

Abstract
Loss of memory is among the first symptoms reported by patients suffering from Alzheimer's disease (AD) and by their caretakers. Working memory and long-term declarative memory are affected early during the course of the disease. The individual pattern of impaired memory functions correlates with parameters of structural or functional brain integrity. AD pathology interferes with the formation of memories from the molecular level to the framework of neural networks. The investigation of AD memory loss helps to identify the involved neural structures, such as the default mode network, the influence of epigenetic and genetic factors, such as ApoE4 status, and evolutionary aspects of human cognition. Clinically, the analysis of memory assists the definition of AD subtypes, disease grading, and prognostic predictions. Despite new AD criteria that allow the earlier diagnosis of the disease by inclusion of biomarkers derived from cerebrospinal fluid or hippocampal volume analysis, neuropsychological testing remains at the core of AD diagnosis.
Keywords: memory, Alzheimer's disease, neuropsychological test, brain atrophy, default mode network
Abstract
La pérdida de memoria está entre los primeros síntomas referidos por los pacientes que padecen Enfermedad de Alzheimer (EA) y por sus cuidadores. La memoria de trabajo y la memoria declarativa de largo plazo se afectan precozmente durante el curso de la enfermedad. El perfil individual de deterioro de las funciones de la memoria se correlaciona con parámetros de integridad cerebral estructural o funcional. La patología de la EA interfiere con la formación de memorias desde el nivel molecular hasta el sistema de redes neurales. La investigación de la pérdida de memoria en la EA ayuda a identificar las estructuras neurales involucradas, como la red neural por defecto, la influencia de factores epigenéticos y genéticos, como el estado de la ApoE4, y aspectos evolucionistas de la cognición humana. Clinicamente, el análisis de la memoria ayuda a la definición de los subtipos de EA, a la clasificación de la enfermedad y a las predicciones pronósticas. A pesar de los nuevos criterios de EA que permiten un diagnóstico más precoz de la enfermedad mediante la incorporación de biomarcadores derivados del liquido céfalo raquideo o del análisis del volumen del hipocampo, las pruebas neuropsicológicas persisten en to nuclear del diagnóstico de EA.
Abstract
La perte de mémoire est un des premiers symptômes rapportés par les patients souffrant de maladie d'Alzheimer (MA) et leurs soignants. La mémoire de travail et la mémoire déclarative à long terme sont touchées de façon précoce au cours de l'évolution de la maladie. Le schéma individuel de la détérioration des fonctions mnésiques correspond aux paramètres de l'intégrité cérébrale fonctionnelle ou structurelle. La pathologie de la MA interfère avec la formation de souvenirs allant du niveau moléculaire jusqu'au cadre des réseaux neuronaux. La recherche de la perte de mémoire de la MA aide à identifier les structures neuronales impliquées, comme le réseau du « mode par défaut », l'influence des facteurs épigénétiques et génétiques, comme l'état de l'ApoE4 et les aspects évolutifs de la cognition humaine. Cliniquement, l'analyse de la mémoire facilite la définition des sous-types de MA, le stade de la maladie et la prévision du pronostic. Malgré les nouveaux critères de MA permettant le diagnostic précoce de la maladie en faisant appel à des biomarqueurs dérivés du liquide céphalo-rachidien ou à l'analyse du volume de l'hippocampe, les tests neuropsychologiques restent au centre du diagnostic de la MA.
Introduction
Asked to name a disease that affects “memory,” most physicians would probably choose Alzheimer's disease (AD). AD is the most common cause of dementia. Currently, 30 million people worldwide suffer from Alzheimer's dementia and the World Health organization projects that this number will triple over the next 20 years.1 The cumulative incidence of Alzheimer dementia has been estimated to rise from about 5% by age 70 to 50% by age 90, making it a very common disease.2 Increasing longevity and demographic shifts in many societies will stress the health systems unless a cure for AD is found—or at least any therapy that postpones the onset of the dementia by 5 to 10 years for the time being. AD is a polygenetic neurodegenerative brain disorder characterized by neocortical atrophy developing over decades, showing the increasing loss of synapses and neurons first described by Alois Alzheimer in 1907.3 Variations in the genes encoding the amyloid precursor proteins, presenilin-1 and presenilin-2, can directly cause Alzheimer's disease. The patients often display an early onset of dementia in their forties, and these deterministic genes affect whole families and patients with trisomy 21. They, however, account for only 1% of all cases of AD. The remaining 99% of sporadic AD occurs predominantly in patients older than 65 years;4 These patients also inherit 60% to 80% of their risk for late-onset AD, although it is not connected to the aforementioned genes, with the remainder being environmental.4,5 In late-onset AD many different genes—some still unidentified—are involved, each only attributing a minor fraction of the individual's overall risk, making it a possible example of antagonistic pleiotropy.6 Recently, several genome-wide association studies have identified new candidates (for reviews and databases see http://www.alzgene.org). The best characterized genetic risk factor in late-onset AD is the apolipoprotein E4 (ApoE4) genotype with ApoE4 carriers having a 4- to 10-fold increased odds ratio of developing AD.7 The ApoE4 allele is an ancestral variant. The human ApoE2 and ApoE3 alleles are fairly recent mutations—less than 200 000 years old.8 ApoE3 is the most frequent allele, with frequencies of about 60% in human populations. In contrast, ApoE4 has a frequency of about 10%. This implies that ApoE3 and ApoE2 must possess an advantage in order to have gained prevalence in human populations so quickly; clinical and preclinical data point to higher synaptic plasticity and repair capacities in carriers of ApoE2 and ApoE3, which may explain the selection bias.9 The consumption and adaption of humans to a diet rich in meat and animal fats can also be a reason for the selection of ApoE3.10
Atrophy in AD
The neuropathological hallmarks of the atrophy process in AD are the presence of senile plaques (amyloid deposits) and neurofibrillary tangles in autopsied brains.11 Neurofibrillary tangles are composed of hyperphosphorylated tau protein located within neurons, whereas senile plaques are made up largely of amyloid-P species aggregating in the extracellular space. These neuropathological changes start in the entorhinal cortex and hippocampal formations, later spreading into other temporal, parietal, and finally frontal association cortices.12-14 The first lesions characteristic of AD appear in poorly myelinated limbic neurons in system areas related to memory and learning, such as the hippocampus and the association cortex. Highly myelinated neurons are only affected in the final phases of the disease.15 Low myelinization increases the overall energy expenditure of neurons. In addition, subcortical neuron loss occurs in the nucleus basalis of Meynert and the locus ceruleus, impairing the cholinergic and noradrenergic transmitter systems in the neocortex.16,17 The parietal lobe, along with certain areas of the prefrontal lobe, is one of the last areas of the human brain to myelinate, and many of its neurons remain poorly myelinated for the entire lifespan, which may explain their vulnerability to factors capable of triggering AD.18-20 The atrophy runs slowly, but while in healthy aging only 0.2% to 0.41% of the brain volume vanishes per year, the rates in AD may be ten times that, and in especially vulnerable regions like the hippocampal formation atrophy rates might be even more devastatingly high and surpass 10% per year (see also Figure 1).21-23 In terms of neuropsychological tests, regional atrophy, and glucose metabolism correlate well with test results.24,25 Left hippocampal gray matter volume, for example, significantly correlates with performance in memory tasks, and left temporal gray-matter volume is related to performance in language tasks. The rate of change in the left hippocampus correlates with decline of performance in the Boston Naming Test Mini-Mental Status Examination, and the trailmaking test B.24 Such analyses help with the definition of special AD subtypes like posterior cortical atrophy, or the logopenic variant of AD.24-31 On the molecular level we find a downregulation of synaptic genes across multiple brain regions and widespread proteomic signs of synaptic stress or decay in the cerebrospinal fluid (CSF) or blood.32-34 Changes in the molecular fine structure of AD brains also arise independently of atrophy as resonance spectroscopic investigations in AD imply.35
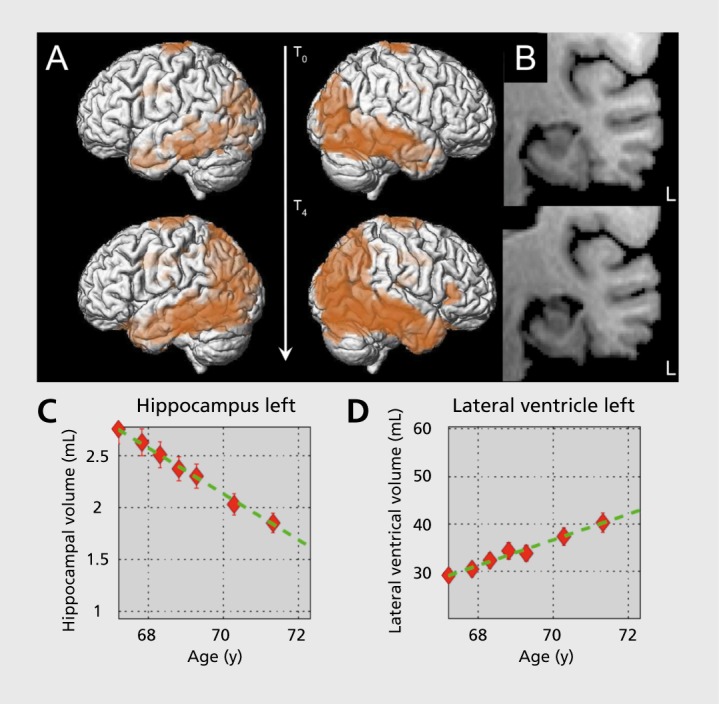
Figure 1. Atrophy in a case of AD over 4 years. (A) Reduction of gray matter (lateral view; corrected for age; P<0.05), (B) coronal view of the left hippocampus at baseline and after 4 years (hippocampal gray matter volume at T0 5.3±0.4 mL, at T4 3.5±0.2 ml), (C) average hippocampal volume reduction of 0.2 mL per year (-12%/year), (D) average increase of lateral ventricle volume of 2.7 mL/per year (+ 6.7%/year). (Courtesy of L. Spies, Jung-diagnostics GmbH, Hamburg, Germany).
This review aims to highlight some aspects concerning the development of memory deficits in AD that recently have or should have gained attention.
Impact of new diagnostic criteria
Recently workgroups of the Alzheimer's Association and the National Institute on Aging have issued new criteria and guidelines to diagnose Alzheimer's disease supplanting the previous guidelines first published in 1984.36-40 This marks a complete overhaul, and attemps to implement advances in our understanding of the disease in the way we diagnose the disease. Hie most notable differences are the use of biomarkers such as hippocampal atrophy, and the formalization of earlier disease stages before dementia is apparent, such as mild cognitive impairment due to AD and the newly defined preclinical AD stage.38,39 While the recommendations of the preclinical AD workgroup are intended purely for research purposes and the aim of diagnosing the disease earlier appears sensible since it is likely that any intervention has to be started early to be successful, it is also clear that we would almost all be defined as having the disease using this definition, given the increasing prevalence of AD in the very old. From a scientific point of view, it might be more interesting to know why a few of us might not develop AD, even when we are not dying from other diseases. As clinicians AD patients may first approach us with mere subjective concerns about cognitive decline. This can develop into mild cognitive impairment with pathological neuropsychological test results and progress into dementia, at which time daily activities can no longer be performed properly. When brain atrophy progresses other psychiatric and neurological symptoms arise, and typically AD patients lose weight and frequently develop difficulties in swallowing. This may lead to aspiration and subsequently pneumonia, which is often the final cause of death in demented patients.
The neuropsychology of AD: tests and what they indicate
Consensus exists that AD starts clinically with memory complaints, which may affect episodic memory, speech production, with naming or semantic problems, or visual orientation. Memory can be defined as a process of encoding, storing, and retrieving information about outer and inner stimuli, or presentation of information to the nervous system of an organism that can be used to react and position the organism towards new stimuli. Different categories of memory have been defined which also have different neuroanatomical and neurophysiological correlates: short-term memory vs long-term memory or implicit versus declarative memory. Short-term memory is limited to just a few “chunks” in capacity, and lasts only seconds to minutes.41 It depends on regions of the frontal lobe and the parietal lobe. In contrast, long-term memory seems almost limitless regarding its storage capacities, for a potentially unlimited duration. It depends on de novo protein synthesis and changes in the molecular components of the neuronal networks involved in the specific cortical areas that can be attributed to different memory types. Declarative memory, for example, can be further subdivided into semantic memory, where context-independent information is stored, and episodic memory, which stores information specific to a particular context, mainly time and place. Semantic memory is at first impaired in the language of AD patients, affecting verbal fluency and naming. Semantic loss in AD may occur several years prior to diagnosis.42 The hippocampus is essential to the consolidation of information from short-term to long-term memory. Destruction of the hippocampal formation makes the storage of new memories impossible. In the clinical context we use neuropsychological test batteries like the CERAD (Consortium to Establish a Registry for Alzheimer's Disease) examination, the Mini-Mental State examination, and various other test constructs and scales, like the clinical dementia rating scale, that investigate different aspects of memory over a broad range of various cognitive domains.43-45 Patients get profiled in relation to tests they show abnormalities on, compared with a healthy reference group adjusted for age and education. AD patients typically display a cognitive profile with impairments in multiple cognitive domains. This cognitive profile develops over time, and AD patients often start to show a progressive decay of working memory. The patients display increased sensitivity to distraction in memory tasks, the capacity of working memory measured, eg, digit span is, however, at first still intact. Interestingly, the medications used currently to treat AD like acetylcholinesterase inhibitors or memantine work partly by increasing attention and concentration and work mainly in mild-to-moderate AD.46,47
The deficits in attention and working memory associated with damage to frontal subcortical circuits also influence executive functions in AD, impairing planning, problem solving, and goal-directed behavior such as the ability to deploy response alternatives or modify behavior. AD patients show impaired results in tests that require planning, problem solving, or cognitive flexibility, eg, the Wisconsin Card Sorting Test, the Stroop test, or the Tower of London Test. The manifestation of impairment in such tests of executive functioning corresponds to the onset of difficulties in the performance of daily activities in these patients and marks the progression to the state of full dementia. The Boston Naming test assesses the ability to name pictures of objects through spontaneous responses, and the need for various types of cueing. Cued recall deficits are most closely associated with CSF biomarkers indicative of AD in subjects with mild cognitive impairment. This novel finding complements results from prospective clinical studies and provides further empirical support for cued recall as a specific indicator of prodromal AD, in line with recently proposed research criteria.48 Another surprisingly simple test is the Clock Drawing test. AD patients show early difficulties in visuospatial processing and conceptual errors like misrepresentation of numbers in the command, but not in the copy, condition, pointing to deficits in semantic memory. The Trail Making Test A+B is a neuropsychological test of visual attention measuring mental processing speed, and the ability to switch between different tasks. It consists of two parts in which the subject is asked to connect a set of 25 dots as fast as possible while maintaining accuracy. Visual search speed, scanning and processing abilities, mental flexibility, and executive functioning can be assessed with this test.
Regarding animal models, there are plenty of paradigmata available to test memory functions, but there is an overall lack of validated animal data that can be aligned with similar tests in human settings. Snigdha et al started with the comprehensive toolbox for Neurologic Behavioral Function from the National Institutes of Health (NIH) which contains evaluated tests for cognitive, motor, sensory, and emotional function for use in epidemiologic and clinical studies spanning 3 to 85 years of age and analyzed strengths and limitations of available animal behavioral tests to find matches. They defined a preclinical battery that aims to parallel the NIH Toolbox, and may help to close the gap between data from different species.49
Subjective cognitive impairment
Subjective cognitive impairment without detectable objective memory deficit may no longer merely regarded as “normal aging” since it has been shown that it is a major risk factor for the development of dementia.50 A clear definition of what subjective memory impairment or subjective cognitive impairment actually mean is currently lacking. An international task force is, however, working on standard operating procedures that would enable comparable study designs. A consensus regarding naming the concept “subjective cognitive impairment” in view of previously used terminology such as “subjective memory impairment” seems to be arising. Subjective cognitive impairment is defined as the individual coming up with the mere feeling that something is not in order, without any objective parameters supporting that notion in the first place. Such a stage labeled subjective cognitive impairment may precede mild cognitive impairment in the continuum of Alzheimer disease manifestation. Using such a definition and without objective neuropsychological test alterations, the atrophy pattern of patients with subjective cognitive impairment seem to be related to the atrophy pattern seen in AD.51 Individuals with subjective memory impairment showed greater similarity to an AD gray matter pattern, and episodic memory decline was associated with an AD gray matter pattern in probands with subjective memory impairment.52 Patients with subjective memory impairment also already showed hypometabolism in the right precuneus and hypermetabolism in the right medial temporal lobe using (fludeoxyglucose positron emission tomography, FDG-PET). Their gray matter volume was reduced in the right hippocampus. At follow-up, these patients showed poorer performance on measures of episodic memory. The observed memory decline was associated with reduced glucose metabolism in the right precuneus at baseline. The authors conclude that their concept of subjective memory impairment may define the earliest clinical manifestation of AD.53 In another study patients with subjective memory underwent an associative episodic memory task matching faces to professions, including encoding, recall, and recognition, and a working memory task during functional magnetic resonance imaging (f'MRI). They showed a reduction in right hippocampal activation during episodic memory recall, still in the absence of performance deficits. This was accompanied by increased activation of the right dorsolateral prefrontal cortex. No such differences in performance and brain activation were detected for working memory. This may indicate subtle early neuronal dysfunction on the hippocampal level and compensatory mechanisms that preserve memory performance.54
Regarding ApoE4, cognitively unimpaired young elderly with and without subjective memory impairment were tested on episodic memory and on tasks of speed and executive function. Medial temporal lobe volumetric measures were calculated from MRI images. In the subjective memory impairment group, ApoE4 carriers performed worse on the episodic memory and showed smaller left hippocampal volumes. In the individuals without memory complaints, the ApoE4 carriers performed better on episodic memory and had larger right hippocampal volumes (P=0.039). The interaction of group and ApoE genotype was significant for episodic memory and right and left hippocampal volumes. The negative effect of ApoE4 on episodic memory and hippocampal volume in the group suffering from subjective memory decline also supports the notion that this may be a prodromal condition of AD.55 In conclusion, the mere subjective feeling of being cognitively altered compared with the individual's reference past can already be accompanied by subtle brain changes that if ongoing may herald increasing memory decline in the future.
Memory of smell
Impaired sense of smell or hyposmia is one of the earliest clinical features in neurodegenerative disorders like both AD or Parkinson's disease.56 This has been known for decades and relates well to the finding that, for example, plaque formation in AD starts in the entorhinal cortex, the region also responsible for processing of information on smell. A recent meta-analysis of 81 studies indicated that AD and PD patients are more impaired on odor identification and recognition tasks than on odor detection threshold tasks. AD patients were found to be more impaired on higher-order olfactory tasks involving specific cognitive processes.57 Odor identification and recognition tests can be easily implemented in cognitive test batteries to detect already subclinical cases of AD. The impairment of smell recognition is of clinical importance, as patients often report malodorous sensations and changes in, eg, the taste of foods leading to behavioral alterations. Consequences may range from increasing malnutriton to the development of delusions of poisoning that may trigger aggressive behavior. The deterioration of the neural network in the entorhinal cortex leads to an impairment in the ability to store and retrieve different representations of smell, with the decaying network yielding increasingly “default values” that have the tendency to be of rather unpleasant character. This feature of the neural network of smell memory reflects the evolutionary pressure towards the secure recognition of “bad” smells pointing to poisonous or rotten food that is pivotal for the survival of the organism.
AD and epileptic activity
The incidence of unprovoked seizures is clearly higher in sporadic AD than in reference populations with implications for memory functions. Nonconvulsive epileptiform activity could underlie at least some of the cognitive impairments observed in AD. Up to 1 in 5 patients with sporadic AD has at least 1 unprovoked clinically apparent seizure during their illness, and clinical guidelines recommend obligatory treatment for this condition. The risk of epileptic activity is greater in early-onset AD. Many mutations in the presenilin-1 gene are associated with epilepsy. Trisomy-21 patients with early-onset AD also have frank seizures in 84% of cases. Many patients with AD show fluctuations in cognitive functions such as transient episodes of amnestic wandering or disorientation. While an intermittent inability to retrieve memories cannot be easily explained by relatively protracted processes such as neuronal loss, plaque deposition, or tangle formation, an abnormal epileptic activity of neuronal networks can. Extensive work in this field was published by the group of Lennart Mucke. They see the possibility that high levels of β-amyloid induce epileptiform activity, which triggers compensatory inhibitory responses to counteract overexcitation that lead to changes in synaptic circuitry and an increase in inhibitory activity in, eg, the temporal cortex. This leads to changes in the texture of the neural networks involved and might explain disruptions of the networks as seen in the default mode network (DMN) in AD.58-60 Transynaptic progression of toxicity effects of β-amyloid inducing epileptic activity from the entorhinal cortex to other brain regions may explain cognitive dysfunctions in AD.61
The default mode network
AD affects the default mode network (DMN). This network comprises brain regions that are active and interconnected in a wakeful state when the mind is not focused on something specific. Anatomically it includes part of the medial temporal lobe, the medial prefrontal cortex, the posterior cingulate cortex, ventral precuneus, and the medial, lateral, and inferior parietal cortex. This networks develops during childhood and adolescence and reaches full integration in adults, characterized by coherent infraslow EEG oscillations smaller than 0.1 Hz. The DMN is linked to other low-frequency resting state networks in the brain and is anti-correlated with the ventral and dorsal attention network. Measurements of glucose metabolism with positron emission tomography (PET), of structural atrophy with MRI, and intrinsic and task-evoked brain activity with fMRI in AD all suggest an increasing disruption in the DMN.62
When AD patients undergo a FDG-PET the pattern of hypometabolism often mirrors the same regions that belong to the posterior parts of the DMN, namely the posterior cingulate cortex, the retrosplenial cortex, inferior parietal lobule, and the lateral temporal cortex.63 Such hypometabolism correlates with the mental status while AD progresses.64 Probands with a genetic risk for AD of being homozygous for ApoE4 develop this hypometabolism already quite early in the course of the disease.62,65 Disruption in the DMN at the preclinical stages of the disease by accelerated cortical atrophy affects the medial temporal lobe and the posterior cingulum and the retrosplenial cortex.63,66 Also, analysis of task-induced deactivation and analysis of intrinsic activity correlations show an impaired DMN consistent with metabolic and structural changes.67-69 The DMN is coupled with hippocampus during memory retrieval but not during memory encoding, pointing to the special positioning of the hippocampus between short-term and long-term memory.70 Encoding structures of the DMN are among the first to show accumulation of β-amyloid even before symptoms emerge and images of β-amyloid plaques taken at the earliest stages of AD show a distribution that is remarkably similar to the anatomy of the default network.71 Buckner et al speculate that AD pathology forms preferentially throughout the DMN and may be linked to DMN activity.63 Their basic idea is that the DMN's continuous activity augments an activity-dependent or metabolism-dependent cascade that starts the β-amyloid cascade in these brain regions. Hence, memory would be affected preferentially by the disease because the DMN is mainly relying on cortical structures that are also vital to memory functions and “burns” them during activity. Interestingly, ApoE4 carriers have also been found to have a higher rate of activity in the DMN at rest compared with ApoE2 or ApoE3 carriers, and decreased connectivity.72-74 A successful connection of this hypothesis with the β-amyloid hypothesis of AD may require any kind of upregulation of β-amyloid during neural activity. Indeed, there are some studies that may support such a link. Cirrito et al showed that β-amyloid increased following stimulation of the brain in mice expressing human amyloid precursor protein. They demonstrated that β-amyloid in the brain interstitial fluid was dynamically and directly influenced by synaptic activity on a timescale of minutes to hours.75 This observation suggests that synaptic activity can increase the presence of extracellular β-amyloid. A further supporting observations come from a new PET method of mapping glycolysis based on measuring the ratio of oxygen to glucose consumption. Vlassenko et al calculated the spatial distribution of the regional glucose use apart from that entering oxidative phosphorylation.76 The so-called “aerobic glycolysis,” the process by which glucose is metabolized into cellular energy, might be more closely associated with neuronal or synaptic activity than the mere glucose utilization. In humans, aerobic glycolysis represents 35% of the glucose turnover in the brain of a newborn and 19% of the glucose used in the brain of an alert adult.77 Certain association areas in the human brain retain elevated levels of aerobic glycolysis in adulthood related to cognitive functions such as the dorsolateral prefrontal cortex, which is associated with working memory, and the ventromedial prefrontal cortex, the dorsomedial prefrontal cortex, the posterior cingulate cortex, the inferior parietal lobe, the lateral temporal cortex, and the hippocampus.62,76-79 In normal young adults aerobic glycolysis correlated positively and spatially with β-amyloid deposition observed in individuals with Alzheimer's dementia and cognitively normal participants with already elevated β-amyloid levels, suggesting a possible link between regional aerobic glycolysis in young adulthood and later development of AD pathology. The map of resting-state glycolysis correlated remarkably well with the distribution of amyloid plaques.76 On the other side DMN activity and coherence is diminished during deep sleep. Here only partial network involvement was observed, with apparent decoupling of frontal areas from the DMN80 An important study by Kang et al used in vivo micro-dialysis in mice and found that the amount of pamyloid in the interstitial brain fluid correlated positively with wakefulness. The amount of interstitial β-amyloid also significantly increased during acute sleep deprivation. Furthermore, chronic sleep restriction significantlyincreased, and a dual orexin receptor antagonist decreased β-amyloid plaque formation in human amyloid precursor protein transgenic mice.81 Interestingly, sleep deprivation, which is known to impair memory storage and retrieval, reduces default mode network connectivity and anti-correlation with the default attention networks during rest and task performance.82 Regarding AD subtypes, Lehmann et al report that the posterior DMN and precuneus network are commonly affected in all AD variants, whereas syndrome-specific neurodegenerative patterns are driven by the involvement of specific networks outside the DMN and characterize differentially early-onset AD (anterior salience network), logopenic variant of AD (language network), and posterior cortical atrophy (visual network).83
Sleep and memory
Sleep and the functional connectome are overlapping research areas. Neuroimaging studies of sleep based on EEG-PET and EEG-fMRI are revealing the brain networks that support sleep. Such infraslow oscillations may organize sleep-dependent neuroplastic processes including consolidation of episodic memory, for example. Picchioni et al found positive correlations between the power in the infraslow EEG band and MRI blood oxygen level-dependent (BOLD) response in subcortical regions and negative correlations in the cortex. Robust negative correlations were detected principally in paramedian heteromodal cortices whereas positive correlations were seen in cerebellum, thalamus, basal ganglia, lateral neocortices, and hippocampus.84 Sleep has adaptive and recreating functions that uphold waking activity in humans and mammals in general. Our understanding of DMN activity and its regulation during sleep may be also important for our general understanding of phenomena like memory, arousal, and consciousness.80,84-86 Clinically sleep-wake disturbances such as increased inadvertent daytime napping and insomnia at night affect 25% to 40% of patients with mild-to-moderate AD.87 Even in mild cognitive impairment there are already abnormalities in sleep architecture and electroencephalography measures. Sleep changes in patients with amnestic mild cognitive impairment may contribute to memory deficits by interfering with sleep-dependent memory consolidation.88
In a small study, Ju investigated sleep in 145 cognitively healthy probands older than 45 years. Amyloid deposition, as assessed by β-amyloid levels, was present in 32 participants. This group had worse sleep quality, as measured by sleep efficiency compared with those without amyloid deposition, after correction for age, sex, and ApoE4 allele carrier status, while the quantity of sleep did not differ between groups. Frequent napping, 3 or more days per week, was associated with amyloid deposition. The authors concluded that indices for amyloid deposition in the preclinical stage of AD appears to be associated with worse sleep quality.89
Taken together the brain activity patterns may directly modulate the molecular cascades that are relevant to diseases. In the case of AD, increased resting-state activity may accelerate the formation of amyloid pathology. This opens up perspectives for new interventions that may take the form of a therapy that attempts to modify glycolysis or other aspects of brain metabolism or to boost prophylaxis by the promotion of healthy sleep behavior or working behavior.
Conclusion
For the clinician the easily applicable neuropsychological test batteries augmented by a test for olfactory recognition continue to be at the heart of the diagnostic process for dementias, although new methods like CSF analyses or MRI volumetry are increasingly available and validated for clinical use. The use of amyloid-tracer PET or fMRI to investigate, for example, the DMN are still more preclinical than clinical in character. Despite enormous progress in our knowledge of some pathophysiologic mechanisms in the last decade, we are still far from understanding AD and also probably from finding a cure. The available medications or medications may, however, help us to manage some symptoms for our patients, and gain some time for them. To date the recent scientific advancements primarily offer earlier diagnosis. With new diagnostic criteria properly applied, we will likely be able to diagnose Alzheimer's disease many years before any relevant impairment is experienced by the patient. In light of the fact that most of us would suffer from AD, were we only to get old enough, one question arises: when the gods put the diagnosis before the therapy, how much “before” did they actually mean? As a clinician, I would appreciate any measure of “momentum” that would help me to translate biomarker findings, eg, a positive amyloid scan in PET, to the patient. Perhaps CSF tau concentrations could be helpful, but at the moment only time will tell us the individual course of the disease.
Selected abbreviations and acronyms
- DMN
default mode network
- DTI
diffusion tensor imaging
- FA
fractional anisotropy
- FX
fragile X
- HARDI
high angular resolution diffusion imaging
- ICA
independent components analysis
- TBM
tensor-based morphometry
- VBM
voxel-based morphometry
REFERENCES
- 1.Wimo A., Jonsson L., Winblad B. An estimate of the worldwide prevalence and direct costs of dementia in 2003. Dement Geriatr Cogn Dis. 2006;21:175–181. doi: 10.1159/000090733. [DOI] [PubMed] [Google Scholar]
- 2.Hebert LE., Scherr PA., Beckett LA., et al Age-specific incidence of Alzheimer's disease in a community population. JAMA. 1995;273:1354–1359. [PubMed] [Google Scholar]
- 3.Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift für Psychiatrie. 1907;64:146–148. [Google Scholar]
- 4.Blennow K., de Leon MJ., Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 5.Gerrish A., Russo G., Richards A., et al The role of variation at A beta PP, PSEN1, PSEN2, and MAPT in late onset Alzheimer's disease. J Alzheimers Dis. 2012;28:377–387. doi: 10.3233/JAD-2011-110824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- 7.Mayeux R. Gene-environment interaction in late-onset Alzheimer disease: the role of apolipoprotein-epsilon4. Alzheimer Dis Assoc Disord. 1998;12(suppl3):S10–S15. [PubMed] [Google Scholar]
- 8.Fullerton SM., Clark AG., Weiss KM., et al Apolipoprotein E variation at the sequence haplotype level:implications for the origin and maintenance of a major human polymorphism. Am J Hum Genet. 2000;67:881–900. doi: 10.1086/303070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bufill E., Carbonell E. Apolipoprotein E polymorphism and neuronal plasticity. Am J Hum Biol. 2006;18:556–558. doi: 10.1002/ajhb.20516. [DOI] [PubMed] [Google Scholar]
- 10.Finch CE., Stanford CB. Meat-adaptive genes and the evolution of slower aging in humans. Q Rev Biol. 2004;79:3–50. doi: 10.1086/381662. [DOI] [PubMed] [Google Scholar]
- 11.Braak H., Braak E. Neurofibrillary changes confined to the entorhinal region and an abundance of cortical amyloid in cases of presenile and senile dementia. Acta Neuropathologica. 1990;80:479–486. doi: 10.1007/BF00294607. [DOI] [PubMed] [Google Scholar]
- 12.Braak H., Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathology. 1996;92:197–201. doi: 10.1007/s004010050508. [DOI] [PubMed] [Google Scholar]
- 13.Braak H., Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 14.Hyman BT., Vanhoesen GW., Damasio AR., Barnes CL. Alzheimers-disease - cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 15.Braak H., Del Tredici K., Schultz C., Braak E. Vulnerability of select neuronal types to Alzheimer's disease. Ann N Y Acad Sci. 2000;924:53–61 . doi: 10.1111/j.1749-6632.2000.tb05560.x. [DOI] [PubMed] [Google Scholar]
- 16.Bondareff W., Mountjoy CQ., Roth M. Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology. 1982;32:164–168. doi: 10.1212/wnl.32.2.164. [DOI] [PubMed] [Google Scholar]
- 17.Mann DM., Yates PO., Marcyniuk B. A comparison of changes in the nucleus basalis and locus caeruleus in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1984;47:201–203. doi: 10.1136/jnnp.47.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobs HI., Van Boxtel MP., Uylings HB., Gronenschild EH., Verhey FR., Jolles J. Atrophy of the parietal lobe in preclinical dementia. Brain Cogn. 2011;75:154–163. doi: 10.1016/j.bandc.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Jacobs HI., Van Boxtel MP., Jolles J., Verhey FR., Uylings HB. Parietal cortex matters in Alzheimer's disease: an overview of structural, functional and metabolic findings. Neurosci Biobehav Rev. 2012;36:297–309. doi: 10.1016/j.neubiorev.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs HI., van Boxtel MP., Gronenschild EH., et al Patterns of gray and white matter changes in individuals at risk for Alzheimer's disease. Curr Alzheimer Res. 2012;9:1097–1105. doi: 10.2174/156720512803568993. [DOI] [PubMed] [Google Scholar]
- 21.Fox NC., Freeborough PA., Rossor MN. Visualisation and quantification of rates of atrophy in Alzheimer's disease. Lancet. 1996;348:94–97. doi: 10.1016/s0140-6736(96)05228-2. [DOI] [PubMed] [Google Scholar]
- 22.Fox NC., Cousens S., Scahill R., Harvey RJ., Rossor MN. Using serial registered brain magnetic resonance imaging to measure disease progression in Alzheimer disease: power calculations and estimates of sample size to detect treatment effects. Arch Neurol. 2000;57:339–344. doi: 10.1001/archneur.57.3.339. [DOI] [PubMed] [Google Scholar]
- 23.Andrews KA., Modat M., Macdonald KE., et al Atrophy rates in asymptomatic amyloidosis: implications for Alzheimer prevention trials. PLoS One. 2013;8:e58816. doi: 10.1371/journal.pone.0058816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arlt S., Buchert R., Spies L., Eichenlaub M., Lehmbeck JT., Jahn H. Association between fully automated MRI-based volumetry of different brain regions and neuropsychological test performance in patients with amnestic mild cognitive impairment and Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci. 2013;263:335–344. doi: 10.1007/s00406-012-0350-7. [DOI] [PubMed] [Google Scholar]
- 25.Arlt S., Brassen S., Wilke F., et al Association between FDG uptake, CSF biomarkers and cognitive performance in patients with probable Alzheimer's disease. Pharmacopsychiatry. 2009;42:210–210. doi: 10.1007/s00259-009-1063-7. [DOI] [PubMed] [Google Scholar]
- 26.Crutch SJ., Lehmann M., Schott JM., Rabinovici GD., Rossor MN., Fox NC. Posterior cortical atrophy. Lancet Neurol. 2012;11:170–178. doi: 10.1016/S1474-4422(11)70289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crutch SJ., Schott JM., Rabinovici GD., et al Shining a light on posterior cortical atrophy. Alzheimers Dement. 2013;9:463–465. doi: 10.1016/j.jalz.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Crutch SJ., Lehmann M., Warren JD., Rohrer JD. The language profile of posterior cortical atrophy. J Neurol Neurosurg Psychiatry. 2013;84:460–466. doi: 10.1136/jnnp-2012-303309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehmann M., Crutch SJ., Ridgway GR., et al Cortical thickness and voxelbased morphometry in posterior cortical atrophy and typical Alzheimer's disease. Neurobiol Aging. 2011;32:1466–1476. doi: 10.1016/j.neurobiolaging.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 30.Gorno-Tempini ML., Dronkers NF., Rankin KP., et al Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55:335–346. doi: 10.1002/ana.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorno-Tempini ML., Hillis AE., Weintraub S., et al Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berchtold NC., Coleman PD., Cribbs DH., Rogers J., Gillen DL., Cotman CW. Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer's disease. Neurobiol Aging. 2013;34:1653–1661. doi: 10.1016/j.neurobiolaging.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jahn H., Wittke S., Zuerbig P., et al Peptide fingerprinting of Alzheimer's disease in cerebrospinal fluid: identification and prospective evaluation of new synaptic biomarkers. Plos One. 2011;6 doi: 10.1371/journal.pone.0026540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zuerbig P., Jahn H. Use of proteomic methods in the analysis of human body fluids in Alzheimer research. Electrophoresis. 2012;33:3617–3630. doi: 10.1002/elps.201200360. [DOI] [PubMed] [Google Scholar]
- 35.Jessen F., Guer O., Block W., et al A multicenter H-1-MRS study of the medial temporal lobe in AD and MCI. Neurology. 2009;72:1735–1740. doi: 10.1212/WNL.0b013e3181a60a20. [DOI] [PubMed] [Google Scholar]
- 36.Jack CR., JR, Albert MS., Knopman DS., et al Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dementia. 2011;7:257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKhann GM., Knopman DS., Chertkow H., et al The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dementia. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sperling RA., Aisen PS., Beckett LA., et al Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dementia. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albert MS., DeKosky ST., Dickson D., et al The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dementia. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hyman BT., Phelps CH., Beach TG., et al National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dementia. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller GA. The magical number seven, plus or minus two: some limits on our capacity for processing information. Psychol Rev. 1956;63:81–97. [PubMed] [Google Scholar]
- 42.Verma M., Howard RJ. Semantic memory and language dysfunction in early Alzheimer's disease: a review. Int J Geriatr Psychiatry. 2012;27:1209–1217. doi: 10.1002/gps.3766. [DOI] [PubMed] [Google Scholar]
- 43.Morris JC. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr. 1997;9(suppl 1):173–176-discussion 177-178. doi: 10.1017/s1041610297004870. [DOI] [PubMed] [Google Scholar]
- 44.Morris JC., Heyman A., Mohs RC., et al The consortium to establish a registry for Alzheimer's Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurology. 1989;39:1159–1165. doi: 10.1212/wnl.39.9.1159. [DOI] [PubMed] [Google Scholar]
- 45.Morris JC., Mohs RC., Rogers H., Fillenbaum G., Heyman A. Consortium to establish a registry for Alzheimer's disease (CERAD): clinical and neuropsychological assessment of Alzheimer's disease. Psychopharmacol Bull. 1988;24:641–652. [PubMed] [Google Scholar]
- 46.Pepeu G., Giovannini MG. Cholinesterase inhibitors and memory. Chem Biol Interact. 2010;187:403–408. doi: 10.1016/j.cbi.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 47.Pepeu G., Giovannini MG., Bracco L. Effect of cholinesterase inhibitors on attention. Chem Biol Interact. 2013;203:361–364. doi: 10.1016/j.cbi.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 48.Wagner M., Wolf S., Reischies FM., et al Biomarker validation of a cued recall memory deficit in prodromal Alzheimer disease. Neurology. 2012;78:379–386. doi: 10.1212/WNL.0b013e318245f447. [DOI] [PubMed] [Google Scholar]
- 49.Snigdha S., Milgram NW., Willis SL., et al A preclinical cognitive test battery to parallel the National Institute of Health Toolbox in humans: bridging the translational gap. Neurobiol Aging. 2013;34:1891–1901. doi: 10.1016/j.neurobiolaging.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jessen F., Wiese B., Bachmann C., et al Prediction of dementia by subjective memory impairment effects of severity and temporal association with cognitive impairment. Arch Gen Psychiatry. 2010;67:414–422. doi: 10.1001/archgenpsychiatry.2010.30. [DOI] [PubMed] [Google Scholar]
- 51.Jessen F., Feyen L., Freymann K., et al Volume reduction of the entorhinal cortex in subjective memory impairment. Neurobiol Aging. 2006;27:1751–1756. doi: 10.1016/j.neurobiolaging.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 52.Peter J., Scheef L., Abdulkadir A., et al Gray matter atrophy pattern in elderly with subjective memory impairment. Alzheimers Dement. 2013 Jul 15. [Epub ahead of print]. doi: 10.1016/j.jalz.2013.05.1764. [DOI] [PubMed] [Google Scholar]
- 53.Scheef L., Spottke A., Daerr M., et al Glucose metabolism, gray matter structure, and memory decline in subjective memory impairment. Neurology. 2012;79:1332–1339. doi: 10.1212/WNL.0b013e31826c1a8d. [DOI] [PubMed] [Google Scholar]
- 54.Erk S., Spottke A., Meisen A., Wagner M., Walter H., Jessen F. Evidence of neuronal compensation during episodic memory in subjective memory impairment. Arch Gen Psychiatry. 2011;68:845–852. doi: 10.1001/archgenpsychiatry.2011.80. [DOI] [PubMed] [Google Scholar]
- 55.Striepens N., Scheef L., Wind A., et al Interaction effects of subjective memory impairment and ApoE4 genotype on episodic memory and hippocampal volume. Psychol Med. 2011;41:1997–2006. doi: 10.1017/S0033291711000067. [DOI] [PubMed] [Google Scholar]
- 56.Peters JM., Hummel T., Kratzsch T., Lötsch J., Skarke C., Frölich L. Olfactory function in mild cognitive impairment and Alzheimer's disease: an investigation using psychophysical and electrophysiological techniques. Am J Psychiatry. 2003;160:1995–2002. doi: 10.1176/appi.ajp.160.11.1995. [DOI] [PubMed] [Google Scholar]
- 57.Rahayel S., Frasnelli J., Joubert S. The effect of Alzheimer's disease and Parkinson's disease on olfaction:a meta-analysis. Behav Brain Res. 2012;231:60–74. doi: 10.1016/j.bbr.2012.02.047. [DOI] [PubMed] [Google Scholar]
- 58.Palop JJ., Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palop JJ., Mucke L. Synaptic depression and aberrant excitatory network activity in Alzheimer's disease: two faces of the same coin? Neuromolecular. Med. 2010;12:48–55. doi: 10.1007/s12017-009-8097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palop JJ., Mucke L., Roberson ED. Quantifying biomarkers of cognitive dysfunction and neuronal network hyperexcitability in mouse models of Alzheimer's disease: depletion of calcium-dependent proteins and inhibitory hippocampal remodeling. Methods Mol Biol. 2011;670:245–262. doi: 10.1007/978-1-60761-744-0_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harris JA., Devidze N., Verret L., et al Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron. 2010;68:428–441. doi: 10.1016/j.neuron.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buckner RL., Andrews-Hanna JR., Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 63.Buckner RL., Snyder AZ., Shannon BJ., et al Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Herholz K., Salmon E., Perani D., et al Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage. 2002;17:302–316. doi: 10.1006/nimg.2002.1208. [DOI] [PubMed] [Google Scholar]
- 65.Reiman EM., Caselli RJ., Yun LS., et al Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 66.Scahill Rl., Schott JM., Stevens JM., Rossor MN., Fox NC. Mapping the evolution of regional atrophy in Alzheimer's disease: unbiased analysis of fluid-registered serial MRI. Proc Natl Acad Sci USA. 2002;99:4703–4707. doi: 10.1073/pnas.052587399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lustig C., Snyder AZ., Bhakta M., et al Functional deactivations: change with age and dementia of the Alzheimer type. Proc Natl Acad Sci USA. 2003;100:14504–14509. doi: 10.1073/pnas.2235925100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Celone KA., Calhoun VD., Dickerson BC., et al Alterations in memory networks in mild cognitive impairment and Alzheimer's disease: an independent component analysis. J Neurosci. 2006;26:10222–10231. doi: 10.1523/JNEUROSCI.2250-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Greicius MD., Srivastava G., Reiss AL., Menon V. Default-mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101:4637–4642. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huijbers W., Pennartz CM., Cabeza R., Daselaar SM. The hippocampus is coupled with the default network during memory retrieval but not during memory encoding. PloS One. 2011;6:e17463. doi: 10.1371/journal.pone.0017463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klunk WE., Engler H., Nordberg A., et al Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 72.Sheline Yl., Morris JC., Snyder AZ., et al APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Aβ42. J Neurosci. 2010;30:17035–17040. doi: 10.1523/JNEUROSCI.3987-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patel KT., Stevens MC., Pearlson GD., et al Default mode network activity and white matter integrity in healthy middle-aged ApoE4 carriers. Brain imaging Behav. 2013;7:60–67. doi: 10.1007/s11682-012-9187-y. [DOI] [PubMed] [Google Scholar]
- 74.Filippini N., Macintosh BJ., Hough MG., et al Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc. Natl Acad Sci U S A. 2009;106:7209–7214. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cirrito JR., Yamada KA., Finn MB., et al Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 76.Vlassenko AG., Vaishnavi SN., Couture L., et al Spatial correlation between brain aerobic glycolysis and arnyloid-β (Aβ) deposition. Proc Natl Acad Sci U S A. 2010;107:17763–17767. doi: 10.1073/pnas.1010461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vaishnavi SN., Vlassenko AG., Rundle MM., Snyder AZ., Mintun MA., Raichle ME. Regional aerobic glycolysis in the human brain. Proc Natl Acad Sci U S A. 2010;107:17757–17762. doi: 10.1073/pnas.1010459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Buckner RL. Human functional connectivity: new tools, unresolved questions. Proc Natl Acad Sci U S A. 2010;107:10769–10770. doi: 10.1073/pnas.1005987107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Buckner RL. The role of the hippocampus in prediction and imagination. Annu Rev Psychol. 2010;61:27–48, C21-C28. doi: 10.1146/annurev.psych.60.110707.163508. [DOI] [PubMed] [Google Scholar]
- 80.Horovitz SG., Braun AR., Carr WS., et al Decoupling of the brain's default mode network during deep sleep. Proc Natl Acad Sci U S A. 2009;106:11376–11381. doi: 10.1073/pnas.0901435106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kang JE., Lim MM., Bateman RJ., et al Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De Havas JA., Parimal S., Soon CS., Chee MW. Sleep deprivation reduces default mode network connectivity and anti-correlation during rest and task performance. Neuroimage. 2012;59:1745–1751. doi: 10.1016/j.neuroimage.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 83.Lehmann M., Madison CM., Ghosh PM., et al Intrinsic connectivity networks in healthy subjects explain clinical variability in Alzheimer's disease. Proc Natl Acad Sci U S A. 2013;110:11606–11611. doi: 10.1073/pnas.1221536110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Picchioni D., Horovitz SG., Fukunaga M., et al Infraslow EEG oscillations organize large-scale cortical-subcortical interactions during sleep: a combined EEG/fMRI study. Brain Res. 2011;1374:63–72. doi: 10.1016/j.brainres.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Picchioni D., Duyn JH., Horovitz SG. Sleep and the functional connectome. Neuroimage. 2013;80:387–396. doi: 10.1016/j.neuroimage.2013.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chow HM., Horovitz SG., Carr WS., et al Rhythmic alternating patterns of brain activity distinguish rapid eye movement sleep from other states of consciousness. Proc Natl Acad Sci U S A. 2013;110:10300–10305. doi: 10.1073/pnas.1217691110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moran M., Lynch CA., Walsh C., Coen R., Coakley D., Lawlor BA. Sleep disturbance in mild to moderate Alzheimer's disease. Sleep Med. 2005;6:347–352. doi: 10.1016/j.sleep.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 88.Westerberg CE., Mander BA., Florczak SM., et al Concurrent impairments in sleep and memory in amnestic mild cognitive impairment. J Int Neuropsychol Soc. 2012;18:490–500. doi: 10.1017/S135561771200001X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ju YE., McLeland JS., Toedebusch CD., et al Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70:587–593. doi: 10.1001/jamaneurol.2013.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]