

Abstract
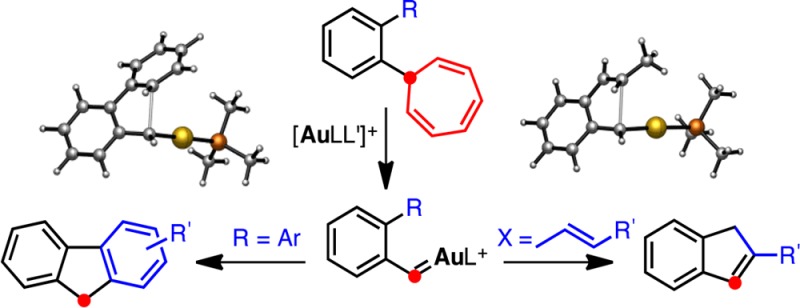
The fate of the aryl gold(I) carbenes generated by retro-Buchner reaction of ortho-substituted 7-aryl-1,3,5-cycloheptatrienes is dependent on the constitution of the ortho substituent. Indenes and fluorenes are obtained by intramolecular reaction of highly electrophilic gold(I) carbenes with alkenes and arenes. According to density functional theory calculations, the gold-catalyzed retro-Buchner process occurs stepwise, although the two carbon–carbon cleavages occur on a rather flat potential energy surface.
1. Introduction
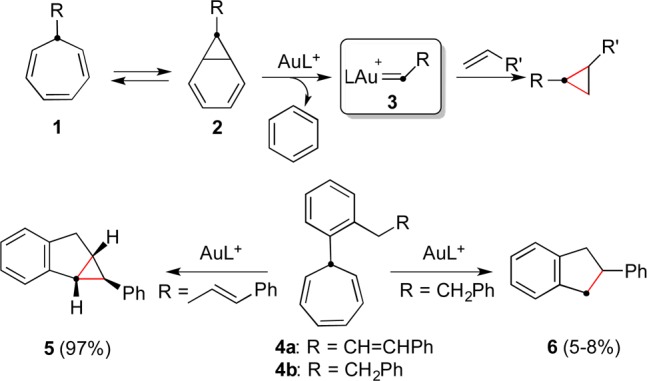
The Buchner reaction of metal carbenes generated from diazocarboxylates and related substrates has been observed under catalytic or stoichiometric conditions using copper(I),1−3 silver(I),4,5 gold(I),3,6 rhodium(II),7 and ruthenium complexes.8 Chloro(tetraphenylporphyrinato)iron has also been reported as a catalyst for the Buchner reaction.9 The copper- and rhodium-catalyzed processes have often been used in total synthesis.10,11 In contrast, we recently found that 7-substituted 1,3,5-cycloheptatrienes 1 react with cationic gold(I) complexes under catalytic conditions through their norcaradiene tautomers 2 to generate gold(I) carbenes [LAu=CHR]+ (3) in an overall retro-Buchner process (a form of retro-cyclopropanation) (Scheme 1).12 Our discovery of this reaction was prompted by the observation of a gold(I)-promoted retro-cyclopropanation in the context of a synthesis of 1,3-disubstituted naphthalenes.13 The retro-Buchner reaction proceeds readily with 7-aryl- and 7-alkenyl-1,3,5-cycloheptatrienes, whereas 7-alkynyl-1,3,5-cycloheptatrienes react differently with gold(I) or gold(III) catalysts to form indenes by a complex cycloisomerization that proceeds through gold-stabilized barbaralyl cations.14
Scheme 1.
Other related gold(I)-promoted retro-cyclopropanations have also been shown to occur in the gas phase by cleavage of 1-ethoxy-2-methoxycyclopropane with [AuIMes]+ [IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene].15,16 The gold(I)-catalyzed retro-Buchner reaction obviates the use of diazo derivatives to generate reactive gold(I) carbenes. Indeed, some diazo compounds are shock-sensitive.17 Although in certain cases the use of tosylhydrazone salts offers a safer alternative for handling diazo compounds,18 the generation of the carbenes requires the use of a strong base, which is not compatible with Lewis acidic [LAuL′]X complexes.
Gold(I) carbenes 3 undergo inter- or intramolecular reactions with alkenes to give cyclopropanes, as shown in the formation of 5. Insertion into a C(sp3)–H bond was also observed in the retro-Buchner reaction of 4b to form 2-phenylindane (6), although with very low efficiency.12
Here we report new results that demonstrate the highly electrophilic character of gold(I) carbenes 3 by their reactions with alkenes and arenes. In these reactions, indenes and fluorenes are obtained by novel annulation processes. The formation of indenes involves an electrophilic addition of the gold(I) carbene to the alkene, which can be followed by an unprecedented 1,4-metallotropic shift. Significantly, the intermediates generated by the retro-Buchner reaction behave more like simple carbenes than benzylic carbocations, as demonstrated in the fluorene annulation.
The mechanism of the Buchner reaction of silver(I)5 and ruthenium(II)19 carbenes has been recently examined computationally. Here we also report a theoretical study of the generation of donor gold(I) carbenes 3 by retro-Buchner reactions of 7-substituted 1,3,5-cycloheptatrienes 1 showing that the retro-cyclopropanation most probably occurs in a stepwise manner, although the barrier for the second C–C cleavage is small.
2. Results and Discussion
Annulation of o-Styrene Derivatives
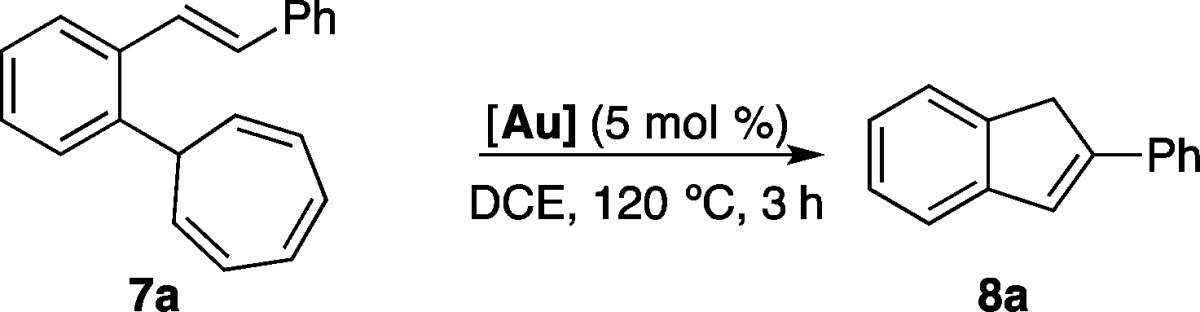
(E)-7-(2-Styrylphenyl)cyclohepta-1,3,5-triene (7a) reacted with cationic gold(I) complexes to give 2-phenyl-1H-indene (8a) (Table 1). Nearly identical good yields were obtained using cationic phosphine–gold(I) complexes A and B and carbene complex E (Table 1, entries 1, 2, and 5), whereas catalyst D led to poor results (entry 4). We identified JohnPhos–gold(I) complex A as the catalyst of choice because of its ready availability and robustness for the synthesis of 2-substituted indenes from o-alkenyl cycloheptatrienes (Scheme 2). This reaction proceeds in a rather general manner with substrates substituted with alkyl, alkenyl, or aryl substituents at C2 to give indenes 8a–l in moderate to good yields.
Table 1. Gold(I)-Catalyzed Synthesis of Indene 8a via Retro-Buchner Reactiona.
Reaction at 120 °C (0.1 M in 1,2-dichloroethane), catalyst (5 mol %), 3 h.
Yields determined by 1H NMR analysis.
Isolated yield.
Scheme 2.

Other cationic gold(I) complexes B–E were also active as catalysts but usually led to lower yields, although for the synthesis of indene 8j IPr–gold(I) catalyst E was used. In this case, only the closest double bond reacted with the carbene intermediate. The cyclopropyl group in 7k did not suffer ring expansion. 1,4-Bis(1H-inden-2-yl)benzene (8l) could also be obtained in moderate yield in a double annulation reaction.
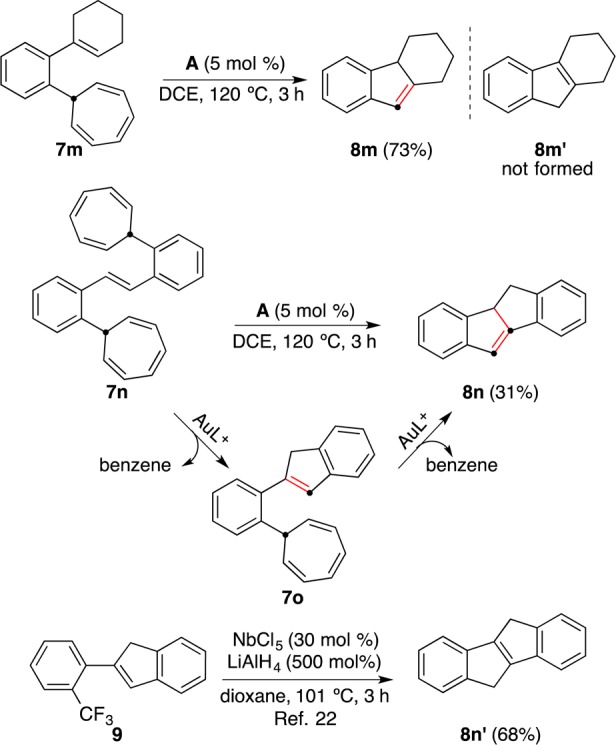
The annulation of o-cyclohexenyl derivative 7m under the standard conditions gave exclusively 2,3,4,4a-tetrahydro-1H-fluorene (8m), which has not been reported before (Scheme 3). The known and more stable isomer20 2,3,4,9-tetrahydro-1H-fluorene (8m′), which would have been formed by C–H insertion, was not observed. The symmetrical derivative 7n reacted by a double retro-Buchner reaction via 7o to give 4b,5-dihydroindeno[2,1-a]indene (8n), a member of a class of compounds that have attracted certain attention lately (Scheme 3).21 This method is thus complementary to the synthesis of indenes by annulation of 2-alkenyl-2-(trifluoromethyl)benzenes with NbCl5 and LiAlH4, since 2-arylindene 9 leads exclusively to 5,10-dihydroindeno[2,1-a]indene (8n′).22
Scheme 3.
Although at first glance indenes 8a–l appear to have been formed by a C(sp2)–H insertion of a gold(I) carbene intermediate, the formation of 8m and 8n strongly suggests that electrophilic addition of a gold(I) carbene to the alkene operates in these systems.
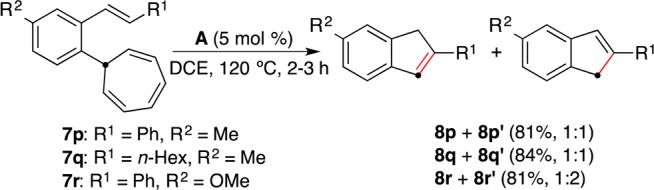
The exclusive formation of 8m and 8n from 7m and 7n contrasts with the observed reactivity of substrates 7p–r (Scheme 4). The reactions of substrates 7p and 7q bearing a p-Me substituent led to 1:1 mixtures of indenes 8p/8p′ and 8q/8q′, respectively. A 1:1 regioisomeric ratio was also obtained when the reaction was performed at 100 °C for 3 h (57% conversion as determined by 1H NMR analysis). Even more surprising was the result obtained in the cyclization of 7r bearing a p-MeO substituent, which led to a 1:2 mixture of 8r and 8r′.
Scheme 4.
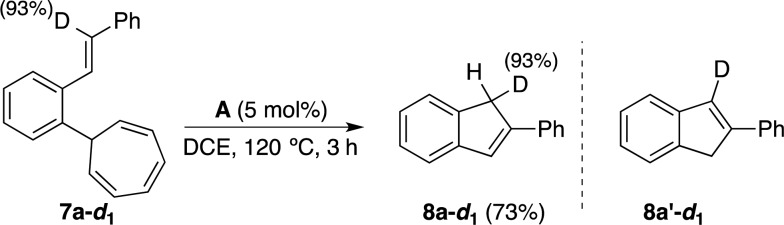
Regioisomeric indenes 8p′, 8q′, and 8r′ could have arisen by isomerization of 8p, 8q, and 8r by two consecutive [1,5]-H sigmatropic migrations.23 However, the reaction of 7a-d1 with catalyst A led exclusively to 8a-d1 with the deuterium label at the methylene, which is not consistent with an isomerization via [1,5]-H sigmatropic migrations, which would have also formed 8a′-d1 (Scheme 5). Additionally, no deuterium incorporation was observed when the reaction of 7a was performed in 1,2-dichloroethane saturated with D2O. Finally, no kinetic isotope effect was observed in the reaction of a 1:1 mixture of 7a and 7a-d1 with gold(I) complex A at 100 °C.24
Scheme 5.
A closer mechanistic inspection of these puzzling results suggested that an unexpected 1,4-gold(I) migration could be involved in the formation of mixtures of regioisomers in some of the indene syntheses. First, we studied computationally the formation of model indenes 8s and 8t from the corresponding gold(I) carbenes using density functional theory (DFT) at the M06 level including solvent effects for 1,2-dichloroethane (Scheme 6). After the retro-Buchner reaction, highly electrophilic gold(I) carbenes Ia and Ib (L = PMe3) react intramolecularly with the alkene through TSIa–IIa and TSIb–IIb in highly exothermic processes to form benzylic carbocations IIa and IIb, respectively. Despite being formally a 5-endo-trig cyclization from the perspective of the alkene,25 this process is rendered kinetically and thermodynamically favorable by the high electrophilicity of the gold(I) carbenes. As expected, the p-OMe group in Ib attenuates the electrophilicity of the gold(I) carbene, raising the energy of TSIb–IIb and resulting in a less exothermic cyclization. Carbocationic intermediates IIa and IIb can then evolve by a 1,2-H shift to form directly the indenes η2-coordinated to AuPMe3+ (IIIa and IIIb, respectively).26,27 Mechanistically, the cyclization of Ia/Ib to IIa/IIb is formally related to the intramolecular reaction of rhodium nitrenes with alkenes in the rhodium-catalyzed synthesis of indoles from o-alkenylaryl azides.28
Scheme 6. DFT Calculations on the Cyclization of Ia and Ib (Free Energies in kcal mol–1).
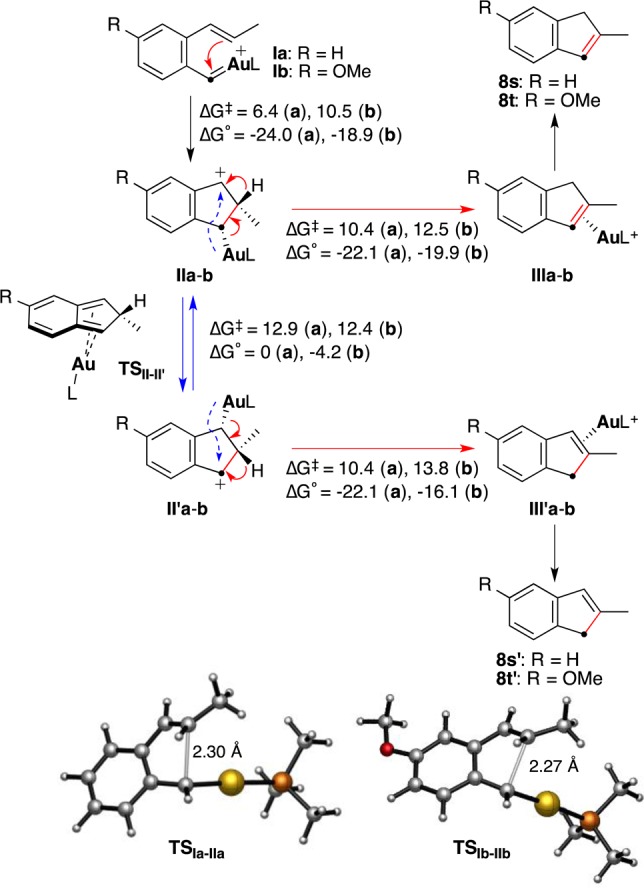
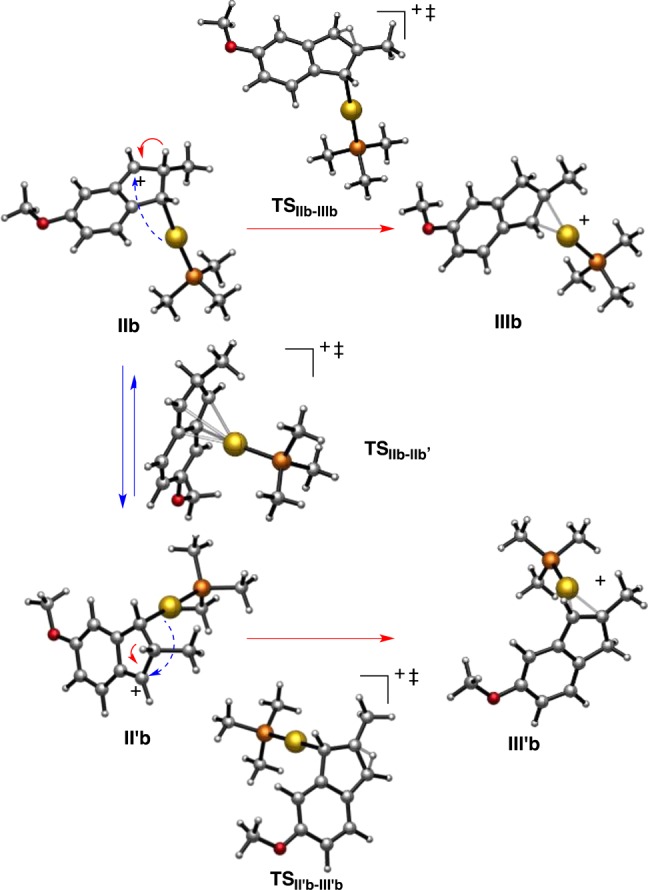
The DFT calculations show that intermediates II can evolve into II′ by a formal metal migration from C1 to C3, which actually corresponds to a suprafacial 1,4-metallotropic migration.29 Another type of gold(I) migration has also been proposed in a metathesis-type process occurring in competition with the cyclopropanation of enol ethers by gold(I) carbenes generated in the gas phase.15c Whereas for intermediate IIa this migration is degenerate, the p-OMe-substituted substrate II′b is 4.2 kcal mol–1 more stable than IIb as a result of the stabilization of the benzylic carbocation by the p-MeO group (Scheme 6). The transition states TSII–II′ for the 1,4-metallotropic migration show an η4-(2H-indene)Au(I) structure, with shorter distances from the metal center to the internal carbons C4a–C7a (2.51–2.54 Å) than to C1 and C3 (2.86–2.88 Å).
The 1,4-migration of AuPMe3 in IIb requires almost the same activation energy (12.4 vs 12.5 kcal mol–1) as the competitive 1,2-H shift to form IIIb (Scheme 7). The difference in activation energies is higher for the unsusbtituted system IIa/IIIa (2.5 kcal mol–1). The computed tendencies are in close agreement with those found experimentally in the cyclizations of 7p–r, which led to 1:1 mixtures in the first two cases (p-Me as the donor substituent), whereas substrate 7r (p-MeO substituent) gave 8r′ preferentially as a result of a more favorable migration of IIb to II′b.30
Scheme 7. Details of 1,4-Metallotropic Migration versus Formation of (η2-Indene)gold(I) Complexes in the Formation of IIIb/IIIb′.
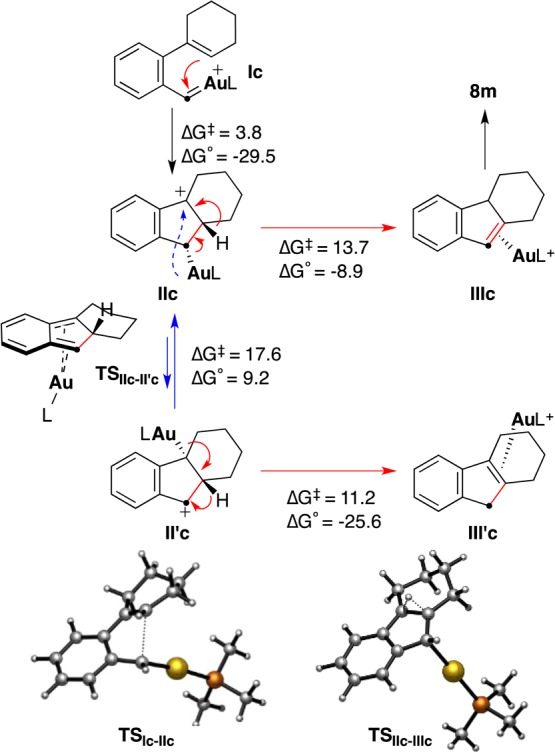
According to DFT calculations, the formation of 2,3,4,4a-tetrahydro-1H-fluorene (8m) followed a similar pathway involving intramolecular electrophilic attack of the gold(I) carbene on the alkene in Ic to form intermediate IIc, which undergoes 1,2-H shift to give (η2-alkene)Au(I) complex IIIc (Scheme 8). In this case, however, the transition state required for the suprafacial 1,4-metallotropic migration (IIc to II′c) was found to lie 3.9 kcal mol–1 higher than that of the 1,2-H shift, and moreover, II′c is destabilized with respect to IIc, which explains the selective formation of 8m over the more stable 8m′ from 7m (Scheme 3).
Scheme 8. DFT Calculations on the Formation of 8m (Free Energies in kcal mol–1).
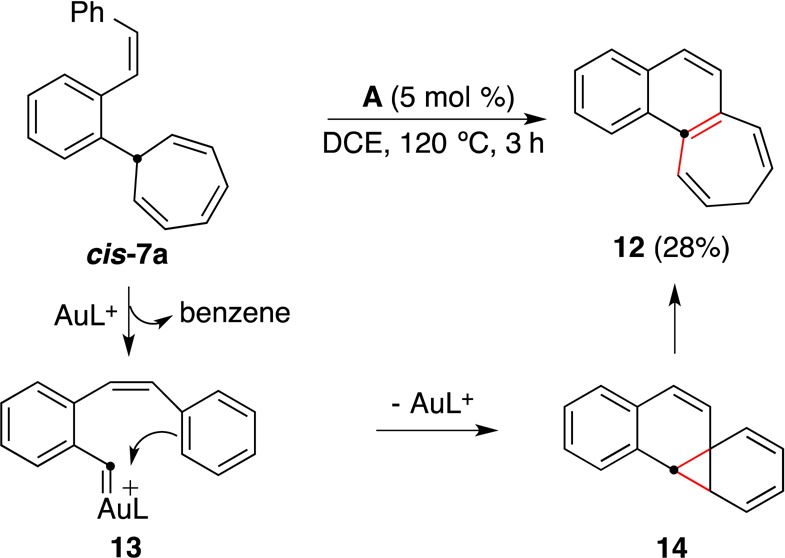
Surprisingly, cis-7a did not behave in the same manner as its trans isomer in the presence of catalyst A (Scheme 9). Presumably as a result of the proximity of the phenyl ring to the gold carbene in intermediate 13, an intramolecular Buchner reaction takes place to form 14, which then undergoes disrotatory norcaradiene-to-cycloheptatriene opening followed by a 1,5-H shift to give 12. A similar transformation had been observed in the thermal decomposition of diazo compounds to form naphtho[a]cycloheptenes of type 12.31
Scheme 9.
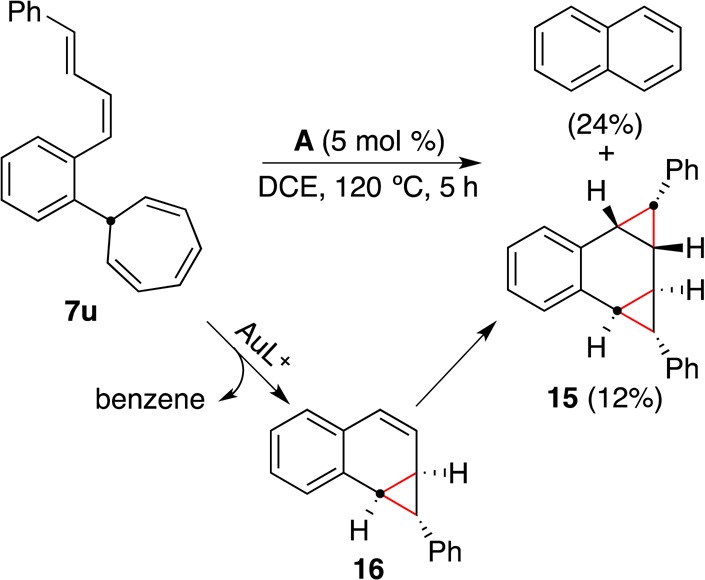
An even more elaborate cascade of retro-cyclopropanation/cyclopropanation reactions was observed starting from diene 7u (Scheme 10). The initially formed intermediate 16, resulting from a gold-catalyzed retro-Buchner reaction followed by intramolecular cyclopropanation, evolves by a second retro-cyclopropanation to form naphthalene with the concomitant generation of the reactive gold carbene [LAu=CHPh]+,13 which reacts with 16 to give 15.
Scheme 10.
In our previous study,12 we found that indenes are suitable substrates for intermolecular cyclopropanation by gold carbenes generated in a retro-Buchner reaction. An efficient tandem transformation can be achieved by combining indene formation and intermolecular cyclopropanation with [LAu=CHPh]+ by reacting 7a with 1a to give 5a and 5a′ in a 4:1 ratio (67% yield) (Scheme 11).
Scheme 11.

Annulation of o-Biphenyl Derivatives
Free o-biphenylcarbene has been demonstrated to undergo cyclization to form fluorene,32,33,40c whereas o-phenylbenzyl carbocation failed to undergo a similar cyclization.34o-Biphenyl gold(I) carbenes generated by retro-Buchner reactions behave like free carbenes, leading to fluorenes. Thus, reaction of 2-cycloheptatrienyl biphenyls 17a–i with catalyst A or E gave fluorenes 18a–i in moderate to good yields by a Friedel–Crafts-type methylenation reaction (Scheme 12). The annulation proceeded satisfactorily with substituents at different positions, although fluorene 18g was obtained in low yield. As expected, in the case of m-ClC6H4 derivative 17f, a mixture of two regioisomers 18f and 18f′ was obtained.
Scheme 12.
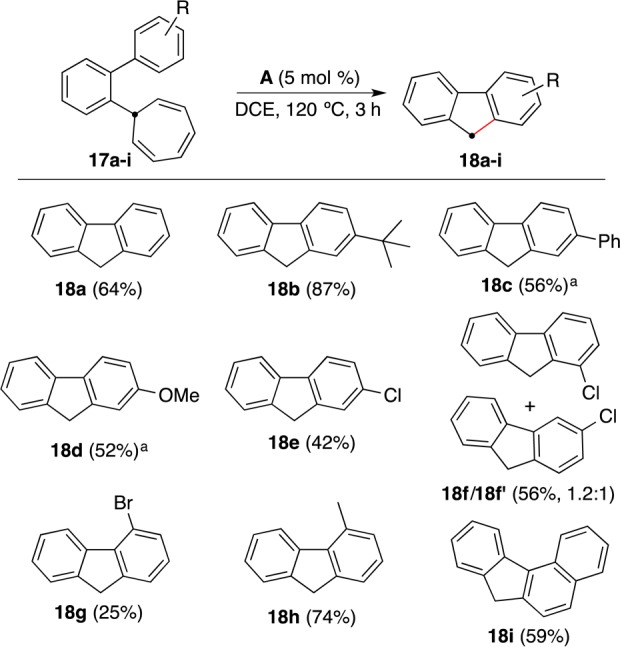
This new fluorene synthesis complements other recently developed methods based on transition-metal-catalyzed annulations.35−37 Interestingly, related rhodium- or copper-catalyzed annulation of biaryl diazoacetates leads to fluorene carboxylates,38 which emphasizes the similar reactivity of rhodium, copper, and gold carbenes.
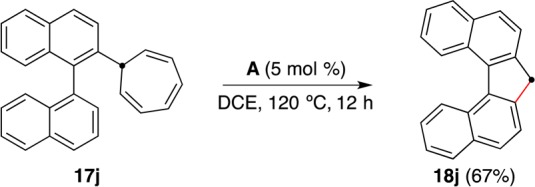
With this method, 2-binaphthylcycloheptatriene (17j), prepared in one step from commercially available 2-bromo-1,1′-binaphthalene,39 was converted into nonplanar 7H-dibenzo[c,g]fluorene (18j) (Scheme 13).40 This is the shortest synthesis of 18j, whose anion, dibenzo[c,g]fluorenide, has attracted recent interest for its particular aromatic character41 and as a six-π-electron donor ligand in organometallic chemistry.42
Scheme 13.
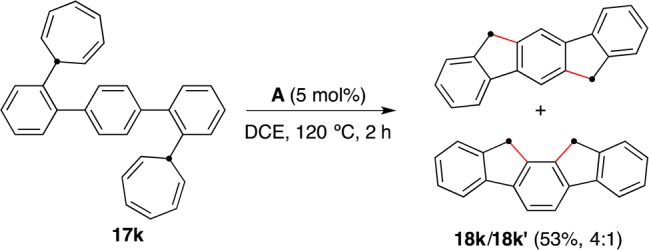
Indenofluorenes (IFs) have found various applications in organic electronics on account of the conjugation along their aromatic framework and their high rigidity, which stems from the methylene bridging unit of their biphenyl core.43 By extending our new method to a double annulation, we prepared indeno[1,2-b]fluorene (18k) as a mixture with indeno[2,1-a]fluorene (18k′) in 53% isolated yield from 17k (Scheme 14).
Scheme 14.
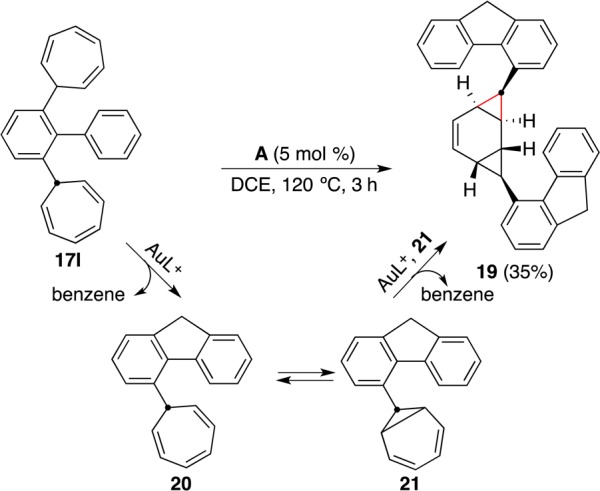
During our attempts to form 4,8-dihydrocyclopenta[def]fluorene by generating two gold(I) carbenes on the same aromatic ring, we found that the rigidity of the fluorene backbone makes the second annulation unfavorable. Instead, intermolecular dimerization to give 19 occurred, with no indication of other diastereoisomers (Scheme 15). The reaction proceeds by retro-Buchner reaction of 17l to form 20 followed by a second retro-Buchner reaction of its tautomer 21 to form a fluorenyl gold(I) carbene intermediate that cyclopropanates one of the double bonds of 21.
Scheme 15.
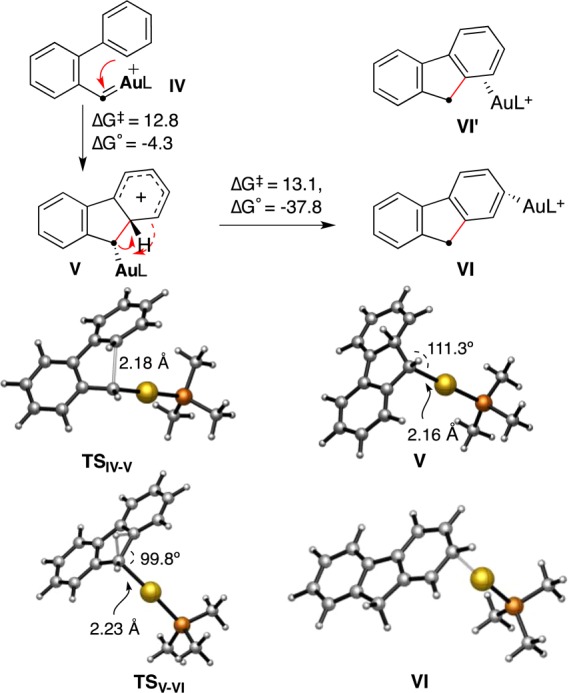
For the reactions leading to fluorenes by retro-Buchner reactions of 2-cycloheptatrienyl biphenyls 17, DFT calculations (M06 level, 1,2-dichloroethane) are consistent with a Friedel–Crafts-type reaction of gold(I) carbene IV (L = PMe3) through transition state TSIV–V in a moderately exothermic process to form Wheland intermediate V (Scheme 16). Intermediate V evolves by a 1,2-H shift via transition state TSV–VI to form (η1-fluorene)gold(I) complex VI instead of VI′ with gold(I) coordinated to the C1–C9a bond. All of the calculations led directly to VI as the global minimum by a further shift of [Au(PMe3)]+ toward C2, which suggests that a very low barrier exists for the isomerization of VI′ into V. In this process, the [Au(PMe3)]+ fragment migrates toward the aryl ring in a concerted but highly asynchronous diatropic-type transformation. The calculated shortest Au–C bond distance of 2.30 Å with C2 in VI is very similar to the shortest contacts determined by X-ray diffraction in complexes [LAu(ArH)]SbF6 (ArH = toluene, p-xylene).44
Scheme 16. DFT Calculations on Intermediates IV–VI (Free Energies in kcal mol–1).
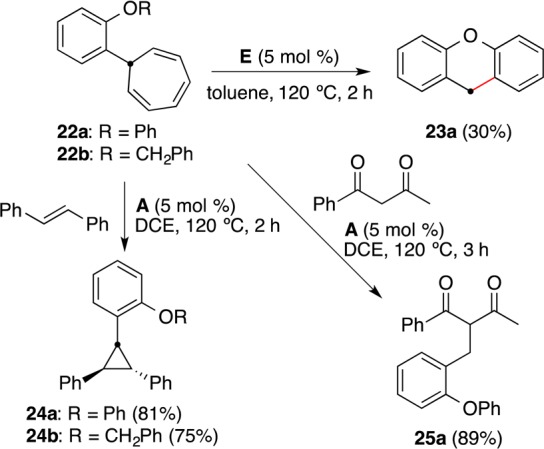
Aryl carbenes generated by pyrolysis of aryl diazomethanes undergo formal insertion into the adjacent ortho position of the XC6H5 ring to form dihydroanthracenes (X = CH2) and dihydroacridines (X = NH), whereas substrates with X = O or S lead to products of the Buchner reaction.45 In our case, the reaction of cycloheptatrienyl derivative 22a gave 9H-xanthene (23a), the product of a formal insertion into the ortho position of the phenyl group, albeit in low yield (Scheme 17). Despite the steric hindrance introduced by the ortho OR substituents, the ortho-substituted aryl gold(I) carbenes generated from 22a and 22b could be trapped efficiently with trans-stilbene to form the corresponding cyclopropanes 24a and 24b. Similarly, 25a could be obtained by trapping with 1-phenylbutane-1,3-dione.
Scheme 17.
Mechanism of the Retro-Buchner Reaction
Cleavage of cyclopropanes to form metal carbenes had only been achieved electrophilically by using a mixture of highly electrophilic PhWCl3 and RAlCl2 (R = Et, Cl).46 On the other hand, highly strained bicyclo[1.1.0]butanes undergo ring opening by oxidative addition to Ni(0) or Rh(I) complexes.47
Prior to our initial report,12 the metal-catalyzed retro-Bucher reaction was unknown, although the reverse process, formation of cycloheptatrienes, is a minor reaction in the gold(I)-catalyzed reaction between ethyl diazoacetate and arenes.48 In contrast to the cleavage of substituted cycloheptatriene derivatives observed in the presence of Au(I), strong electrophiles such as TeCl4 lead to the cleavage of a single C–C bond to form benzylic chlorides.49 The reaction of 7-ethynylcyclohepta-1,3,5-triene with trifluoroacetic acid leads to phenylallene by protonation of the alkyne in the norcaradiene tautomer followed by cyclopropane cleavage to form the arenium cation.50 The reaction of 7-ethoxycarbonyl-1,3,5-cycloheptatriene with an equimolecular amount of Pd(OAc)2 at 80 °C in MeCN had been reported to give ethyl 2- and 4-formylbenzoate (8% each) by cleavage of one cyclopropane C–C bond of the corresponding norcaradienes.51 In addition, diethyl maleate (14% yield) was formed, presumably by a dimetization of a Pd(II) carbene formed by a retro-Buchner process.52 Formation of benzyl-Rh(II) from cycloheptatrienyl-Rh(I) occurs by a metalloradical process. Cycloheptatriene has also been found to react on the metal carbide W(100)-(5 × 1)-C surface to generate benzene at 600 K,53 whereas (tricarbonyl)(tropylium)chromium perchlorate forms benzene(tricarbonyl)chromium by reaction with sodium cyclopentadienide or diethyl sodium malonates by an unknown mechanism.54 Carbenes have been generated by photochemical cleavage of a cyclopropanated phenanthrene.55
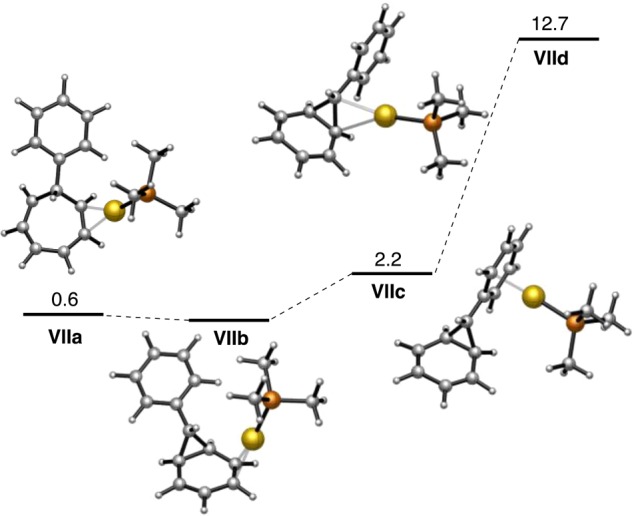
Our DFT computational study of the gold-catalyzed retro-Buchner reaction (M06 level, 1,2-dichloroethane) was complicated by the likely existence of several η2-coordinated gold(I) species in solution. Thus, in addition to η2-coordinated cycloheptatriene (VIIa) and norcaradiene (VIIb), the (η1-arene)gold(I) complex VIIc was also found as a local minimum (Scheme 18). An intermediate norcaradiene in which gold(I) is η2-coordinated to the cyclopropane C–C bond (VIId) was also found at a free energy 12.7 kcal mol–1 higher than that of VIIb. Previously, on the basis of related DFT calculations, we proposed the formation of related edge- or corner-metalated cyclopropanes as products in intra- and intermolecular gold(I)-catalyzed cyclopropanations of alkenes with 1,6-enynes56 and intramolecular cyclopropanation of 1,5-enynes.57
Scheme 18. DFT Calculations on η2-Coordinated Gold(I) Species (Free Energies in kcal mol–1).
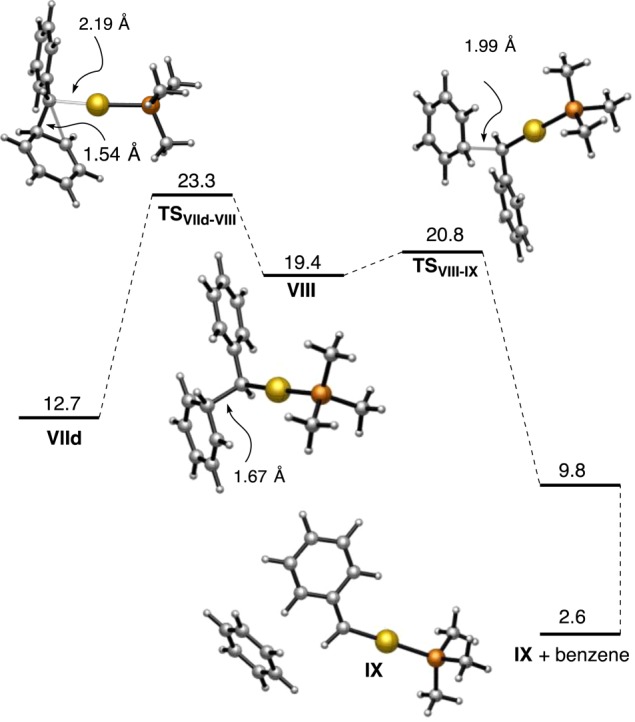
A transition state (TSVIId–VIII) was found for the electrophilic cleavage of intermediate VIId (Scheme 19), which lies 23.3 kcal mol–1 higher than the most stable initial complex VIIb. This value for the activation energy of the retro-Buchner reaction is consistent with the range of temperatures required for these reactions (100–120 °C). Transition state TSVIId–VIII leads to Wheland-type intermediate VIII, which is in a shallow minimum that smoothly evolves through TSVIII–IX to form phenyl gold(I) carbene IX and benzene. Although the overall process of the retro-Buchner reaction is moderately endothermic, further reactions of gold(I) carbene IX with alkenes (including intermolecular cyclopropanations56) or arenes are highly exothermic processes.
Scheme 19. DFT Calculations on the Mechanism (Free Energies in kcal mol–1).
3. Conclusions
In summary, gold(I) carbenes generated by the retro-Buchner reaction of 1,3,5-cycloheptatrienes catalyzed by cationic gold(I) complexes can be trapped intramolecularly by alkenes or arenes to form indenes or fluorenes. This methodology provides a new synthetic approach to indenes and fluorenes and may be applied to the synthesis of indenofluorenes used in organic electronics. These reactions proceed via intramolecular Friedel–Crafts-type attack of the highly electrophilic gold(I) carbenes to the alkenes and arenes. The reactivity displayed by the cationic intermediates generated by the retro-Buchner reaction is more similar to that of metal carbenes of rhodium or copper or even free carbenes than that of carbocations.
Closer scrutiny of the mechanisms of these reactions has revealed some intriguing details. Thus, in the indene synthesis, we have found that a novel 1,4-metallotropic migration competes with the primary pathway for the formation of the (η2-indene)gold(I) complexes by a concerted 1,2-H migration/gold(I) elimination. The formation of fluorenes involves a diatropic-type process in the formation of an (η1-fluorene)gold(I) complex. Finally, the mechanism of the gold(I)-catalyzed retro-Buchner reaction of substituted cycloheptatrienes proceeds by electrophilic cleavage of two C–C bonds of the norcaradiene tautomers. Although the cleavage occurs stepwise, the potential energy surface connecting the two transition states is rather shallow.
Acknowledgments
We thank MEC (Project CTQ2010-16088/BQU), the European Research Council (Advanced Grant 321066), EU Project ICT (ATMOL, Contract FP7-270028), the AGAUR (Project 2009 SGR 47), and the ICIQ Foundation for financial support.
Supporting Information Available
Additional data, experimental details, and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- a Scott L. T. Chem. Commun. 1973, 882–883. [Google Scholar]; b Scott L. T.; Minton M. A.; Kirms M. A. J. Am. Chem. Soc. 1980, 102, 6311–6314. [Google Scholar]
- Morilla M.; Díaz-Requejo M. M.; Belderrain T. R.; Nicasio M. C.; Trofimenko S.; Pérez P. J. Organometallics 2004, 23, 293–295. [Google Scholar]
- Flores J. A.; Dias H. V. R. Inorg. Chem. 2008, 47, 4448–4450. [DOI] [PubMed] [Google Scholar]
- Lovely C.; Browning G.; Badarinarayana V.; Dias H. V. R. Tetrahedron Lett. 2005, 46, 2453–2455. [Google Scholar]
- Komine N.; Flores J. A.; Pal K.; Caulton K. G.; Mindiola D. J. Organometallics 2013, 32, 3185–3191. [Google Scholar]
- a Fructos M.; Belderrain T. R.; Fremont P.; Scott N.; Nolan S. P.; Díaz-Requejo M. M.; Pérez P. J. Angew. Chem., Int. Ed. 2005, 44, 5284–5288. [DOI] [PubMed] [Google Scholar]; b Rivilla I.; Gómez-Emeterio B.; Fructos M.; Díaz-Requejo M. M.; Pérez P. J. Organometallics 2011, 30, 2855–2860. [Google Scholar]; c Pérez P. J.; Díaz-Requejo M. M.; Rivilla I. Beilstein J. Org. Chem. 2011, 7, 653–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Anciaux A. J.; Demonceau A.; Hubert A. J.; Noels A. F.; Petiniot N.; Teyssié P. Chem. Commun. 1980, 765–766. [Google Scholar]; b Anciaux A. J.; Demonceau A.; Noels A. F.; Hubert A. J.; Warin R.; Teyssié P. J. Org. Chem. 1981, 46, 873–876. [Google Scholar]; c Manitto P.; Monti D.; Speranza G. J. Org. Chem. 1995, 60, 484–485. [Google Scholar]
- Galan B.; Gembicky M.; Dominiak P.; Keister J.; Diver S. J. Am. Chem. Soc. 2005, 127, 15702–15703. [DOI] [PubMed] [Google Scholar]
- Mbuvi H. M.; Keith L. W. J. Porphyrins Phthalocyanines 2009, 13, 136–152. [Google Scholar]
- Lead references:; a McDowell P. A.; Foley D. A.; O’Leary P.; Ford A.; Maguire A. R. J. Org. Chem. 2012, 77, 2035–2040. [DOI] [PubMed] [Google Scholar]; b Levin S.; Nani R. R.; Reisman S. E. Org. Lett. 2010, 12, 780–783. [DOI] [PubMed] [Google Scholar]; c Levin S.; Nani R. R.; Reisman S. E. J. Am. Chem. Soc. 2011, 133, 774–776. [DOI] [PubMed] [Google Scholar]; d Nani R. R.; Reisman S. E. J. Am. Chem. Soc. 2013, 135, 7304–7311. [DOI] [PubMed] [Google Scholar]
- Review:Reisman S. E.; Nani R. R.; Levin S. Synlett 2011, 2437–2442. [Google Scholar]
- Solorio-Alvarado C. R.; Wang Y.; Echavarren A. M. J. Am. Chem. Soc. 2011, 133, 11952–11955. [DOI] [PubMed] [Google Scholar]
- Solorio-Alvarado C. R.; Echavarren A. M. J. Am. Chem. Soc. 2010, 132, 11881–11883. [DOI] [PubMed] [Google Scholar]
- McGonigal P. R.; de León C.; Wang Y.; Homs A.; Solorio-Alvarado C. R.; Echavarren A. M. Angew. Chem., Int. Ed. 2012, 51, 13093–13096. [DOI] [PubMed] [Google Scholar]
- a Batiste L.; Fedorov A.; Chen P. Chem. Commun. 2010, 46, 3899–3901. [DOI] [PubMed] [Google Scholar]; b Fedorov A.; Chen P. Organometallics 2010, 29, 2994–3000. [Google Scholar]; c Fedorov A.; Batiste L.; Bach A.; Birney D. M.; Chen P. J. Am. Chem. Soc. 2011, 133, 12162–12171. [DOI] [PubMed] [Google Scholar]; d Related generation of gold(I) carbenes in solution:Ringger D. H.; Chen P. Angew. Chem., Int. Ed. 2013, 52, 4686–4689. [DOI] [PubMed] [Google Scholar]
- Other metal-promoted gas-phase retro-cyclopropanations:Eller K.; Schwarz H. Chem. Rev. 1991, 91, 1121–1177. [Google Scholar]
- a Creary X. Org. Synth. 1986, 64, 207–216. [Google Scholar]; b Closs G. L.; Moss R. A. J. Am. Chem. Soc. 1964, 86, 4042–4053. [Google Scholar]
- a Aggarwal V. K.; Alonso E.; Fang G. Y.; Ferrara M.; Hynd G.; Porcelloni M. Angew. Chem., Int. Ed. 2001, 40, 1433–1436. [DOI] [PubMed] [Google Scholar]; b Fulton J. R.; Aggarwal V. K.; de Vicente J. Eur. J. Org. Chem. 2005, 1479–1492. [Google Scholar]; c Barluenga J.; Valdés C. Angew. Chem., Int. Ed. 2011, 50, 7486–7500. [DOI] [PubMed] [Google Scholar]; d Shao Z.; Zhang H. Chem. Soc. Rev. 2012, 41, 560–572. [DOI] [PubMed] [Google Scholar]; e Xiao Q.; Zhang Y.; Wang J. Acc. Chem. Res. 2013, 46, 236–247. [DOI] [PubMed] [Google Scholar]
- a Poater A.; Ragone F.; Correa A.; Cavallo L. J. Am. Chem. Soc. 2009, 131, 9000–9006. [DOI] [PubMed] [Google Scholar]; b Poater A.; Cavallo L. Theor. Chem. Acc. 2012, 131, 1155–1160. [Google Scholar]
- On the basis of DFT calculations [B3LYP, 6-31G(d)], isomer 8m′ is 5.6 kcal mol–1 more stable than 8m.
- a Saito M. Symmetry 2010, 2, 950–969. [Google Scholar]; b Hopf H. Angew. Chem., Int. Ed. 2013, 52, 12224–12226. [DOI] [PubMed] [Google Scholar]
- Fuchibe K.; Mitomi K.; Akiyama T. Chem. Lett. 2007, 36, 24–25. [Google Scholar]
- a Roth W. R. Tetrahedron Lett. 1964, 5, 1009–1013. [Google Scholar]; b Miller L. L.; Greisinger R.; Boyer R. F. J. Am. Chem. Soc. 1969, 91, 1578–1580. [Google Scholar]; c Spangler C. W. Chem. Rev. 1976, 76, 187–217. [Google Scholar]
- A high barrier (free energy of activation = 38.7 kcal mol–1) was calculated for the first [1,5]-H migration to form the intermediate isoindene. This barrier is higher than that required for the generation of the gold(I) carbene by the retro-Buchner reaction.
- 5-Endo-trig cyclizations for the formation of indenes are very rare processes. See:Ichikawa J.; Sakoda K.; Mihara J.; Ito N. J. Fluorine Chem. 2006, 127, 489–504. [Google Scholar]
- Cyclization of the gold carbene to the alkene by an anti approach requires slightly higher initial activation energies. See the Supporting Information for the complete reaction profiles and coordinates.
- Reviews of two-coordinate gold π complexes:; a Schmidbaur H.; Schier A. Organometallics 2010, 29, 2–23. [Google Scholar]; b Brooner R. E. M.; Widenhoefer R. A. Angew. Chem., Int. Ed. 2013, 52, 11714–11724. [DOI] [PubMed] [Google Scholar]
- Shen M.; Leslie B. E.; Driver T. G. Angew. Chem., Int. Ed. 2008, 47, 5056–5059. [DOI] [PubMed] [Google Scholar]
- For other 1,n-metal migrations that are mechanistically unrelated, see the following lead references:; a Zhang J.; Liu J.-F.; Ugrinov A.; Pillai A. F. X.; Sun Z. M.; Zhao P. J. Am. Chem. Soc. 2013, 135, 17270–17273. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ikeda Y.; Takano K.; Kodama S.; Ishii Y. Chem. Commun. 2013, 49, 11104–11106. [DOI] [PubMed] [Google Scholar]
- Despite the isolobal analogy between H and AuL, the shorter C–H bond imposes a prohibitively high barrier for the alternative 1,4-H shift. The generation of the corresponding gold(I) carbene was calculated to have activation energies of 51.5 kcal mol–1 from IIa and 48.2 kcal mol–1 from IIaa.26
- a Munro D. P.; Sharp J. T. Tetrahedron Lett. 1980, 21, 4109–4110. [Google Scholar]; b Munro D. P.; Sharp J. T. J. Chem. Soc., Perkin Trans. 1 1984, 849–858. [Google Scholar]
- Denney D. B.; Klemchuck P. P. J. Am. Chem. Soc. 1958, 80, 3289–3290. [Google Scholar]
- Régimbald-Krnel M. J.; Wentrup C. J. Org. Chem. 2013, 78, 8789–8795and references cited therein. [DOI] [PubMed] [Google Scholar]
- Kirmse W.; Kund K.; Ritzer E.; Dorigo A. E.; Houk K. N. J. Am. Chem. Soc. 1986, 108, 6045–6046. [DOI] [PubMed] [Google Scholar]
- a Campeau L. C.; Parisien M.; Jean A.; Fagnou K. J. Am. Chem. Soc. 2006, 128, 581–590. [DOI] [PubMed] [Google Scholar]; b Dong C.-G.; Hu Q.-S. Angew. Chem., Int. Ed. 2006, 45, 2289–2292. [DOI] [PubMed] [Google Scholar]; c Tobisu M.; Kita Y.; Ano Y.; Chatani N. J. Am. Chem. Soc. 2008, 130, 15982–15989. [DOI] [PubMed] [Google Scholar]; d Hwang S. J.; Kim H. J.; Chang S. Org. Lett. 2009, 11, 4588–4591. [DOI] [PubMed] [Google Scholar]
- Synthesis of fluorenes by annulation from ortho-arylated trifluorotoluenes using NbCl5:Fuchibe K.; Akiyama T. J. Am. Chem. Soc. 2006, 128, 1434–1435. [DOI] [PubMed] [Google Scholar]
- Pd-catalyzed synthesis of fluorenones by annulation:Shi Z.; Glorius F. Chem. Sci. 2013, 4, 829–833. [Google Scholar]
- Kim J.; Ohk Y.; Park S. H.; Jung S.; Chang S. Chem.—Asian J. 2011, 6, 2040–2047. [DOI] [PubMed] [Google Scholar]
- 2-Bromo-1,1′-binaphthalene can be easily prepared in 72% yield by monolithiation of 2,2′-dibromo-1,1′-binaphthyl followed by protonation:Schilling B.; Kaufmann D. E. Eur. J. Org. Chem. 1998, 701–709. [Google Scholar]
- a Martin R. H. J. Chem. Soc. 1941, 679–685. [Google Scholar]; b Harvey R. G.; Pataki J.; Cortez C.; Di Raddo P.; Yang C. J. Org. Chem. 1991, 56, 1210–1217. [Google Scholar]; c Régimbald-Krnel M.; Wentrup C. J. Org. Chem. 1998, 63, 8417–8423. [DOI] [PubMed] [Google Scholar]
- Pammer F.; Sun Y.; Weismann D.; Sitzmann H.; Thiel W. R. Chem.—Eur. J. 2010, 16, 1265–1270. [DOI] [PubMed] [Google Scholar]
- a Pammer F.; Sun Y.; Sieger M.; Fiedler J.; Sarkar B.; Thiel W. R. Organometallics 2010, 29, 6165–6168. [Google Scholar]; b Pammer F.; Sun Y.; May C.; Wolmershäuser G.; Kelm H.; Krüger H.-J.; Thiel W. R. Angew. Chem., Int. Ed. 2007, 46, 1270–1273. [DOI] [PubMed] [Google Scholar]
- a Thirion D.; Poriel C.; Rault-Berthelot J.; Barrière F.; Jeannin O. Chem.—Eur. J. 2010, 16, 13646–13658. [DOI] [PubMed] [Google Scholar]; b Poriel C.; Liang J.-J.; Rault-Berthelot J.; Barrière F.; Cocherel N.; Slawin A. M. Z.; Horhant D.; Virboul M.; Alcaraz G.; Audebrand N.; Vignau L.; Huby N.; Wantz G.; Hirsch L. Chem.—Eur. J. 2007, 13, 10055–10069and references cited therein. [DOI] [PubMed] [Google Scholar]
- a Herrero-Gómez E.; Nieto-Oberhuber C.; López S.; Benet-Buchholz J.; Echavarren A. M. Angew. Chem., Int. Ed. 2006, 45, 5455–5459. [DOI] [PubMed] [Google Scholar]; b Pérez-Galán P.; Delpont N.; Herrero-Gómez E.; Maseras F.; Echavarren A. M. Chem.—Eur. J. 2010, 16, 5324–5332. [DOI] [PubMed] [Google Scholar]
- Crow W. D.; McNab H. Aust. J. Chem. 1981, 34, 1037–1350. [Google Scholar]
- a Gassman P. G.; Johnson T. H. J. Am. Chem. Soc. 1976, 98, 6057–6058. [Google Scholar]; b Gassman P. G.; Johnson T. H. J. Am. Chem. Soc. 1976, 98, 6058–6059. [Google Scholar]
- a Ni(0):Takaya H.; Suzuki T.; Kumagai Y.; Hosoya M.; Kawauchi H.; Noyori R. J. Org. Chem. 1981, 46, 2854–2861. [Google Scholar]; b Rh(I):Walczak M. A. A.; Wipf P. J. Am. Chem. Soc. 2008, 130, 6924–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fructos M. R.; Belderrain T. R.; de Frémont P.; Scott N. M.; Nolan S. P.; Díaz-Requejo M. M.; Pérez P. J. Angew. Chem., Int. Ed. 2005, 44, 5284–5288. [DOI] [PubMed] [Google Scholar]; b Rivilla I.; Gómez-Emeterio B. P.; Fructos M. R.; Díaz-Requejo M. M.; Pérez P. J. Organometallics 2011, 30, 2855–2860. [Google Scholar]
- Albeck M.; Tamari T.; Sprecher M. J. Org. Chem. 1983, 48, 2276–2278. [Google Scholar]
- a Kitagawa T.; Kamada J.; Minegishi S.; Takeuchi K. Org. Lett. 2000, 2, 3011–3013. [DOI] [PubMed] [Google Scholar]; b Minegishi S.; Kamada J.; Takeuchi K.; Komatsu K.; Kitagawa T. Eur. J. Org. Chem. 2003, 3497–3504. [Google Scholar]
- Saito K.; Kozaki M.; Takahashi K. Chem. Pharm. Bull. 1993, 41, 2187–2189. [Google Scholar]
- Chan Y. W.; Chan K. S. Chem. Commun. 2011, 47, 4802–4804. [DOI] [PubMed] [Google Scholar]
- Pearlstine K. A.; Friend C. M. J. Am. Chem. Soc. 1985, 107, 5898–5901. [Google Scholar]
- Munro J. D.; Pauson P. L. J. Chem. Soc. 1961, 3479–3483. [Google Scholar]
- a Richardson D. B.; Durrett L. R.; Martin J. M.; Putnam W. E.; Slaymaker S. C.; Dvoretzky I. J. Am. Chem. Soc. 1965, 87, 2763–2765. [Google Scholar]; b Graves K. S.; Thamattoor D. M.; Rablen P. R. J. Org. Chem. 2011, 76, 1584–1591. [DOI] [PubMed] [Google Scholar]; c Moore K. A.; Vidaurri-Martinez J. S.; Thamattoor D. M. J. Am. Chem. Soc. 2012, 134, 20037–20040. [DOI] [PubMed] [Google Scholar]
- a Nieto-Oberhuber C.; López S.; Muñoz M. P.; Jiménez-Núñez E.; Buñuel E.; Cárdenas D. J.; Echavarren A. M. Chem.—Eur. J. 2006, 12, 1694–1702. [DOI] [PubMed] [Google Scholar]; b Pérez-Galán P.; Herrero-Gómez E.; Hog D. T.; Martin N. J. A.; Maseras F.; Echavarren A. M. Chem. Sci. 2011, 2, 141–149. [Google Scholar]
- López-Carrillo V.; Huguet N.; Mosquera Á.; Echavarren A. M. Chem.—Eur. J. 2011, 17, 10972–10978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.