyrrolidine dithiocarbamate (PDTC) or IL-6r antibody (Ab) to mice with LLC tumors.
MATERIALS AND METHODS
Animals
Male mice on a C57BL/6 background were bred with gp130fl/fl mice provided by Dr. Colin Stewart's laboratory [Laboratory of Cancer and Developmental Biology, National Cancer Institute, U.S. National Institutes of Health (NIH), Frederick, MD, USA] in collaboration with Dr. Lothar Hennighausen (Laboratory of Genetics and Physiology, National Institute of Diabetes and Digestive and Kidney Diseases, NIH, Bethesda, MD, USA; ref. 27). Gp130fl/fl male mice were bred with Cre-expressing mice driven by myosin light chain (MLC), from Dr. Steven Burden (New York University, New York, NY, USA; ref. 28). The resulting fl/fl cre/cre (skm-gp130) mice have a skeletal muscle deletion of gp130. Offspring were genotyped by using tail snips to obtain Cre recombinase (forward, 5′-AAGCCCTGACCCTTTAGATTCCATTT-3′, and reverse, 5′-AAAACGCCTGGCGATCCCTGAAC-3′; and wild type 5′-GCGGGCTTCTTCACGTCTTTCTTT-3′) and floxed gp130 (forward, 5′-ACGTCACAGAGCTGAGTGATGCAC-3′, and reverse, 5′-GGCTTTTCCTCTGGTTCTTG-3′). All animal experimentation was approved by the University of South Carolina's Institutional Animal Care and Use Committee.
IL-6 overexpression
The skm-gp130 and C57BL/6 mice were electroporated at 12 wk of age with either an empty vector or an IL-6 plasmid (12).
LLC cell implantation
C57BL/6 and skm-gp130 mice were injected with 1 × 106 LLC cells subcutaneously in the right flank at 8 wk of age (29). Body weights were measured weekly, starting at 8 wk. Tumors were allowed to grow for ∼30 d, and the mice were euthanized at 13 wk of age.
IL-6r Ab administration
After 4 wk of tumor growth, a subset of mice were treated with 100 μg of IL-6r Ab (Chugai Pharmaceutical Co., Ltd., Tokyo, Japan), according to a published method (30), with modifications. The animals were treated with i.p. injections every 3 d for 1 wk, receiving IL-6r Ab once every 3 d.
PDTC
After 4 wk of tumor growth, a subset of mice was treated with daily i.p. injections with 10 mg/kg PDTC in PBS (31) for 1 wk.
Tissue sampling
At the study end points, the mice were anesthetized with a ketamine/xylazine/acepromazine cocktail, and tissues were removed, weighed, and frozen at −80°C until further analysis. Blood samples obtained from the retroorbital sinus were collected in heparinized capillary tubes.
C2C12 cells
C2C12 myoblasts (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin, and differentiation was induced (26). Fully differentiated myotubes were treated with LLC-conditioned medium (LCM) for 4 or 72 h, with the medium replaced every 24 h in the control and treatment groups (24). IL-6r Ab (Chugai Pharma) or gp130 Ab (dialyzed in PBS at 4°C overnight; Santa Cruz Biotechnology, Santa Cruz CA, USA) was added to the LCM for 72 h at a 1:1000 dilution. PDTC (50 μM; Sigma-Aldrich, St. Louis, MO, USA) and LLL12 (BioVision, Milpitas, CA, USA), a STAT3 inhibitor (100 nM), were added to the LCM for the duration of the study (26). The rate of protein synthesis was determined by adding 1 μM puromycin to the culture medium 30 min before protein collection (32) and then harvesting the cells (26).
Myotube diameter measurement
C2C12 myotube diameter was quantified as previously published (26). All measurements were conducted in a blinded fashion.
Plasma IL-6
Plasma IL-6 was quantified as previously published (14). Briefly, blood samples were centrifuged at 10,000 g for 10 min at 4°C. Plasma was collected and stored at −80°C until analysis. An ultrasensitive mouse IL-6 ELISA (Invitrogen, Carlsbad, CA, USA) was performed according to the manufacturer's instructions.
RNA isolation and PCR
RNA isolation, cDNA synthesis, and real-time PCR were performed as previously described (26) with reagents from ABI (Foster City, CA, USA). Gp130 (forward, 5′-CAGCGTACACTGATGAAGGTGGGAAA-3′, and reverse, 5′-GCTGACTGCAGTTCTGCTTGA-3′), IGF-1 (33), REDD1 (33), IL-6 (34), and GAPDH primers were purchased from IDT (Coralville, IA, USA). Data were analyzed by the cycle threshold (CT) method with ABI software.
Western blot analysis
Western blot analysis was performed as previously described (35). Briefly, gastrocnemius muscle was homogenized, and protein concentration was determined by the Bradford method (36). Homogenates were fractionated on SDS-polyacrylamide gels and transferred to PVDF membrane. After the membranes were blocked, Abs for phosphorylated and total 4EBP1, AMP-activated protein kinase (AMPK), S6RP, STAT3, Akt, ubiquitin, GAPDH (Cell Signaling Technology, Danvers, MA, USA), p38 (Santa Cruz Biotechnology), FOXO3a, anti-puromycin (Millipore, Billerica, MA, USA), and atrogin-1 (ECM Biosciences, Versailles, KY, USA) were incubated at dilutions of 1:2000 to 1:6000 overnight at 4°C in 1% TBST milk. Anti-rabbit or anti-mouse IgG-conjugated secondary Abs (Cell Signaling Technology) were incubated with the membranes at 1:2000 to 1:5000 dilutions for 1 h in 1% TBST milk. Enhanced chemiluminescence developed by autoradiography was used to visualize the Ab–antigen interactions. Blots were analyzed by measuring the integrated optical density (IOD) of each band with ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
A 2-way ANOVA was used to examine the effects of LLC and genotype. Post hoc analyses were performed with Student-Newman-Keuls methods. Preplanned t tests were used to examine the effects of gp130 loss in control mice to define the phenotype. All C2C12 experiments were analyzed by 1-way ANOVA. Values of P < 0.05 were considered significant.
RESULTS
Expression of gp130 in C57BL/6 mice
Gp130 mRNA expression was examined in adult C57BL/6 mice. Although gp130 is expressed in all cell types, muscle had significantly greater expression than did liver or kidney (Fig. 1A). Skeletal muscle had the greatest expression level, and expression in heart was 67% lower than that in gastrocnemius skeletal muscle. Expression was greatest in glycolytic tibialis anterior (TA) muscle, and the lowest expression was found in oxidative soleus muscle (Fig. 1B). These data indicate that, although gp130 was ubiquitously expressed in these mice, there was differential expression in tissues and in skeletal muscle phenotypes.
Figure 1.
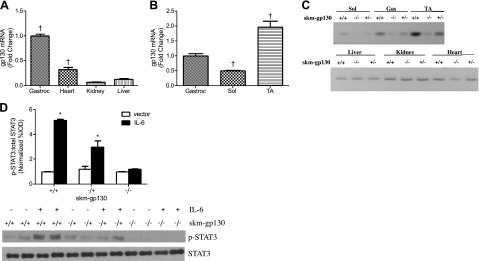
Mouse tissue expression of gp130. A) gp130 was differentially expressed in tissues. B) Differential expression of gp130 mRNA in the soleus (Sol), gastrocnemius (Gastroc), and TA muscles of C57BL/6 mice. C) PCR analysis of gp130 mRNA (643 bp product size) in the gastrocnemius (Gas), soleus (Sol), and TA muscles and the liver, kidney, and heart of skm-gp130+/+, skm-gp130+/−, and skm-gp130−/− mice. D) STAT3 protein phosphorylation in heterozygous and homozygous skm-gp130-KO mice after 2 wk of IL-6 overexpression. All values are means ± sem. *P < 0.05 vs. genotype vector control; †P < 0.05 vs. all other groups.
Characterization of skm-gp130 mice
The Cre-loxP approach was used to generate a muscle-specific KO of gp130 in C57BL/6 mice (Fig. 1C, D) by using MLC Cre-expressing mice (28) and gp130 fl/fl mice (27). In skm-gp130 mice, gp130 mRNA expression was significantly reduced in all hind-limb muscles examined and heart, but was not altered in liver or kidney (Fig. 1C). Heart Cre activity has been reported in this Cre mouse (28). Since all cell types expressed gp130, cells in skeletal muscle other than myofibers continue to express gp130. There was no effect of genotype on overall body size, measured by tibia length (Table 1). The body weight increase in the skm-gp130 mice was associated with an increase in lean mass. This finding may be partially attributable to increased organ mass; for example, the skm-gp130 mice had significantly larger heart and testes mass. Genotype had no effect on gastrocnemius muscle and epididymal fat mass.
Table 1.
Changes in body weight, fat mass, and muscle mass with LLC-induced cachexia
| Parameter | BL/6 |
skm-gp130 |
||
|---|---|---|---|---|
| Control | LLC | Control | LLC | |
| n | 5 | 6 | 5 | 6 |
| Body weight | ||||
| BW at death (g) | 25.2 ± 0.4 | 24.4 ± 1.1 | 27.2 ± 0.4@ | 27.5 ± 0.8@ |
| BW minus tumor (g) | 25.2 ± 0.5 | 22.4 ± 0.5# | 27.2 ± 0.4 | 24.7 ± 0.5* |
| Lean mass (g) | 18.8 ± 0.3 | 17.3 ± 0.4# | 20.2 ± 1.2@ | 19.3 ± 0.6@ |
| Tibia length (mm) | 16.7 ± 0.1 | 16.8 ± 0.1 | 16.9 ± 0.1 | 16.8 ± 0.0 |
| Epididymal fat (mg) | 376 ± 32 | 239 ± 55§ | 372 ± 25 | 258 ± 26§ |
| Gastrocnemius (mg) | 133.5 ± 2.4 | 106.8 ± 5.3# | 125.8 ± 4.9 | 120.6 ± 5.6 |
| Organs | ||||
| Heart (mg) | 96.8 ± 0.9 | 99.2 ± 6.1.6 | 117.0 ± 4.6@ | 105.7 ± 2.6@ |
| Testes (mg) | 182 ± 3 | 178 ± 7 | 215 ± 6@ | 210 ± 5@ |
| Spleen (mg) | 87.6 ± 3.7 | 247.0 ± 58.4§ | 91.8 ± 1.8 | 244.0 ± 44.2§ |
| BMD (g/cm2) | 0.0514 ± 0.001 | 0.0467 ± 0.001* | 0.0482 ± 0.001 | 0.0493 ± 0.001 |
LLC cells (1×106) were implanted in 8-wk-old skm-gp130 mice. After death, the muscle, epididymal fat, and organs of each mouse were excised and weighed, and a DEXA scan was performed to determine bone mineral density (BMD) and tumor-free lean mass. All values are means ± sem.
P < 0.05 vs. control within genotype;
P < 0.05 vs. all other groups;
P < 0.05, main effect of skm-gp130,
P < 0.05, main effect of LLC.
Skm-gp130 loss attenuated Il-6-induced STAT3 phosphorylation
IL-6 was systemically overexpressed to examine gp130 function. Circulating IL-6 was not detectable in vector control mice, but was increased by overexpression in all genotypes (skm-gp130+/+, 86.2±12.1 pg/ml; skm-gp130+/−, 70.2±14.2 pg/ml; and skm-gp130−/−, 71.0±25.9 pg/ml). Skeletal muscle STAT3 phosphorylation (Y705) in the quadriceps of the wild-type mice was increased 5-fold by IL-6 overexpression. This induction was suppressed in skm-gp130 heterozygous mice and blocked in skm-gp130 homozygous mice (Fig. 1D).
LLC induced body weight loss in skm-gp130 mice
We examined the role of muscle gp130 in cancer-induced muscle loss by implanting LLC cells in wild-type and skm-gp130 mice (Table 1 and Fig. 2). LLC-induced cachexia decreased muscle gp130 mRNA expression by 50% (Fig. 2A). We did not detect gp130 mRNA expression in the control or LLC cell–implanted skm-gp130 mice. Loss of muscle gp130 had no effect on tumor mass (Fig. 2B). LLC decreased body weight by 11% in the wild-type mice, and the loss was not attenuated in the skm-gp130 mice (Fig. 2C). Cachexia-related lean body mass loss, measured by DEXA scanning, was attenuated in the skm-gp130 mice when compared to that in the wild-type mice (Table 1). LLC induced a 20% reduction in gastrocnemius muscle mass, which was attenuated in the skm-gp130 mice (Fig. 2D). LLC cell implantation decreased epididymal fat mass by 36% and induced splenomegaly in the wild-type mice. Muscle gp130 loss had no effect on LLC-induced loss of epididymal fat mass or spleen size (Table 1). Plasma IL-6 was increased by LLC cell implantation, and there was no effect of gp130 loss on circulating IL-6 levels (BL/6, 0.0±0.0 pg/ml; BL/6+LLC, 38.7±13.9 pg/ml; skm-gp130, 0.0±0.0 pg/ml; and skm-gp130+LLC 16.7±8.1 pg/ml). LLC had no effect on heart mass. However, the skm-gp130 mice had heart enlargement that was not affected by the LLC tumor (Table 1). LLC cell implantation decreased bone mineral density in the wild-type mice but not in the skm-gp130 mice, suggesting a muscle–bone interaction (Table 1).
Figure 2.

Effect of skm-gp130 on development of cancer-induced cachexia. LLC tumor cells were implanted in 8-wk-old wild-type BL/6 and skm-gp130−/− (skm-gp130) mice. Tumors were allowed to grow until 13 wk of age. A) Skm-gp130 expression changed with LLC-induced cachexia in wild-type and skm-gp130 mice in the gastrocnemius. ND, not detected. B) Tumor mass was measured at the time of death. C, D) Percentage change in body weight (BW) from genotype control (C) and percentage change in gastrocnemius muscle mass (D) were calculated from weights collected at the time of death. All values are means ± sem. ***P < 0.05 vs. BL/6 group.
LLC-induced muscle signaling pathways in skm-gp130-KO mice
The signaling that regulates muscle mass was examined in the gastrocnemius muscle of the wild-type and skm-gp130 mice (Fig. 3). LLC cell implantation increased muscle STAT3 and p65 phosphorylation, which was attenuated by skm-gp130 loss (Fig. 3A). LLC cell implantation induced muscle AMPK phosphorylation regardless of genotype, suggesting that LLC-induced AMPK phosphorylation does not require muscle gp130. The phosphorylation of p38 was induced by LLC cell implantation in the wild-type mice, and skm-gp130 loss ablated this induction (Fig. 3A). Basal STAT, AMPK, and p38 phosphorylation in muscle were not altered by gp130 loss, whereas Akt phosphorylation increased (Fig. 3B). LLC induced Akt (T308) phosphorylation in the wild-type mice, but phosphorylation of Akt (S473) was unaltered. The LLC induction of Akt (T308) was not affected by skm-gp130 loss.
Figure 3.
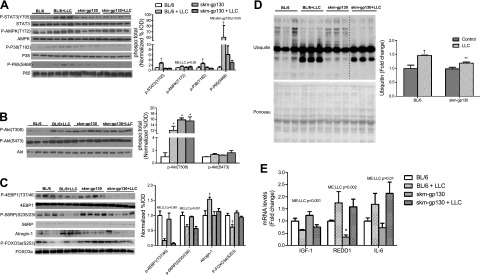
Effect of skm-gp130 on LLC-induced signaling. A) Western blot analysis of p-STAT3, total STAT3, p-AMPK, total AMPK, p-p38, total p38, p-NF-κB (p-p65), and total NF-κB protein expression. Ponceau staining verified equal loading. B) Western blot analysis of p-Akt (T308), p-Akt(S473), and total Akt. C) Western blot analysis of the mTOR signaling proteins p-4EBP1, total 4EPB1, p-S6, total S6, p-FOXO3, total FOXO3, and atrogin. Ponceau staining verified equal loading. B, C) Graphs represent the ratio of phosphorylated to total protein levels. D) Levels of ubiquitinated proteins were quantified by Western blot analysis, with equal loading verified by Ponceau stain. Dashed line indicates different sections of the same gel. E) Skeletal muscle mRNA expression of IGF-1, REDD1, and IL-6 was measured in the gastrocnemius. All values are means ± se. Two-way ANOVA was used to analyze the effect of skm-gp130 and LLC. *P < 0.05 vs. BL/6 control; **P < 0.05 vs. BL/6 LLC; †P < 0.05 vs. all other groups.
Regulation of the LLC-induced signaling that mediates protein turnover through mTOR and FOXO3a was examined in gastrocnemius muscle (Fig. 3C). In the wild-type mice, LLC suppressed the phosphorylation of the mTOR substrates p-4EBP-1 and p-S6RP, and this suppression was not affected by skm-gp130 loss (Fig. 3C). LLC cell implantation reduced FOXO3a phosphorylation, which was attenuated by skm-gp130 loss. LLC also induced atrogin-1 expression and the abundance of ubiquitinated proteins, both of which were attenuated by skm-gp130 loss (Fig. 3C, D). We measured the mRNA levels of IGF-1 and REDD1, which are established regulators of muscle anabolic signaling through mTOR (Fig. 3E). LLC cachexia decreased muscle IGF-1 mRNA expression, and gp130 loss did not reverse this suppression. Basal REDD1 expression was suppressed in the skm-gp130 mice. However, during LLC-induced cachexia, REDD1 expression was induced regardless of genotype (Fig. 3E). LLC-induced cachexia increased muscle IL-6 mRNA expression in both the wild-type and skm-gp130 gastrocnemius muscles (Fig. 3E).
Effect of LCM on the regulation of C2C12 myotube growth
To examine the direct role of LLC-secreted factors on the regulation of C2C12 myotube growth, myotubes were incubated in 15 or 25% LCM. LCM decreased myosin heavy chain expression (Fig. 4A), whereas only the higher dosage of LCM (25%) was sufficient (P≤0.01) to reduce myotube diameter (control, 32.3±0.7 μm; 15% LCM, 30.9±0.7 μm; and 25% LCM, 29.0±0.6 μm). High-dose LCM administration increased IL-6 mRNA levels, whereas 15% LCM had no effect on IL-6 mRNA levels (control: 1.00±0.10-fold; 15% LCM, 1.04±0.11-fold; and 25% LCM, 1.52±0.16-fold change from control). LCM induced p-STAT3 at 4 h, but it returned to baseline by 72 h (Fig. 4B). The phosphorylation of Akt (S473) was unaffected by LCM; however, LCM decreased mTOR phosphorylation and phosphorylation of the substrates 4EBP1 and S6RP in a dose-dependent manner (Fig. 4C). AMPK was induced after a 72 h exposure to LCM (Fig. 4C). LCM treatment suppressed FOXO3a phosphorylation, but induced atrogin-1 protein expression (Fig. 4D).
Figure 4.

Effect of LCM on myosin heavy chain level, STAT3 signaling, and protein turnover regulation in C2C12 myotubes. A) Myotube atrophy induced by 72 h LCM exposure was measured with protein levels of myosin heavy chain. B) Inflammatory signaling of STAT3 induced by administration of LCM for 4 or 72 h. C) Protein expression of p-Akt, total Akt, p-AMPK, total AMPK, p-mTOR, total mTOR, p-S6RP, total S6RP, p-4EBP1, and total 4EPB1 after 72 h treatment with LCM was measured by Western blot analysis. D) Protein expression of p-FOXO3, total FOXO3, atrogin, and GAPDH with various LLC levels was measured by Western blot analysis. Graphs represent the ratio of phosphorylated to total protein levels. Values are means ± sem. Data were analyzed with 1-way ANOVA. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005 vs. control group.
IL-6 inhibition in LLC-mediated C2C12 and in vivo atrophy
In another study, we demonstrated that IL-6 can induce C2C12 myotube atrophy (26), and we found circulating IL-6 to be significantly elevated (P=0.03) with LLC-induced cachexia (C57BL/6 0.0±0.0 pg/ml vs. C57BL/6 LLC 38.07±13.9 pg/ml). C2C12 myotubes were treated with either IL-6 or LCM, and then IL-6r Ab was administered. As previously reported, p-STAT3 was induced by IL-6 (26). IL-6r Ab blocked IL-6-induced STAT3 phosphorylation, but had no effect on LCM-induced STAT3 phosphorylation (Fig. 5A). LCM reduced the rate of protein synthesis in C2C12 myotubes by 31%, and IL-6r Ab treatment was not sufficient to reverse this decrease (Fig. 5B). IL-6r Ab had no effect on LLC-induced AMPK, Akt phosphorylation, or suppression of 4-EBP-1 (Fig. 5C). LLC activation of protein degradation pathways, measured by FOXO3a phosphorylation and atrogin-1 expression was unaffected by IL-6r Ab (Fig. 5C).
Figure 5.

Effect of IL-6 inhibition on LLC-induced signaling. A) p-STAT3 and total STAT3 protein expression was measured via Western blot analysis in fully differentiated C2C12 cells treated with IL-6 (100 ng/ml) or LCM (25%) for 4 h, with or without IL-6r Ab (1:1000). B) Protein synthesis was measured by the incorporation of puromycin into proteins in C2C12 cells treated for 72 h with LCM (25%), with or without IL-6r Ab (1:1000). Loading was verified with Ponceau staining. C) Protein expression of p-AMPK, total AMPK, p-Akt, total Akt, p-4EBP1, total 4EBP1, p-FOXO3, total FOXO3, atrogin, and GAPDH was measured by Western blot in C2C12 cells treated with LCM (25%) for 72 h, with or without IL-6r Ab (1:1000). Graph represents the ratio of phosphorylated to total protein levels. D) IL-6r Ab was administered acutely in vivo in mice with LLC tumors for 1 wk after tumor development. Western blot analysis measured LLC-associated signaling of STAT3, p38, Akt, S6RP, and atrogin-1. E) Protein synthesis was determined by Western blot 30 min after injection of puromycin, with loading verified by Ponceau staining. Dashed line indicates different sections of the same gel. Values are means ± se. Cell culture data were analyzed with 1-way ANOVA and in vivo data with the t test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005 vs. control group.
We examined the role of systemic IL-6 signaling during LLC-induced cachexia in vivo. Acute administration of IL-6r Ab to LLC cell–implanted mice (Table 2) did not reverse the induction of p-STAT3, p-p38, or atrogin-1 (Fig. 5D). Systemic IL-6 inhibition resulted in a reduction in the mTOR substrate S6RP from the already suppressed levels in LLC-cell–implanted mice (Fig. 5D). LLC decreased protein synthesis, and IL-6r Ab had no effect on the suppression (Fig. 5E).
Table 2.
Body weight, fat mass, and muscle mass in LLC-induced cachexia with acute administration of IL-6r-Ab or PDTC
| Parameter | Control | LLC | LLC+IL-6r Ab | LLC+PDTC |
|---|---|---|---|---|
| n | 5 | 4 | 4 | 4 |
| BW at death (g) | 27.3 ± 1.2 | 23.4 ± 2.4 | 26.2 ± 0.9 | 24.4 ± 1.3 |
| BW minus tumor (g) | 27.3 ± 1.2 | 20.1 ± 1.4* | 23.1 ± 0.6* | 22.1 ± 0.8* |
| Tumor (g) | — | 3.27 ± 1.20 | 3.08 ± 0.94 | 2.26 ± 0.98 |
| Epididymal fat (mg) | 429 ± 48 | 111 ± 16* | 166 ± 46* | 165 ± 47* |
| Gastrocnemius (mg) | 141.6 ± 7.2 | 92.5 ± 6.8* | 113.3 ± 5.8* | 99.3 ± 5.3* |
| Spleen (mg) | 104 ± 22 | 397 ± 59* | 354 ± 87* | 277 ± 70* |
| Tibia length (mm) | 16.9 ± 0.2 | 16.5 ± 0.1 | 16.6 ± 0.1 | 16.5 ± 0.1 |
LLC cells (1×106) were implanted in 8-wk-old skm-gp130 mice. After 4 wk of tumor growth, the mice were treated with PBS, IL-6r-Ab, or PDTC for 1 wk. Body weights were measured. Muscles and fat were excised and weighed at the time of death. All values are means ± sem. One-way ANOVA was used to compare data across treatments.
P < 0.05 vs. control.
IL-6 signaling inhibition in LLC-mediated C2C12 and in vivo atrophy
The effects of STAT3 inhibition on LLC-induced myotube signaling were examined by administration of PDTC (a STAT3 and NF-κB inhibitor), LLL12 (a STAT-specific inhibitor), or gp130 receptor (gp130r) Ab. LLC-induced STAT3 phosphorylation was blocked by all three (Fig. 6A). Myotube diameter decreased with LLC treatment (Fig. 6B). Inhibition of gp130 attenuated LLC-induced myotube atrophy, but IL-6 inhibition did not restore myotube diameter (Fig. 6B). LLC-suppressed protein synthesis was not restored by administration of PDTC, LLL12, or gp130 Ab (Fig. 6C). Increased p65 phosphorylation was blocked by gp130 Ab administration, and p38 phosphorylation was attenuated. LCM-induced AMPK phosphorylation was not affected by gp130r Ab (Fig. 6D). Reduction of gp130 expression did not stop inhibition of the mTOR target, 4-EBP1, but did suppress the induction of atrogin-1 expression (Fig. 6D). Specific STAT3 inhibition by LLL12 did not prevent LLC-induced NF-κB phosphorylation, p38 activation, or activation of AMPK (Fig. 6E). LLL12 was unable to preserve mTOR signaling through 4EBP1; however, it suppressed LLC-induced atrogin-1 (Fig. 6E). LCM activation of NF-κB was blocked by PDTC (Fig. 6F). PDTC had no effect on LLC-induced AMPK activation, p38 phosphorylation, or 4EBP1, but attenuated atrogin-1 expression (Fig. 6F).
Figure 6.

Effect of gp130/STAT signaling inhibition on LLC-induced signaling. Fully differentiated C2C12 cells were treated with LCM, with or without PDTC (a STAT/NF-kB inhibitor, 50 μM), LLL12 (a STAT3 specific inhibitor, 100 nM), and gp130r Ab (1:1000) for 4 h (A) or 72 h (B–F). A) p-STAT3 and total STAT3 protein expression was measured by Western blot analysis after 4 h treatment. B) Diameter of C2C12 myotubes after 72 h of LLC, PDTC, LLL12, or gp130r Ab administration. C) Relative protein synthesis rates were measured by Western blot after the incorporation of puromycin into proteins after 72 h of exposure to PDTC, LLL12, or gp130 Ab. Loading was verified by Ponceau staining. D–F) C2C12 signaling protein was measured by Western blot analysis after 72 h of exposure to gp130r Ab (D), LLL12 (E), or PDTC (F) in LCM. G) PDTC was administered for 1 wk to mice bearing LLC tumors. LLC-associated signaling of STAT3, P65, AMPK, P38, and Akt, was measured by Western blot analysis. Graphs represent ratio of phosphorylated to total protein levels. Values are means ± sem. Cell culture data were analyzed with 1-way ANOVA, and in vivo data were analyzed with the t test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005 vs. control group; #P ≤ 0.05 vs. LLC group.
We examined acute administration of PDTC in vivo (Table 2) in cachectic mice bearing LLC tumors. PDTC reduced muscle STAT3 and p65 phosphorylation (Fig. 6G). It did not alter LLC-induced AMPK or Akt phosphorylation. Of interest, phosphorylation of p38 showed a trend (P=0.07) toward increasing with PDTC treatment. Taken together, these results demonstrate a role for gp130/STAT3 in the regulation of muscle protein degradation in LLC-induced cachexia; however, the regulation of LLC-induced suppression of MPS requires further investigation.
DISCUSSION
Chronic inflammation, a hallmark of cancer-associated cachexia, is also a potential target for treating the devastating condition. IL-6 and muscle JAK/STAT signaling can regulate muscle mass in tumor-bearing mice (13, 21, 26). Although no treatments for cachexia in cancer are currently approved, two separate clinical case studies recently published demonstrate improved cachexia symptoms in cancer patients who were treated with IL-6r Ab (37, 38). However, significant gaps remain in our understanding of how cancer-induced systemic inflammation regulates the disruption of skeletal muscle protein turnover. To this end, our study provides novel information on the role of the skm-gp130 for the regulation of muscle protein turnover during LLC-induced cachexia. We report that skm-gp130 signaling is an important mediator of LLC-induced skeletal muscle mass loss, as the loss was attenuated in the skm-gp130 mice without altered tumor size or fat mass loss. We also report that loss of gp130 signaling attenuated the LLC activation of both STAT3 and p38, while activating Akt phosphorylation. Although LLC-induced gp130 signaling was associated with activation of FOXO3/atrogin signaling, we also provide in vivo and cell culture data indicating that cancer-induced suppression of protein synthesis was independent of these signaling pathways.
Several IL-6 family cytokines are elevated during cancer-related cachexia and have a potential regulatory role for LLC-induced muscle mass loss in mice (11, 39). Although gp130 is ubiquitously expressed, in our current study, we report that gp130 mRNA expression was higher in skeletal muscle than in liver or kidney. We also found differential expression of gp130 mRNA in the oxidative and glycolytic muscle. Muscle gp130 mRNA expression also showed plasticity, decreasing with the progression of cachexia. In contrast with the situation in cancer, elevated skm-gp130 expression has been reported in diabetic mice (40), and gp130 has been identified as a potential therapeutic target for obesity (41). Intracellular signaling initiated by gp130 can regulate growth, differentiation, and apoptosis (19). In addition, gp130 activation can phosphorylate AMPK and enhance glucose uptake and fatty acid oxidation, independent of STAT3 (42, 43), thus pointing to the potential for gp130 to have a dynamic role in the muscle response to the cancer environment. Further work is needed to determine whether gp130 alters muscle metabolic regulation during different stages of cachexia and whether muscle phenotype has a role in the response.
STAT3 has a role in the regulation of muscle protein degradation during wasting in cancer (10, 21) and can regulate atrogin-1 expression in C-26 tumor-bearing mice (21). However, the mechanism by which the cachectic environment, circulating IL-6, or both activate muscle STAT3 is not well understood. STAT3 can be activated by proinflammatory cytokines in the IL-6 family, but also can be activated by the signaling of leptin (44), IFNγ (45), and epidermal growth factor (46).We have extended these findings to demonstrate a role for muscle gp130 signaling for the induction of FOXO3 activation and atrogin-1 expression, both in vivo and in vitro. LLC-induced muscle STAT3 phosphorylation was attenuated by muscle-specific loss of gp130, and gp130r Ab blocked LCM-induced STAT3 phosphorylation in cultured myotubes. Our data suggest that muscle STAT3 induction during LLC-induced cachexia may be, at least in part, independent of circulating IL-6. IL-6r Ab administration to LLC tumor-bearing mice and LCM-treated myotubes was not sufficient to attenuate STAT3 phosphorylation, as in ApcMin/+ mice (13). Although STAT3 has been closely linked with muscle mass loss through regulation of degradation pathways, there may be redundancy in the activation of protein degradation cachectic mediators, such as myostatin (47), FOXO (48), and C/EBPβ (24). Also of interest, we have shown that chronically activated STAT3 does not lead to muscle mass loss in treadmill-exercising mice (35), suggesting that STAT3 regulation of skeletal muscle mass can be circumvented by other signaling pathways.
p38, one of the mitogen-activated protein kinases, serves as a nexus for signal transduction and is involved in a large variety of cellular processes. In skeletal muscle, p38 can be activated by many stress signals, including oxidative stress, inflammatory cytokines, and exercise. Activation of this pathway can regulate many functions in muscle, including myogenesis (49), exercise-induced PGC-1α transcription (45), and exercise-induced glucose uptake (46). However, chronic p38 activation in skeletal muscle has been implicated in pathologies such as muscle wasting via the FOXO3/atrogin-1 protein-degradation pathway (24, 25). p38 MAPK signaling may participate in local activation of NF-κB (44), which is another upstream activator of atrogin-1/MAFbx expression and muscle wasting (50). We report that LLC-induced muscle p38 phosphorylation is dependent on the presence of muscle gp130. Consistent with previous findings (24), we found p38 activation in both in vivo LLC-induced cachexia and in LLC-treated myotubes. Inhibition of gp130 caused the suppression of p38, while PDTC administration potentiated p38 activation by LLC, as was found in another study (51). Taken together, our results demonstrate that gp130 is necessary for p38 activation in LLC-induced cachexia, independent of STAT3. Further work is needed to integrate our understanding of gp130/p38/STAT3 cross-talk for the regulation of protein degradation in LLC-induced cachexia.
There has been considerable progress in understanding muscle protein degradation regulation during cachexia, and as a result, degradation processes are considered a control point of muscle mass loss in cachexia (6). However, MPS is suppressed with cancer cachexia in humans (52) and mice (13), and this anabolic repression most likely has physiological ramifications. mTOR functions as an integration point for hormone, nutrition, and contraction regulation of MPS and is subject to complex regulation (53, 54). IGF-1 can activate PI3K/Akt signaling to induce mTOR activity and protein synthesis through TSC1/2 phosphorylation (49, 55). Muscle IGF-1 expression is suppressed in several models of cachexia (13, 54, 56), and the regulation of this suppressed expression is not well understood. We report that loss of muscle gp130 signaling, a reduction in STAT3 signaling, and inhibited p38 signaling were not sufficient to protect against suppressed expression of muscle IGF-1. Similarly, systemic IL-6 inhibition in ApcMin/+ mice after the onset of cachexia did not completely restore muscle IGF-1 expression (13). Despite suppressed IGF-1, Akt phosphorylation can increase during cachexia (13), and cachexia-induced mTOR suppression is independent of Akt activation in ApcMin/+ mouse and the LLC implantation models (13). We conclude that gp130/STAT3 signaling is not a direct regulator of cachexia-induced suppression of protein synthesis.
AMPK is a cellular sensor of nutrient stress and can negatively regulate skeletal muscle mass (57). It inhibits mTORC1 formation through phosphorylation of raptor and TSC1/2, resulting in suppressed protein synthesis. In addition, AMPKα1 and AMPKα2 KOs produce skeletal muscle and cultured myotube hypertrophy (56). As it relates to the cachectic condition, AMPK can be activated by IL-6 in cultured myotubes and suppress protein synthesis (26), and gp130 in the presence of IL-6r is sufficient for STAT3-independent AMPK activation (42). During LLC-induced cachexia in muscle, AMPK phosphorylation increased, and LCM induced AMPK phosphorylation in C2C12 myotubes. However, inhibition of gp130, IL-6, and STAT3 signaling was not sufficient to attenuate LLC induction of AMPK, suggesting that gp130/STAT3-independent mechanisms regulate AMPK during LLC-induced cachexia in cancer. In prior work, we have shown that treatment of C2C12 myotubes with compound C, an AMPK inhibitor, can alleviate IL-6 suppression of protein synthesis (26). REDD1 is a potent suppressor of mTOR, which is increased in the cachectic condition (13). It can suppress mTORC1 through the dissociation of TSC2/14-3-3 complex (58) and can suppress mTOR independent of AMPK (53). The inhibition of skm-gp130 signaling did not alter LLC-induced REDD1 expression. A better understanding of the mechanistic relationship between the regulation of protein degradation and synthesis in cachectic muscle is needed.
In summary, this is the first investigation of the role of skm-gp130 regulation in protein turnover during cachexia. Inhibition of skm-gp130 attenuated LLC-induced muscle atrophy. In addition, activation of STAT3, p38, and NF-κB was suppressed with skm-gp130 inhibition. The LLC-induced activation of FOXO3a protein degradation signaling was suppressed by gp130/STAT3 inhibition, both in vivo and in vitro. Of note, inhibition of IL-6 was not sufficient to repress LLC-induced atrophy signaling. We report a gp130/STAT3-independent suppression of mTOR signaling. Further work is necessary to determine the LLC inhibition of protein synthesis during cachexia. In addition, the LLC tumor appears not to be an IL-6-dependent cachexia model; consequently, further work is needed to establish a more IL-6-dependent role of muscle gp130 in the regulation of cachexia. Targeted gp130 therapies may be useful in combination with treatments that alleviate the suppression of mTOR signaling.
Acknowledgments
The authors thank Ms. Tia Davis and Dr. Marj Peña for technical assistance with the animal breeding. The skeletal muscle-specific Cre-expressing mice were a gift from Dr. Steven Burden (New York University, New York, NY, USA). The gp130 fl/fl mice were a gift from Drs. Colin Stuart [Laboratory of Cancer and Developmental Biology, National Cancer Institute (NCI), U.S. National Institutes of Health (NIH), Frederick, MD, USA] and Lothar Hennighausen (Laboratory of Genetics and Physiology, National Institute of Diabetes and Digestive and Kidney Diseases, NIH, Bethesda, MD, USA).
The research described in this report was supported by U.S. NIH/NCI research grant R01 CA121249 to J.A.C.
Footnotes
- Ab
- antibody
- AMPK
- AMP-activated protein kinase
- gp130
- glycoprotein 130
- gp130r
- glycoprotein 130 receptor
- IL-6r
- interleukin-6 receptor
- KO
- knockout
- LCM
- LLC-conditioned medium
- LLC
- Lewis lung carcinoma
- MLC
- myosin light chain
- MPS
- muscle protein synthesis
- PDTC
- pyrrolidine dithiocarbamate
- skm-gp130
- skeletal muscle–specific glycoprotein 130
- TA
- tibialis anterior
REFERENCES
- 1. Evans W. J., Morley J. E., Argiles J., Bales C., Baracos V., Guttridge D., Jatoi A., Kalantar-Zadeh K., Lochs H., Mantovani G., Marks D., Mitch W. E., Muscaritoli M., Najand A., Ponikowski P., Rossi Fanelli F., Schambelan M., Schols A., Schuster M., Thomas D., Wolfe R., Anker S. D. (2008) Cachexia: a new definition. Clin. Nutr. 27, 793–799 [DOI] [PubMed] [Google Scholar]
- 2. Fearon K., Strasser F., Anker S. D., Bosaeus I., Bruera E., Fainsinger R. L., Jatoi A., Loprinzi C., MacDonald N., Mantovani G., Davis M., Muscaritoli M., Ottery F., Radbruch L., Ravasco P., Walsh D., Wilcock A., Kaasa S., Baracos V. E. (2011) Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495 [DOI] [PubMed] [Google Scholar]
- 3. Muscaritoli M., Anker S. D., Argiles J., Aversa Z., Bauer J. M., Biolo G., Boirie Y., Bosaeus I., Cederholm T., Costelli P., Fearon K. C., Laviano A., Maggio M., Rossi Fanelli F., Schneider S. M., Schols A., Sieber C. C. (2010) Consensus definition of sarcopenia, cachexia and pre-cachexia: joint document elaborated by special interest groups (SIG) “cachexia-anorexia in chronic wasting diseases” and “nutrition in geriatrics”. Clin. Nutr. 29, 154–159 [DOI] [PubMed] [Google Scholar]
- 4. Bruera E. (1997) ABC of palliative care: anorexia, cachexia, and nutrition. BMJ 315, 1219–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tisdale M. J. (2002) Cachexia in cancer patients. Nat. Rev. Cancer 2, 862–871 [DOI] [PubMed] [Google Scholar]
- 6. Tisdale M. J. (2009) Mechanisms of cancer cachexia. Physiol. Rev. 89, 381–410 [DOI] [PubMed] [Google Scholar]
- 7. Argiles J. M., Busquets S., Lopez-Soriano F. J. (2003) Cytokines in the pathogenesis of cancer cachexia. Curr. Opin. Clin. Nutr. Metab. Care 6, 401–406 [DOI] [PubMed] [Google Scholar]
- 8. Fortunati N., Manti R., Birocco N., Pugliese M., Brignardello E., Ciuffreda L., Catalano M. G., Aragno M., Boccuzzi G. (2007) Pro-inflammatory cytokines and oxidative stress/antioxidant parameters characterize the bio-humoral profile of early cachexia in lung cancer patients. Oncol. Rep. 18, 1521–1527 [PubMed] [Google Scholar]
- 9. Tazaki E., Shimizu N., Tanaka R., Yoshizumi M., Kamma H., Imoto S., Goya T., Kozawa K., Nishina A., Kimura H. (2011) Serum cytokine profiles in patients with prostate carcinoma. Exp. Ther. Med. 2, 887–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bonetto A., Aydogdu T., Kunzevitzky N., Guttridge D. C., Khuri S., Koniaris L. G., Zimmers T. A. (2011) STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PloS One 6, e22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matthys P., Heremans H., Opdenakker G., Billiau A. (1991) Anti-interferon-gamma antibody treatment, growth of Lewis lung tumours in mice and tumour-associated cachexia. Eur. J. Cancer 27, 182–187 [DOI] [PubMed] [Google Scholar]
- 12. Baltgalvis K. A., Berger F. G., Pena M. M., Davis J. M., Muga S. J., Carson J. A. (2008) Interleukin-6 and cachexia in ApcMin/+ mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R393–R401 [DOI] [PubMed] [Google Scholar]
- 13. White J. P., Baynes J. W., Welle S. L., Kostek M. C., Matesic L. E., Sato S., Carson J. A. (2011) The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PloS One 6, e24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Puppa M. J., White J. P., Sato S., Cairns M., Baynes J. W., Carson J. A. (2011) Gut barrier dysfunction in the Apc(Min/+) mouse model of colon cancer cachexia. Biochim. Biophys. Acta 1812, 1601–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barton B. E., Murphy T. F. (2001) Cancer cachexia is mediated in part by the induction of IL-6-like cytokines from the spleen. Cytokine 16, 251–257 [DOI] [PubMed] [Google Scholar]
- 16. Mori M., Yamaguchi K., Honda S., Nagasaki K., Ueda M., Abe O., Abe K. (1991) Cancer cachexia syndrome developed in nude mice bearing melanoma cells producing leukemia-inhibitory factor. Cancer Res. 51, 6656–6659 [PubMed] [Google Scholar]
- 17. Kamoshida S., Watanabe K., Suzuki M., Mizutani Y., Sakamoto K., Sugimoto Y., Oka T., Fukushima M., Tsutsumi Y. (2006) Expression of cancer cachexia-related factors in human cancer xenografts: an immunohistochemical analysis. Biomed. Res. 27, 275–281 [DOI] [PubMed] [Google Scholar]
- 18. Rose-John S. (2012) IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 8, 1237–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ernst M., Jenkins B. J. (2004) Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet. 20, 23–32 [DOI] [PubMed] [Google Scholar]
- 20. Heinrich P. C., Behrmann I., Muller-Newen G., Schaper F., Graeve L. (1998) Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 334, 297–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bonetto A., Aydogdu T., Jin X., Zhang Z., Zhan R., Puzis L., Koniaris L. G., Zimmers T. A. (2012) JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 303, E410–E421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schiaffino S., Mammucari C. (2011) Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet. Muscle 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stitt T. N., Drujan D., Clarke B. A., Panaro F., Timofeyva Y., Kline W. O., Gonzalez M., Yancopoulos G. D., Glass D. J. (2004) The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 14, 395–403 [DOI] [PubMed] [Google Scholar]
- 24. Zhang G., Jin B., Li Y. P. (2011) C/EBPbeta mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 30, 4323–4335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang G., Li Y. P. (2012) p38beta MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPbeta. Skelet. Muscle 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. White J. P., Puppa M. J., Gao S., Sato S., Welle S. L., Carson J. A. (2013) Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am. J. Physiol. Endocrinol. Metab. 304, E1042–E1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao L., Hart S., Cheng J., Melenhorst J. J., Bierie B., Ernst M., Stewart C., Schaper F., Heinrich P. C., Ullrich A., Robinson G. W., Hennighausen L. (2004) Mammary gland remodeling depends on gp130 signaling through Stat3 and MAPK. J. Biol. Chem. 279, 44093–44100 [DOI] [PubMed] [Google Scholar]
- 28. Bothe G. W., Haspel J. A., Smith C. L., Wiener H. H., Burden S. J. (2000) Selective expression of Cre recombinase in skeletal muscle fibers. Genesis 26, 165–166 [PubMed] [Google Scholar]
- 29. Hariri G., Yan H., Wang H., Han Z., Hallahan D. E. (2010) Radiation-guided drug delivery to mouse models of lung cancer. Clin. Cancer Res. 16, 4968–4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. White J. P., Puppa M. J., Sato S., Gao S., Price R. L., Baynes J. W., Kostek M. C., Matesic L. E., Carson J. A. (2012) IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nai Y. J., Jiang Z. W., Wang Z. M., Li N., Li J. S. (2007) Prevention of cancer cachexia by pyrrolidine dithiocarbamate (PDTC) in colon 26 tumor-bearing mice. JPEN J. Parenter. Enteral Nutr. 31, 18–25 [DOI] [PubMed] [Google Scholar]
- 32. Goodman C. A., Mabrey D. M., Frey J. W., Miu M. H., Schmidt E. K., Pierre P., Hornberger T. A. (2011) Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 25, 1028–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. White J. P., Gao S., Puppa M. J., Sato S., Welle S. L., Carson J. A. (2013) Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol. Cell. Endocrinol. 365, 174–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Washington T. A., White J. P., Davis J. M., Wilson L. B., Lowe L. L., Sato S., Carson J. A. (2011) Skeletal muscle mass recovery from atrophy in IL-6 knockout mice. Acta Physiol. (Oxf.) 202, 657–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Puppa M. J., White J. P., Velazquez K. T., Baltgalvis K. A., Sato S., Baynes J. W., Carson J. A. (2011) The effect of exercise on IL-6-induced cachexia in the Apc (Min/+) mouse. J. Cachexia Sarcopenia Muscle 3, 117–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 37. Ando K., Takahashi F., Motojima S., Nakashima K., Kaneko N., Hoshi K., Takahashi K. (2013) Possible role for tocilizumab, an anti-interleukin-6 receptor antibody, in treating cancer cachexia. J. Clin. Oncol. 31, e69–72 [DOI] [PubMed] [Google Scholar]
- 38. Hirata H., Tetsumoto S., Kijima T., Kida H., Kumagai T., Takahashi R., Otani Y., Inoue K., Kuhara H., Shimada K., Nagatomo I., Takeda Y., Goya S., Yoshizaki K., Kawase I., Tachibana I., Kishimoto T., Kumanogoh A. (2013) Favorable responses to tocilizumab in two patients with cancer-related cachexia. J. Pain Symptom Manage. 46, e9–e13 [DOI] [PubMed] [Google Scholar]
- 39. Kim S., Takahashi H., Lin W. W., Descargues P., Grivennikov S., Kim Y., Luo J. L., Karin M. (2009) Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457, 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Toledo-Corral C. M., Banner L. R. (2012) Early changes of LIFR and gp130 in sciatic nerve and muscle of diabetic mice. Acta Histochem. 114, 159–165 [DOI] [PubMed] [Google Scholar]
- 41. Febbraio M. A. (2007) Gp130 receptor ligands as potential therapeutic targets for obesity. J. Clin. Invest. 117, 841–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Watt M. J., Dzamko N., Thomas W. G., Rose-John S., Ernst M., Carling D., Kemp B. E., Febbraio M. A., Steinberg G. R. (2006) CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat. Med. 12, 541–548 [DOI] [PubMed] [Google Scholar]
- 43. Kelly M., Keller C., Avilucea P. R., Keller P., Luo Z. J., Xiang X. Q., Giralt M., Hidalgo J., Saha A. K., Pedersen B. K., Ruderman N. B. (2004) AMPK activity is diminished in tissues of IL-6 knockout mice: the effect of exercise. Biochem. Biophys. Res. Commun. 320, 449–454 [DOI] [PubMed] [Google Scholar]
- 44. Baeza-Raja B., Muñoz-Cánoves P. (2004) p38 MAPK-induced nuclear factor-κB activity is required for skeletal muscle differentiation: role of interleukin-6. Mol. Biol. Cell 15, 2013–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Akimoto T., Pohnert S. C., Li P., Zhang M., Gumbs C., Rosenberg P. B., Williams R. S., Yan Z. (2005) Exercise stimulates Pgc-1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 280, 19587–19593 [DOI] [PubMed] [Google Scholar]
- 46. Widegren U., Jiang X. J., Krook A., Chibalin A. V., Björnholm M., Tally M., Roth R. A., Henriksson J., Wallberg-henriksson H., Zierath J. R. (1998) Divergent effects of exercise on metabolic and mitogenic signaling pathways in human skeletal muscle. FASEB J. 12, 1379–1389 [DOI] [PubMed] [Google Scholar]
- 47. Murphy K. T., Chee A., Gleeson B. G., Naim T., Swiderski K., Koopman R., Lynch G. S. (2011) Antibody-directed myostatin inhibition enhances muscle mass and function in tumor-bearing mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R716–R726 [DOI] [PubMed] [Google Scholar]
- 48. Reed S. A., Sandesara P. B., Senf S. M., Judge A. R. (2012) Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J. 26, 987–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu Z., Woodring P. J., Bhakta K. S., Tamura K., Wen F., Feramisco J. R., Karin M., Wang J. Y., Puri P. L. (2000) p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol. Cell. Biol. 20, 3951–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cai D., Frantz J. D., Tawa N. E., Jr., Melendez P. A., Oh B.-C., Lidov H. G., Hasselgren P.-O., Frontera W. R., Lee J., Glass D. J. (2004) IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell 119, 285–298 [DOI] [PubMed] [Google Scholar]
- 51. Pfeilschifter W., Czech B., Hoffmann B. P., Sujak M., Kahles T., Steinmetz H., Neumann-Haefelin T., Pfeilschifter J. (2010) Pyrrolidine dithiocarbamate activates p38 MAPK and protects brain endothelial cells from apoptosis: a mechanism for the protective effect in stroke? Neurochem. Res. 35, 1391–1401 [DOI] [PubMed] [Google Scholar]
- 52. Dworzak F., Ferrari P., Gavazzi C., Maiorana C., Bozzetti F. (1998) Effects of cachexia due to cancer on whole body and skeletal muscle protein turnover. Cancer 82, 42–48 [PubMed] [Google Scholar]
- 53. Frost R. A., Lang C. H. (2011) mTor signaling in skeletal muscle during sepsis and inflammation: where does it all go wrong? Physiology 26, 83–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goodman C. A., Frey J. W., Mabrey D. M., Jacobs B. L., Lincoln H. C., You J. S., Hornberger T. A. (2011) The role of skeletal muscle mTOR in the regulation of mechanical load-induced growth. J. Physiol. 589, 5485–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Frost R. A., Huber D., Pruznak A., Lang C. H. (2009) Regulation of REDD1 by insulin-like growth factor-I in skeletal muscle and myotubes. J. Cell. Biochem. 108, 1192–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lantier L., Mounier R., Leclerc J., Pende M., Foretz M., Viollet B. (2010) Coordinated maintenance of muscle cell size control by AMP-activated protein kinase. FASEB J. 24, 3555–3561 [DOI] [PubMed] [Google Scholar]
- 57. Goodman C. A., Mayhew D. L., Hornberger T. A. (2011) Recent progress toward understanding the molecular mechanisms that regulate skeletal muscle mass. Cell. Signal. 23, 1896–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ho R. C., Hirshman M. F., Li Y., Cai D., Farmer J. R., Aschenbach W. G., Witczak C. A., Shoelson S. E., Goodyear L. J. (2005) Regulation of IκB kinase and NF-κB in contracting adult rat skeletal muscle. Am. J. Physiol. Cell Physiol. 289, C794–C801 [DOI] [PubMed] [Google Scholar]