

Abstract
Replication stress is a significant contributor to genome instability. Recent studies suggest that the centromere is particularly susceptible to replication stress and prone to rearrangements and genome damage, as well as chromosome loss. This effect is enhanced by loss of heterochromatin. The resulting changes in genetic organiation including chromosome loss, increased mutation, and loss of heterozygosity are important contributors to malignant growth.
Keywords: fission yeast, S. pombe, centromere, Swi6, heterochromatin, recombination, chromosomes, LOH
Introduction
Certain cancer cells, particularly solid tumors, are known for dramatic chromosome instability (CIN) phenotypes. Aneuploidy and other Numerical disruptions (nCIN) likely occur from malfunction of the chromosome segregation apparatus, while Structural instability (sCIN) is typically manifested as gross chromosome rearrangements, frequently linked replication stress and DNA breakage (rev. in [1]). Both forms of CIN can contribute to loss of heterozygosity (LOH), which is also a contributor to malignant transformation (e.g., [2]).
The role of the centromere in maintaining faithful chromosome segregation and preventing nCIN is self-evident: it is a structural domain on the chromosome that is required for proper attachment of the spindle (rev. in [3]). But it is increasingly clear that the centromere is also a significant contributor to sCIN, particularly in conditions of replication stress. Such stress can be caused by exposure to external DNA damaging agents, including radiation or chemicals, or chemotherapeutic drugs that block S phase or stall fork progression. Endogenous stresses can occur in naturally fragile regions of the genome, including repetitive sequences or pause sites that render the genome vulnerable to replication fork stalling or fork collapse. Indeed, a robust system of DNA damage response and repair has been invoked as the primary barrier to malignant transformation (e.g., [4])
A conserved centromere alternates silencing with synthesis
Fission yeast offers an outstanding genetic model to study how replication stress impacts the centromere, because proteins that respond to replication stress and proteins responsible for centromere maintenance are each highly conserved (rev. in [3, 5, 6] ). As in most eukaryotes, the centromere in fission yeast is a large element, where heterochromatic pericentromere repeats flank a central core domain. This heterochromatin is defined by the presence of histone H3K9 methylation, which is bound by two homologues of Heterochromatin Protein 1, Swi6 and Chp2, via their conserved chromodomains to establishes a rigid, transcriptionally silenced structure (Fig 1A).
Figure 1.
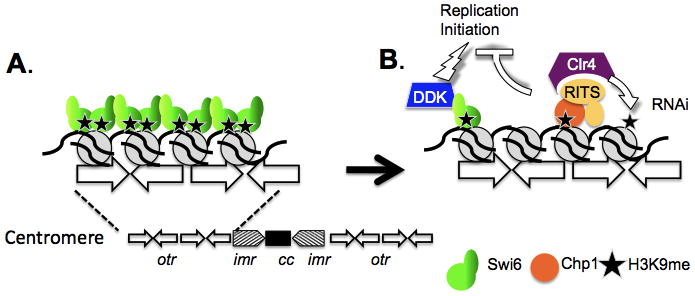
A. The fission yeast centromere, showing the structure of established heterochromatin with Swi6 binding tH3K9me. B. During early S phase, H3K9me is reduced. Swi6 binding to DDK promotes early replication initiation. Unopposed binding of Chp1 (in the absence of Swi6) opposes early initiation.
The methyl-mark is established and maintained by the methyltransferase Clr4 (SuVar3-9). Paradoxically, Clr4 is targeted this domain by transient de-silencing during S phase, which allows a brief wave of convergent transcription [7, 8]. These short RNAs are processed by RNAi mechanisms that are used to target Clr4 back to the site of transcription, re-establishing the methyl mark on newly incorporated histones [9, 10]. Clr4 binding to Swi6 and to components of the replisome provide additional mechanisms to recruitment and spreading of the methyl mark [11–13]. This is a simplified summary, as additional players that fine tune the response continue to be identified (e.g., rev. in [3, 6]. See Fig. 1.)
Regulating the timing of centromere DNA replication
The pericentromere is rich in replication origins, which alternate in position with the transcription units that generate the siRNAs [14, 15]. Initiation of replication in eukaryotes is a well conserved process that requires the orderly assembly of a complex of proteins that mark potential origins for use (rev in [16]). This begins with identification of the origin by ORC, the origin recognition complex. ORC is activated by the Cdc18 (Cdc6) and Cdt1 proteins which load the heterohexameric MCM complex, and together these form the pre-Replication Complex, or preRC, which remains poised for activation. The activation signal depends on the combined effects of the CDK cell cycle kinase, which conveys a global replication signal, and the DDK replication kinase, which will activate the preRC directly through phosphorylation of MCM proteins and other substrates. This activation allows the initiation of unwinding, and recruitment of additional proteins to convert the preRC into an initiation complex and finally an intact replisome that processively synthesizes DNA.
Unlike most heterochromatin domains, the fission yeast outer repeats undergo replication early in S phase [17] and this depends upon Swi6 [18, 19]. Swi6 binds to the DDK regulatory subunit, Dfp1 [20] and this interaction is required for early replication [18]. Indeed, artificially tethering Dfp1 to the chromatin via the Swi6 chromo-domain restores early replication in a swi6Δ cell [18], which indicates that sufficient histone methylation remains in early S phase, despite the transient de-silencing, to recruit chromodomain proteins. The late replication in Δswi6Δ is also rescued in a Δswi6 Δclr4 double mutant, suggesting that the late effect is linked in some way to unopposed H3K9me [18] (Fig 1b.)
In addition to binding to Dfp1, Swi6 also binds to the Cdc18 initiator protein [19] and to ORC (P-C Li & SLF, unpublished). A mutation in Cdc18 that disrupts its association with Swi6 has no effect on centromere silencing or replication in the euchromatin, but leads to even earlier replication in the centromere than in wild type cells, suggesting that Swi6 interactions with Dfp1 and Cdc18 balance the proper timing of replication. In addition to its connection with components of initiation, there is also a role at the replication fork. Swi6 binds to DNA polymerase alpha, which is required for silencing [21]. Clr4, the histone methyltransferase, is also associated with the leading strand DNA polymerase epsilon in fission yeast [11], which provides a direct connection for rapid re-methylation of the heterochromatin domain.
While swi6Δ mutants replicate their centromeres late, we recently showed that clr4Δ mutants replicate early [22]; this is consistent with the observation clr4Δ rescues the late replication phenotype of swi6Δ [18]. Early replication was also observed in chp1Δ mutants [22]. Chp1 is a chromodomain protein that binds to H3K9me and to siRNA as part of the RITS complex, which provides one means of recruiting Clr4 [10, 23]. Loss of chp1Δ leads to a severe reduction in H3K9me but does not eliminate it [24–26].
Unexpectedly, however, we find that other mutations that significantly reduce H3K9me such as the RNAi components dcr1Δ, hrr1Δ, or rdp1Δ nevertheless cause late replication similar to swi6Δ [22]. There is residual H3K9me and Chp1 binding in RNAi mutants [27, 28], so we speculate that it is not H3K9me per se but Chp1 bound to it that results in late replication [22]. Chp1 binding to H3K9me occurs with higher affinity that that of Swi6, and it needs to be removed to allow proper Swi6 association and assembly of the fully repressed heterochromatin structure [29, 30]. (This probably via interactions between adjacent Swi6 chromoshadow domains (see fig;[31]). Together, these results suggest that Chp1 binding in the absence of Swi6 leads to a structure that is refractory to early replication.
The replication timing results are unexpected, given that ChIP analysis suggests that Swi6 is largely removed from the centromere during mitosis and returns in late S phase [7, 8]. However, it seems likely that a subpopulation of Swi6 is recruited earlier than the bulk of the protein that performs DNA silencing; this can be observed by cytological assays comparing the timing of GFP-Swi6 recruitment to the spindle poles (and thus, centromeres) following mitosis. We find that Swi6 is restored to the centromeres very quickly after anaphase, lagging behind the centromere specific histone H3 variant Cnp1 (CENP-A) by only about 3 minutes [22]. In contrast, Chp1-GFP recruitment is more prolonged. We posit that this low level of early Swi6 recruitment allow concentration of Dfp1 (and thus, the origin activating DDK kinase) to facilitate replication directly, and perhaps also works by antagonizing the high-affinity Chp1 protein. By this model, there are two roles for Swi6 in S phase: one in establishing a permissive replication structure early, and then later, a higher local concentration that locks down the chromatin into typically repressive heterochromatin. This may be related to the distinct intra- and inter-molecular interactions of Swi6 molecules that distinguishes different degrees of repression [32]).
Replication as a source of stress
Several aspects of this process expose vulnerabilities that are specific to S phase. First, the pericentromere is made up of repeated sequences, and such sequences are known to be prone to recombination or replication fork pausing (e.g, [33–35]). The presence of heterochromatin is generally refractory to recombination [36]. However, from M to S phase, heterochromatin is partly disrupted [7, 8], suggesting a window of vulnerability. Transcription during S phase to generate the siRNAs used to target Clr4 leads to the possibility of collisions between DNA and RNA polymerase; recent work showed that the RNAi proteins contribute to RNA polymerase eviction to reduce this possibility [15].
That this domain is subject to intrinsic stress even in normal cells with intact heterochromatin is suggested by the constitutive presence of a low-level of histone H2AX phosphorylation; this modification is typically associated with regions of DNA damage or double strand breaks [37]. The SMC5/6 protein complex, which is associated with recombination-mediated replication fork restart, is enriched at the centromere and other repetitive domains (particularly the rDNA; [38–40]). Brc1, a BRCT-motif containing protein that binds gH2AX and is also associated with replication fork restart, is also enriched at the centromere, where its presence depends on Clr4 [41]. The pericentromere may be refractive to repair (e.g., [42, 43]), making it particularly vulnerable to damage from replication stress.
Interestingly, the repeats of the centromere provides a substrate for break-induced replication (BIR) even in wild type cells; this has been demonstrated using an elegant genetic system, in which a non-essential mini-chromosome derivative of chromosome 3 is examined for rearrangements, including iso-chromosome formation and translocations from the intact chromsome 3 udner a variety of conditions ([44, 45]; Fig 2). These results show that recombination through the centromere is a significant contributor to loss of heterozygosity. Intriguingly, in rad51Δ mutants, there is an increased recombination through the imr repeats to generate iso-chromosomes [45]. This suggests that Rad51 functions to limit the scale of rearrangement, possibly promoting a maintenance recombination-based mechanisms to preserve the repetitive centromere structure. There are synthetic growth defects between swi6Δ and rad51Δ [46], which suggests that this mechanism becomes particularly important when heterochromatin formation is impaired. Intriguingly, has been suggested that recombination in the centromere is an intrinsic part of centromere maintenance, particularly of the inner, imr repeat sequences that flank the central core, and are unique to each centromere [47]. Thus, the cell must balance between limited recombination and the potential chromosome rearrangements.
Figure 2.
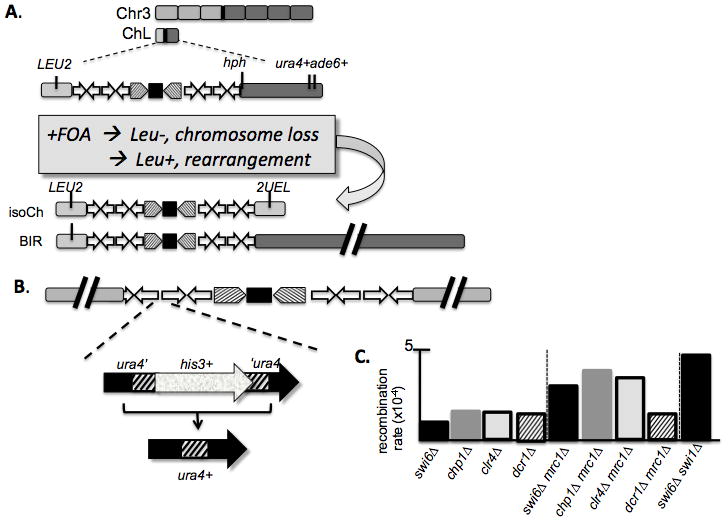
A. Minichromosome model for chromosome rearrangement [Tinline-Purvis, 2009 #5381][Nakamura, 2008 #5382] leads to isochromosome formation or acrocentric deriiatives through BIR. B. Internal model for centromere rearrangement uses an interrupted ura4+ gene to monitor recombination. C. Double mutants that disrupt Swi6 or Chp1 and components of replication fork stability have enhanced defects, using the system in Fig 2b. Median recombination rate reported. Original data and statistical analysis from [Li, 2013 #6465].
Heterochromatin and replication fork stability proteins make independent contributions to centromere stability
We hypothesized that the outer repeats of the centromere are sources of replication stress in the absence of heterochromatin, and reasoned this should be exacerbated by mutations that destabilize the replication fork. Mrc1 is a nonessential component of the replisome that acts as a processivity factor (part of the fork protection complex), and has a genetically separable function in activating the replication checkpoint kinase Cds1 ([48, 49]). We found double mutants between heterochromatin components and mrc1Δ have reduced growth rates and dramatically increased sensitivity to HU, which is not seen in double mutants with the checkpoint-specific mrc1-SA allele [48]. This suggests that replicaiton fork processivity associated with Mrc1 is particularly important when repeated domains are destabilized. We also observed that chp1Δ mrc1Δ shows significantly lower plating efficiency than chp1Δ or swi6Δ mrc1Δ, suggesting that the effect is exacerbated in that strain.
Using the genetic system developed in [45], we examined the frequency of chromosome loss (nCIN) and rearrangements (sCIN) involving the non-essential minichromosome in swi6Δ, swi6Δ mrc1Δ and Δchp1 (the double mutant chp1Δ mrc1Δ could not be maintained with the mini-chromosome) [50]. As expected, the rate of chromosome loss was very high in the heterochromatin mutants relative to wild type, consistent with the structural role of Swi6 and Chp1 in chromosome transmission. Rearrangements occurred at a low frequency in the heteorchromatin mutants, but in contrast to wild type, these were largely isochromosomes that involved a recombination specifically through the outer repeat. This indicates that the outer repeat is destabilized and prone to rearrangement in a mutant that lacks either swi6Δ or chp1Δ. An mrc1Δ single mutant, conversely, shows a low rate of chromosome loss but a high rate of rearrangement not limited to the centromere repeats. Strikingly, the double mutant swi6Δ mrc1Δ has a reduced rate of chromosome loss compared to swi6Δ, accompanied by a dramatic increase in recombination. In at least five of the 12 independent isolates, this results in loss of normal chromosome 3 and recovery of its two arms as separate chromosomes [50], presumably by a break-induced replication mechanism (BIR; eg, [44, 45]).
We extended these observations using a simpler assay in which we scored recombination between two repeats of a partial tandem duplication embedded within the heterochromatin (fig 2B). Results were consistent with those in the minichromosome system; consistent with the effects of swi6Δ mrc1Δ, we found that a double mutant swi6Δ swi1Δ that lacks the fork protection complex also significantly enhanced recombination. Mutants that lack Swi1 are sensitive to fork destabilizing agents such as MMS [51, 52]. Similar results were observed for clr4Δ as for chp1Δ or swi6Δ. Double mutants with cds1Δ had a less severe, but still detectable effect. Unexpectedly, however, the synergy between replication fork protection proteins and heterochromatin mutants in causing genome rearrangements was not found in RNAi mutants such as dcr1Δ.. Further epistatic analysis will be required to determine the source of the differences.
Conclusions
Fission yeast centromeres suffers from endogenous replication stress, and is also susceptible to several different forms of fork stressors including transcriptional collisions and reduced fork processivity. The sensitivity cannot be linked simply to replication timing as both early-replicating mutants (e.g., chp1Δ, clr4Δ) and late replicating mutants (swi6Δ) show similar synthetic defects combined with mrc1Δ, and some late replicators (e.g., dcr1Δ,) do not show the same degree of sensitivity. The centromere suffers intrinsic stress even in heterochromatin-competent cells (e.g., by the recruitment of phosphorylated histone H2A, and the Brc1 and Smc5/6 proteins), which may be related to recombination-mediated mechanisms required for its normal maintenance.
There is evidence that the centromere is structurally unstable in cancer cells, prone to fission or breakage [53–55]. It has been proposed that defects in chromosome segregation can contribute to centromere-linked breaks and fusions in cancer (e.g., [1]). However, it is now clear that replication stress contributes to mitotic mis-segregation, especially if the checkpoint is disrupted [55–57]. Fission yeast provides important insights into the mechanisms involved in centromere-linked genome damage and rearrangements, and how they contribute to loss of heterozygosity (LOH) and aneuploidy. Together, these data emphasize that centromere is a weak link in genome maintenance, and the target of multiple converging pathways to manage intrinsic stress and prevent multiple CINs.
Acknowledgments
Thanks to members of my lab for helpful comments on the manuscript. Apologies to those colleagues whose work could not be cited, for reasons of space.
Funding
These studies were funded by grants from the National Institutes of Health (GM059321) and National Science Foundation (MCB 0640103)
References
- 1.Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–8. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- 2.Lasko D, Cavenee W, Nordenskjold M. Loss of constitutional heterozygosity in human cancer. Annu Rev Genet. 1991;25:281–314. doi: 10.1146/annurev.ge.25.120191.001433. [DOI] [PubMed] [Google Scholar]
- 3.Verdaasdonk JS, Bloom K. Centromeres: unique chromatin structures that drive chromosome segregation. Nat Rev Mol Cell Biol. 2011;12:320–32. doi: 10.1038/nrm3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halazonetis TD, V, Gorgoulis G, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 5.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–19. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 6.Lejeune E, Bayne EH, Allshire RC. On the connection between RNAi and heterochromatin at centromeres. Cold Spring Harb Symp Quant Biol. 2010;75:275–83. doi: 10.1101/sqb.2010.75.024. [DOI] [PubMed] [Google Scholar]
- 7.Kloc A, Zaratiegui M, Nora E, Martienssen R. RNA interference guides histone modification during the S phase of chromosomal replication. Curr Biol. 2008;18:490–5. doi: 10.1016/j.cub.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SI. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature. 2008;451:734–7. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- 9.Noma K, Sugiyama T, Cam H, Verdel A, Zofall M, Jia S, Moazed D, Grewal SI. RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat Genet. 2004;36:1174–80. doi: 10.1038/ng1452. [DOI] [PubMed] [Google Scholar]
- 10.Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303:672–6. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li F, Martienssen R, Cande WZ. Coordination of DNA replication and histone modification by the Rik1-Dos2 complex. Nature. 2011;475:244–8. doi: 10.1038/nature10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 13.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 14.Wohlgemuth JG, Bulboaca GH, Moghadam M, Caddle MS, Calos MP. Physical mapping of origins of replication in the fission yeast Schizosaccharomyces pombe. Mol Biol Cell. 1994;5:839–849. doi: 10.1091/mbc.5.8.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaratiegui M, Castel SE, Irvine DV, Kloc A, Ren J, Li F, de Castro E, Marin L, Chang AY, Goto D, Cande WZ, Antequera F, Arcangioli B, Martienssen RA. RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature. 2011;479:135–8. doi: 10.1038/nature10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forsburg SL. Eukaryotic MCM proteins: beyond replication initiation. Microbiol Mol Biol Rev. 2004;68:109–31. doi: 10.1128/MMBR.68.1.109-131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SM, Dubey DD, Huberman JA. Early-replicating heterochromatin. Genes Dev. 2003;17:330–335. doi: 10.1101/gad.1046203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayashi MT, Takahashi TS, Nakagawa T, Nakayama J, Masukata H. The heterochromatin protein Swi6/HP1 activates replication origins at the pericentromeric region and silent mating-type locus. Nat Cell Biol. 2009;11:357–62. doi: 10.1038/ncb1845. [DOI] [PubMed] [Google Scholar]
- 19.Li PC, Chretien L, Cote J, Kelly TJ, Forsburg SL. S. pombe replication protein Cdc18 (Cdc6) interacts with Swi6 (HP1) heterochromatin protein: region specific effects and replication timing in the centromere. Cell Cycle. 2011;10:323–36. doi: 10.4161/cc.10.2.14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bailis JM, Bernard P, Antonelli R, Allshire R, Forsburg SL. Hsk1/Dfp1 is required for heterochromatin-mediated cohesion at centromeres. Nat Cell Biol. 2003:1111–1116. doi: 10.1038/ncb1069. [DOI] [PubMed] [Google Scholar]
- 21.Nakayama J, Allshire RC, Klar AJ, Grewal SI. A role for DNA polymerase alpha in epigenetic control of transcriptional silencing in fission yeast. EMBO J. 2001;20:2857–2866. doi: 10.1093/emboj/20.11.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li PC, Green MD, Forsburg SL. Mutations disrupting histone methylation have different effects on replication timing in S. pombe centromere. PLOS ONE. 2013;8:e61464. doi: 10.1371/journal.pone.0061464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schalch T, Job G, Shanker S, Partridge JF, Joshua-Tor L. The Chp1-Tas3 core is a multifunctional platform critical for gene silencing by RITS. Nat Struct Mol Biol. 2011;18:1351–7. doi: 10.1038/nsmb.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayashi A, Ishida M, Kawaguchi R, Urano T, Murakami Y, Nakayama J. Heterochromatin protein 1 homologue Swi6 acts in concert with Ers1 to regulate RNAi-directed heterochromatin assembly. Proc Natl Acad Sci U S A. 2012;109:6159–64. doi: 10.1073/pnas.1116972109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadaie M, Iida T, Urano T, Nakayama J. A chromodomain protein, Chp1, is required for the establishment of heterochromatin in fission yeast. EMBO J. 2004;23:3825–35. doi: 10.1038/sj.emboj.7600401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Debeauchamp JL, Moses A, Noffsinger VJ, Ulrich DL, Job G, Kosinski AM, Partridge JF. Chp1-Tas3 interaction is required to recruit RITS to fission yeast centromeres and for maintenance of centromeric heterochromatin. Mol Cell Biol. 2008;28:2154–66. doi: 10.1128/MCB.01637-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Motamedi MR, Hong EJ, Li X, Gerber S, Denison C, Gygi S, Moazed D. HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol Cell. 2008;32:778–90. doi: 10.1016/j.molcel.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugiyama T, Cam H, Verdel A, Moazed D, Grewal SI. RNA-dependent RNA polymerase is an essential component of a self-enforcing loop coupling heterochromatin assembly to siRNA production. Proc Natl Acad Sci U S A. 2005;102:152–7. doi: 10.1073/pnas.0407641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schalch T, Job G, Noffsinger VJ, Shanker S, Kuscu C, Joshua-Tor L, Partridge JF. High-affinity binding of Chp1 chromodomain to K9 methylated histone H3 is required to establish centromeric heterochromatin. Mol Cell. 2009;34:36–46. doi: 10.1016/j.molcel.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xhemalce B, Kouzarides T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 2010;24:647–52. doi: 10.1101/gad.1881710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Canzio D, Chang EY, Shankar S, Kuchenbecker KM, Simon MD, Madhani HD, Narlikar GJ, Al-Sady B. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol Cell. 2011;41:67–81. doi: 10.1016/j.molcel.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Canzio D, Liao M, Naber N, Pate E, Larson A, Wu S, Marina DB, Garcia JF, Madhani HD, Cooke R, Schuck P, Cheng Y, Narlikar GJ. A conformational switch in HP1 releases auto-inhibition to drive heterochromatin assembly. Nature. 2013;496:377–81. doi: 10.1038/nature12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM, Mirkin SM. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat Struct Mol Biol. 2009;16:226–8. doi: 10.1038/nsmb.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci U S A. 2008;105:9936–41. doi: 10.1073/pnas.0804510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizuno K, Lambert S, Baldacci G, Murray JM, Carr AM. Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev. 2009;23:2876–86. doi: 10.1101/gad.1863009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakaseko Y, Kinoshita N, Yanagida M. A novel sequence common to the centromere regions of Schizosaccharomyces pombe chromosomes. Nucleic Acids Res. 1987;15:4705–15. doi: 10.1093/nar/15.12.4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rozenzhak S, Mejia-Ramirez E, Williams JS, Schaffer L, Hammond JA, Head SR, Russell P. Rad3 decorates critical chromosomal domains with gammaH2A to protect genome integrity during S-Phase in fission yeast. PLoS Genet. 2010;6:e1001032. doi: 10.1371/journal.pgen.1001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irmisch A, Ampatzidou E, Mizuno K, O’Connell MJ, Murray JM. Smc5/6 maintains stalled replication forks in a recombination-competent conformation. EMBO J. 2009;28:144–55. doi: 10.1038/emboj.2008.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pebernard S, Schaffer L, Campbell D, Head SR, Boddy MN. Localization of Smc5/6 to centromeres and telomeres requires heterochromatin and SUMO, respectively. Embo J. 2008;27:3011–23. doi: 10.1038/emboj.2008.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tapia-Alveal C, O’Connell MJ. Nse1-dependent recruitment of Smc5/6 to lesion-containing loci contributes to the repair defects of mutant complexes. Mol Biol Cell. 2011;22:4669–82. doi: 10.1091/mbc.E11-03-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SY, Rozenzhak S, Russell P. gammaH2A-binding protein Brc1 affects centromere function in fission yeast. Mol Cell Biol. 2013;33:1410–6. doi: 10.1128/MCB.01654-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng W, Tsao SW, Guan XY, Cheung AL. Pericentromeric regions are refractory to prompt repair after replication stress-induced breakage in HPV16 E6E7-expressing epithelial cells. PLoS One. 2012;7:e48576. doi: 10.1371/journal.pone.0048576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell. 2011;144:732–44. doi: 10.1016/j.cell.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tinline-Purvis H, Savory AP, Cullen JK, Dave A, Moss J, Bridge WL, Marguerat S, Bahler J, Ragoussis J, Mott R, Walker CA, Humphrey TC. Failed gene conversion leads to extensive end processing and chromosomal rearrangements in fission yeast. EMBO J. 2009;28:3400–12. doi: 10.1038/emboj.2009.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura K, Okamoto A, Katou Y, Yadani C, Shitanda T, Kaweeteerawat C, Takahashi TS, Itoh T, Shirahige K, Masukata H, Nakagawa T. Rad51 suppresses gross chromosomal rearrangement at centromere in Schizosaccharomyces pombe. EMBO J. 2008;27:3036–46. doi: 10.1038/emboj.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roguev A, Bandyopadhyay S, Zofall M, Zhang K, Fischer T, Collins SR, Qu H, Shales M, Park HO, Hayles J, Hoe KL, Kim DU, Ideker T, Grewal SI, Weissman JS, Krogan NJ. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science. 2008;322:405–10. doi: 10.1126/science.1162609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McFarlane RJ, Humphrey TC. A role for recombination in centromere function. Trends Genet. 2010;26:209–13. doi: 10.1016/j.tig.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Xu YJ, Davenport M, Kelly TJ. Two-stage mechanism for activation of the DNA replication checkpoint kinase Cds1 in fission yeast. Genes Dev. 2006;20:990–1003. doi: 10.1101/gad.1406706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nitani N, Nakamura K, Nakagawa C, Masukata H, Nakagawa T. Regulation of DNA replication machinery by Mrc1 in fission yeast. Genetics. 2006;174:155–65. doi: 10.1534/genetics.106.060053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li PC, Petreaca RC, Jensen A, Yuan JP, Green MD, Forsburg SL. Replication fork stability is essential for the maintenance of centromere integrity in the absence of heterochromatin. Cell Rep. 2013;3:638–45. doi: 10.1016/j.celrep.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dalgaard JZ, Klar AJS. swi1 and swi3 perform imprinting, pausing and termination of DNA replication in S. pombe. Cell. 2000;102:745–751. doi: 10.1016/s0092-8674(00)00063-5. [DOI] [PubMed] [Google Scholar]
- 52.Noguchi E, Noguchi C, Du LL, Russell P. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol Cell Biol. 2003;23:7861–7874. doi: 10.1128/MCB.23.21.7861-7874.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Slee RB, Steiner CM, Herbert BS, Vance GH, Hickey RJ, Schwarz T, Christan S, Radovich M, Schneider BP, Schindelhauer D, Grimes BR. Cancer-associated alteration of pericentromeric heterochromatin may contribute to chromosome instability. Oncogene. 2012;31:3244–53. doi: 10.1038/onc.2011.502. [DOI] [PubMed] [Google Scholar]
- 54.Martinez AC, van Wely KH. Centromere fission, not telomere erosion, triggers chromosomal instability in human carcinomas. Carcinogenesis. 2011;32:796–803. doi: 10.1093/carcin/bgr069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beeharry N, Rattner JB, Caviston JP, Yen T. Centromere fragmentation is a common mitotic defect of S and G 2 checkpoint override. Cell Cycle. 2013;12:1588–97. doi: 10.4161/cc.24740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, Chew SK, Rowan AJ, Schenk A, Sheffer M, Howell M, Kschischo M, Behrens A, Helleday T, Bartek J, Tomlinson IP, Swanton C. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–6. doi: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng W, Bachant J, Collingwood D, Raghuraman MK, Brewer BJ. Centromere replication timing determines different forms of genomic instability in Saccharomyces cerevisiae checkpoint mutants during replication stress. Genetics. 2009;183:1249–60. doi: 10.1534/genetics.109.107508. [DOI] [PMC free article] [PubMed] [Google Scholar]