

Abstract
Glycogen storage disease type-Ia (GSD-Ia) patients deficient in glucose-6-phosphatase-α (G6Pase-α or G6PC) manifest impaired glucose homeostasis characterized by fasting hypoglycemia, growth retardation, hepatomegaly, nephromegaly, hyperlipidemia, hyperuricemia, and lactic acidemia. Two efficacious recombinant adeno-associated virus pseudotype 2/8 (rAAV8) vectors expressing human G6Pase-α have been independently developed. One is a single-stranded vector containing a 2864-bp of the G6PC promoter/enhancer (rAAV8-GPE) and the other is a double-stranded vector containing a shorter 382-bp minimal G6PC promoter/enhancer (rAAV8-miGPE). To identify the best construct, a direct comparison of the rAAV8-GPE and the rAAV8-miGPE vectors was initiated to determine the best vector to take forward into clinical trials. We show that the rAAV8-GPE vector directed significantly higher levels of hepatic G6Pase-α expression, achieved greater reduction in hepatic glycogen accumulation, and led to a better toleration of fasting in GSD-Ia mice than the rAAV8-miGPE vector. Our results indicated that additional control elements in the rAAV8-GPE vector outweigh the gains from the double-stranded rAAV8-miGPE transduction efficiency, and that the rAAV8-GPE vector is the current choice for clinical translation in human GSD-Ia.
Keywords: glycogen storage disease type I, glucose-6-phosphatase, adeno-associated virus, gene therapy
1. Introduction
Glycogen storage disease type Ia (GSD-Ia, or von Gierke disease, MIM232200) is an autosomal recessive disorder caused by deficiencies in the liver/kidney/intestine-restricted glucose-6-phosphatase-α (G6Pase-α or G6PC) that catalyzes the hydrolysis of glucose-6-phosphate (G6P) to glucose and phosphate in the terminal rate-limiting step of gluconeogenesis and glycogenolysis [1,2]. GSD-Ia patients manifest impaired glucose homeostasis characterized by fasting hypoglycemia, growth retardation, hepatomegaly, nephromegaly, hyperlipidemia, hyperuricemia, and lactic acidemia [1,2]. There is no cure for GSD-Ia. Hypoglycemia can be managed using dietary therapies that enable patients to attain near normal growth and pubertal development [3,4]. However, the longer term clinical complications including osteoporosis, gout, renal disease, pulmonary hypertension, and hepatocellular adenomas (HCA) that may undergo malignant transformation, and their underlying pathological processes, remain uncorrected.
To develop a gene therapy for treating GSD-Ia, recombinant adeno-associated virus (rAAV) vectors carrying G6Pase-α directed by a variety of different promoter/enhancer elements have been investigated. These have included strong constitutive promoters such as the hybrid chicken β-actin (CBA) promoter/CMV enhancer (CBA) as well as native tissue-specific promoters including the canine G6PC gene promoter/enhancer, the 2864-bp of the human G6PC promoter/enhancer (GPE), and the 382-bp minimal human G6PC promoter/enhancer (miGPE) [reviewed in 5]. From these studies the rAAV8-miGPE (originally named rAAV8-G6Pase) [6] and rAAV8-GPE [7,8] were identified as the most efficacious vectors. Compared to the rAAV8-GPE vector, the rAAV8-CBA vector is less efficient in directing persistent in vivo hepatic transgene expression [7]. Moreover, the rAAV8-CBA vector also elicits elevated hepatic CD8+ lymphocyte infiltration which correlates with the rapid decline in transgene expression and low efficacy of this vector [7]. The rAAV8-GPE construct, developed at the National Institutes of Health (NIH), is a single-stranded rAAV8 vector that uses genomic sequences at nucleotides −2864 to +1, relative to the ATG initiation codon at nucleotides +1-3, that contained the human G6PC promoter/enhancer [7]. In contrast, the rAAV8-miGPE construct, developed at Duke University, is a double-stranded rAAV8 vector that uses nucleotides −382 to +1 of the human G6PC minimal promoter/enhancer) [6]. The constructs differed not only in the length of the human G6PC promoter/enhancer but also in the single-stranded or double-stranded nature of the vectors, imposed by the strict packaging size restrictions for rAAV vectors [9]. The double-stranded rAAV vectors have an increased transduction efficiency [9] which arises from bypassing the rate-limiting conversion of single-stranded to double-stranded vector genomes during transduction [10]. However, the trade-off of the double-stranded rAAV vector is a 50% reduction in packaging capacity compared to single-stranded rAAV vector [9] which restricts the size of the promoter/enhancer it can carry, such as the rAAV8-miGPE vector [6]. In short term (24-26 week) studies, both rAAV8-GPE and rAAV8-miGPE vectors demonstrated efficacy in treating GSD-Ia (G6pc−/−) mice [6,7].
In a recent long-term study that followed rAAV8-GPE-mediated gene transfer in G6pc−/− mice for up to 90 weeks showed that mice expressing 3% to128% of wild type hepatic G6Pase-α activity maintained glucose homeostasis and showed no evidence of hepatocellular carcinomas (HCA) [8]. The treated mice displayed normal hepatic fat storage, normal blood metabolite and glucose tolerance profiles, reduced fasting blood insulin levels, maintained normoglycemia over a 24-hour fast, and had no evidence of hepatic abnormalities. The longer term efficacy of the rAAV8-miGPE vector has not been established to determine whether additional control elements contained within the GPE are necessary for optimal efficacy. In this study, a direct comparison of the rAAV8-GPE and the rAAV8-miGPE vectors was initiated to determine the best vector to take forward into clinical trials. Our results indicated that additional control elements in the rAAV8-GPE vector outweigh the gains from the double-stranded rAAV8-miGPE transduction efficiency, and that the rAAV8-GPE vector is the current choice for clinical translation in human GSD-Ia.
2. Materials and methods
2.1. Infusion of G6pc−/− mice with rAAV vectors
All animal studies were conducted under an animal protocol approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Animal Care and Use Committee. All G6pc−/− mice were kept alive with glucose therapy [11]. The rAAV vectors were infused into 2-week-old G6pc−/− mice via the retro-orbital sinus. Age-matched G6pc+/+/G6pc+/− mice were used as controls. For rAAV vector-infused mice, glucose therapy was terminated immediately after infusion.
2.2. Evaluation of the efficacy of rAAV8-GPE and rAAV8-miGPE vectors
Efficacy of gene therapy was independently assessed by both NIH and Duke University using the same G6pc−/− mouse strain [11]. All rAAV vectors were produced at the University of Florida Powell Gene Therapy Center Vector Core Laboratory (Gainesville, FL). The two vectors were tittered independently by University of Florida and Duke University. The experimental protocols described were established by mutual agreement between the two centers and were conducted similarly at each center. All viral transductions were performed on 2-week-old G6pc−/− mice and the efficacy evaluated at age 12 weeks.
2.3. Phosphohydrolase assays
Microsome isolation and phosphohydrolase assays were determined essentially as described previously [11]. For phosphohydrolase assays, reaction mixtures (100 μl) contained 50 mM cacodylate buffer, pH 6.5, 10 mM G6P and appropriate amounts of microsomal preparations were incubated at 30 °C for 10 min. Disrupted microsomal membranes were prepared by incubating intact membranes in 0.2% deoxycholate for 20 min at 0 °C. Non-specific phosphatase activity was estimated by pre-incubating disrupted microsomal preparations at pH 5 for 10 min at 37 °C, to inactivate the acid labile G6Pase-α.
2.4. Quantification of vector DNA and mRNA
Total DNA from mouse tissues was isolated using the GenElute™ Mammalian Genomic DNA Miniprep Kits (Sigma-Aldrich, St Louis, MO) and total RNAs were isolated from mouse tissues using the TRIzol Reagent (Invitrogen, Carlsbad, CA). The vector genome numbers and mRNA expression were quantified by PCR and real-time RT-PCR, respectively in an Applied Biosystems 7300 Real-Time PCR System using Applied Biosystems TaqMan probes (Appied Biosystems, Foster City, CA). The vector genome numbers of human G6PC gene was normalized to mouse β-actin using TaqMan probe sets Hs00609178_m1 for G6PC and Mm00607939_s1 for β-actin. Plasmid DNA corresponding to 0.01 to 100 copies of human G6PC gene was used in a standard curve. To determine the vector genome copy number, the Ct values of sample were compared to the standard curve. G6PC mRNA expression was normalized to Rpl19 RNA using TaqMan probe sets Hs00609178_m1 for G6PC and Mm02601633_g1 for Rpl19.
2.5. Phenotype analyses
Blood glucose was analyzed using kits obtained from Thermo Electron (Louisville, CO). Hepatic glycogen and triglyceride contents were measured as described previously [7,8]. To determine hepatic triglyceride, liver tissues were homogenized in RIPA buffer (50 mM Tris HCl, pH 8.0, 150 mM Nacl, 1% Triton X-100, 0.5% Na-deoxycholate, and 0.1% SDS) (Thermo Scientific, Rockford, IL), and triglycerides were measured using a kit from Sigma Diagnostics (St Louis, MO).
Glucose tolerance testing of mice consisted of fasting for 6 hours, prior to blood sampling, followed by intraperitoneal injection of a glucose solution at 2 mg/g body weight, and repeated blood sampling via the tail vein for 2 hours.
2.6. Statistical analysis
The unpaired t test was performed using the GraphPad Prism Program, version 4 (GraphPad Software Inc., San Diego, CA). Values were considered statistically significant at p <0.05.
3. Results
3.1. Hepatic G6Pase-α expression in rAAV8-GPE- or rAAV8-miGPE-treated G6pc−/− mice
Researchers at the NIH and Duke University have jointly conducted independent, parallel studies to examine the efficacy of the rAAV8-GPE [7] and rAAV8-miGPE [6] vectors in treating G6pc−/− mice over 12-weeks. Each vector was used at two doses, 1 ×1013 viral particles (vp)/kg (high dose) and 2 × 1012 vp/kg (low dose) with 6 mice per group, at each center. Similar results were obtained from both centers. While hepatic G6Pase-α activity in the transduced G6pc−/− mice represents the combined data from the two centers, other reported data are from the NIH study. Studies have shown that the efficiency and persistence of rAAV-mediated hepatic gene transfer are lower during early development because the fast rate of hepatocellular proliferation associated with liver growth, which dilutes out the number of cells effectively infected with rAAV (7,12). In this study, the rAAV vectors were administered to 2-week-old G6pc−/− mice when hepatocellular proliferation remains high. Consequently, hepatic G6Pase-α expression examined at age 12 weeks is significantly lower than that seen in adult mice infused with the same vector dosage. The best time of intervention in the human disease remains to be established.
All treated G6pc−/− mice survived to age 12 weeks with no premature deaths at either center. In the liver of 12-week-old wild type mice, microsomal G6Pase-α activity was 203.5 ± 10.3 nmol/min/mg (n = 24). The combined data (n = 12 per treatment), determined independently at the two centers, showed that at age 12 weeks, the high dose rAAV8-GPE therapy reconstituted about 18% of wild type hepatic G6Pase-α activity which was 3.5-fold more activity than rAAV8-miGPE, while the low dose rAAV8-GPE therapy produced over 3.6 times more activity than rAAV8-miGPE (Fig. 1A). The hepatic G6Pase-α activity increased linearly with hepatic vector genome copy number, and the copy numbers in rAAV8-GPE-treated G6pc−/− mice (n = 6) were significantly higher than the rAAV8-miGPE-treated mice (n = 6) (Fig. 1A).
Fig. 1.

Biochemical analyses in 12-week-old wild type and rAAV8-treated G6pc−/− mice. (A) Hepatic microsomal G6Pase-α activity and its relationship to vector genome copy numbers. (B) Growth curve. (C) BMI values. (D) Blood glucose levels. GPE-high, high dose rAAV8-GPE-treated (○); miGPE-high, high dose rAAV8-miGPE-treated (●); GPE-low, low dose rAAV8-GPE-treated (□); miGPE-low, low dose rAAV8-miGPE-treated (∎) G6pc−/− mice; (+/+), wild type (▾) mice. Hepatic G6Pase activity represents the combined data from the two centers (n = 12 per treatment) and the other data are from the NIH (n = 6 per treatment). Data are mean ± SEM. *P < 0.05.
3.2. Metabolic profiles of rAAV8-GPE- or rAAV8-miGPE-treated G6pc−/− mice
All rAAV8-GPE- and rAAV8-miGPE-treated G6pc−/− mice exhibited growth curves that paralleled their wild type littermates, albeit at lower weights (Fig. 1B). The body mass index (BMI) values of the treated mice were indistinguishable from those of wild type mice irrespective of the vector or dose level (Fig. 1C). All treated G6pc−/− mice had blood glucose levels consistently lower than wild type controls, but still above the lower end of the normal range (Fig. 1D), and none of the infused animals suffered from the frequent hypoglycemic seizures typical of GSD-Ia [1,2,8].
The relative weight of the liver to the body, one measure of liver glycogen and/or neutral fat accumulation [1,2], was higher in all 4 treated G6pc−/− mouse groups than wild type mice, although the high dose rAAV8-GPE-treated mice were significantly closer to normal than the other groups (Fig. 2A). Consistent with this, glycogen contents in rAAV8-GPE- and rAAV8-miGPE-treated G6pc−/− mice were markedly higher than their wild type controls (Fig. 2B) with the higher vector doses lowering glycogen better than the lower vector doses, demonstrating that restoring higher hepatic G6Pase-α expression improves hepatomegaly. However, comparing just high dose therapies, the rAAV8-miGPE-treated mice had 43% more glycogen than rAAV8-GPE-treated mice. Despite the continued elevation in liver glycogen there were no histological abnormalities observed in the liver tissue sections of any of the rAAV-treated G6pc−/− mice at age 12 weeks (data not shown). With the exception of rAAV8-miGPE-treated mice at low dose, hepatic triglyceride contents were not statistically different between wild type and the other 3 groups of rAAV-treated G6pc−/− mice (Fig. 2C).
Fig. 2.
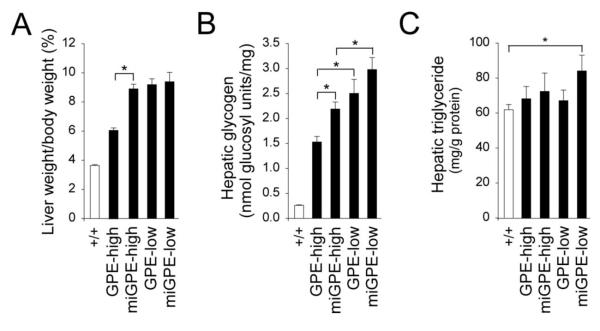
Phenotypic analyses in 12-week-old wild type and rAAV8-treated G6pc−/− mice. (A) Liver weight. (B) Hepatic glycogen contents. (C) Hepatic triglyceride contents. GPE-high (n = 6), high dose rAAV8-GPE-treated; miGPE-high (n = 6), high dose rAAV8-miGPE-treated; GPE-low (n = 6), low dose rAAV8-GPE-treated; miGPE-low (n = 6), low dose rAAV8-miGPE-treated G6pc−/− mice; (+/+), wild type mice. Data are mean ± SEM. *P < 0.05.
3.3 Fasting glucose and glucose tolerance profiles in rAAV8-GPE- or rAAV8-miGPE-treated G6pc−/− mice
For wild type mice (n = 24), the mean blood glucose level before fasting was 172.2 ± 2.6 mg/dl (zero time), which decreased to 134.6 ± 3.8 mg/dl after 8 hours of fast (Fig. 3A). The fasting blood glucose profiles of high dose therapy mice (n = 6 per treatment) were significantly better than low dose therapy mice (n = 6 per treatment), however, even at high dose, rAAV8-GPE-treated mice sustained significantly higher glucose levels that stabilized to wild type levels 6 hours into the fast, while rAAV8-miGPE-treated mice plateaued much lower at 60% of wild type levels (Fig. 3A). The high dose rAAV8-GPE- and rAAV8-miGPE-treated mice could also sustain a 24-hour fast but fasting blood glucose levels were significantly higher in rAAV8-GPE-treated mice than rAAV8-miGPE-treated mice (Fig. 3B). In summary, the rAAV8-GPE-treated G6pc−/− mice were closer to wild type and more capable of tolerating fasting than the rAAV8-miGPE-treated G6pc−/− mice.
Fig. 3.

Fasting blood glucose and glucose tolerance profiles in 12-week-old wild type and rAAV8-treated G6pc−/− mice. (A) Fasting blood glucose profiles. (B) Blood glucose levels following a 24-hour fast. (C) Glucose tolerance profiles. GPE-high (n = 6), high dose rAAV8-GPE-treated (○); miGPE-high (n = 6), high dose rAAV8-miGPE-treated (●); GPE-low (n = 6), low dose rAAV8-GPE-treated (□); miGPE-low (n = 6), low dose rAAV8-miGPE-treated (∎) G6pc−/− mice; (+/+), wild type mice (n = 24) (▾). Data are mean ± SEM. *P < 0.05, **P < 0.005.
Blood glucose tolerance profiles of the treated G6pc−/− mice (n = 6 per treatment) were monitored following an intraperitoneal glucose injection. In general, the profiles paralleled those of the wild-type mice (Fig. 3C) with the high dose treated mice responding better than the low dose treated mice.
The bio-distribution of the human G6PC transgene in liver, kidney, intestine, brain, testis, and ovary in 12-week-old, high dose rAAV8-GPE-treated G6pc−/− mice was analyzed by quantitative PCR (Table 1). In the transduced liver, vector genome copy numbers/μg DNA was 94,440 ± 7,624 (or 0.51 ± 0.04 vector copies/diploid genome). In the transduced kidney and intestine, the numbers were dramatically lower, averaging just 2.57% and 0.64% respectively of liver copy number, showing that the rAAV8 virus did not transduce kidney and intestine efficiently. The genome copy numbers/μg DNA in the brain and testis were even lower at 0.12% and 0.02%, respectively of liver copy number. Only background levels of human G6PC genomes were detected in the ovary.
Table 1.
Human G6PC genome distribution and mRNA expression in 12-week-old high dose rAAV-GPE-treated G6pc−/− mice
| Tissue | Human G6PC Copy number/μg genomic DNA |
Human G6PC mRNA relative to RpI19 mRNA × 105 |
|---|---|---|
| Wild type tissues, no transgene (n = 6) | 8 ± 2 | 8 ± 1 |
| Liver, −/−/rAAV8-GPE (n = 6) | 94,440 ± 7,624 (100) | 62740 ± 4445 (100) |
| Kidney, −/−/rAAV8-GPE (n = 6) | 2,429 ± 626 (2.57) | 22 ± 5 (0.033) |
| Intestine, −/−/rAAV8-GPE (n = 6) | 601 ± 124 (0.64) | 9 ± 1 |
| Brain, −/−/rAAV8-GPE (n = 6) | 121 ± 23 (0.12) | 8 ± 1 |
| Testis, −/−/rAAV8-GPE (n = 6) | 25 ± 8 (0.02) | 9 ± 1 |
| Ovary, −/−/rAAV8-GPE (n = 6) | 7 ± 2 | 9 ± 1 |
Data are means ± SEM. The values of wild type tissues, which contain no transgene, were the mean ± SEM of liver, kidney, intestine, brain, testis, and ovary. Numbers in parentheses are % of liver value.
The rAAV8-GPE vector contains a tissue-specific promoter/enhancer element expressed primarily in the liver, proximal tubules in the kidney, and intestine [1,2]. Quantitative real-time RTPCR analysis of human G6PC transcripts showed a correlation between genome copy number and gene expression (Table 1). In the liver, levels of human G6PC mRNA relative to the RpI19 transcript were 0.62740 ± 0.04445. As expected from the genome copy analysis, the kidney expressed only 0.03% of the liver human G6PC mRNA, and only background levels of human G6PC mRNA were detected in the intestine, brain, testis, and ovary.
4. Discussion
Previous gene therapy studies using the mouse model of GSD-Ia have shown that two human G6Pase-α-expressing rAAV vectors, rAAV8-GPE [7,8] and rAAV8-miGPE [6], show promise treating murine GSD-Ia. The rAAV8-GPE and rAAV8-miGPE vectors differed in the promoter/enhancer with the rAAV8-GPE vector driven by 2864-bp of the human G6PC promoter/enhancer and the rAAV8-miGPE vector driven by a shorter 382-bp of the minimal G6PC promoter/enhancer. The two constructs also differed in the single-stranded or double-stranded nature of the vectors. To select a gene therapy vector for a first human clinical trial of GSD-Ia, we set out to compare the in vivo efficacy of hepatic G6Pase-α gene delivery in G6pc−/− mice using these two vectors. Despite the enhanced transduction efficiency of double-stranded rAAV vectors [9], the packaging constraint limits the size of the promoter/enhancer elements. In the rAAV8-GPE vector the more complete promoter/enhancer elements outweigh the lower single-stranded rAAV transduction efficiency, the construct being more efficacious in correcting all endpoints important to the treatment of murine GSD-Ia. The rAAV8-GPE vector directed significantly higher levels of hepatic G6Pase-α expression, achieved greater reduction in hepatic glycogen accumulation, and led to a better toleration of fasting than the rAAV8-miGPE-treated G6pc−/− mice. In summary, the differences in the promoter/enhancer elements used in directing G6Pase-α expression are key factors. In this regard, additional regulatory elements contained within nucleotides −2864 to −383 of the G6PC 5′ flanking region are critical for optimal G6PC expression in vivo in rAAV-mediated gene transfer for GSD-Ia.
One of the most significant chronic risks in GSD-Ia is HCA, that develops in 70-80% of GSD-I patients over 25 year-old [1,2,13,14]. In 10% of GSD-Ia patients, HCA undergoes malignant transformation to hepatocellular carcinoma [1,2,14,15]. We have recently shown that the rAAV8-GPE-mediated gene transfer restoring more than 3% of wild type hepatic G6Pase-α activity in G6pc−/− mice corrects hepatic G6Pase-α deficiency and prevents HCA formation for up to 90 weeks [8]. Renal disease is another long-term complication in GSD-Ia [1,2]. Correction of renal disease in GSD-I has been less extensively studied. The rAAV8-GPE vector contains a tissue-specific promoter/enhancer element expressed primarily in the liver, proximal tubules in the kidney, and intestine. Analysis of the vector genome copy numbers/μg DNA in rAAV8-GPE-treated G6pc−/− mice showed that the G6PC transgene was delivered primarily to the liver. Different AAV serotypes exhibit different tropisms, due to the distribution of capsid-specific receptors on target cells [16]. Zincarelli et al. [17] compared the tropism of AAV serotypes 1-9, introduced via tail vein injection in mice, by following the distribution and kinetics of a recombinant luciferase gene. They found that for liver expression, rAAV8 and rAAV9 were best, but rAAV9 was more efficient in transducing the kidney. Luo et al. [18] compared the efficacy of AAV serotypes 2, 7-9 for hepatic and renal transduction in G6pc−/− mice. In agreement with Zincarelli et al. [17], they showed that rAAV8-miGPE and rAAV9-miGPE were the most efficacious for liver transduction. However, both rAAV9-miGPE and rAAV8-miGPE vectors achieved similar low levels of biochemical correction in the kidney [18]. The rAAV8-GPE vector also delivers little or no transgene to the kidney [7], like rAAV8-miGPE [6,18]. As expected, the 12-week-old rAAV8-GPE- and rAAV8-miGPE-infused G6pc−/− mice continued manifesting nephromegaly (data not shown). We have shown that the rAAV8-GPE infused mice expressing 3% to 128% of wild type hepatic G6Pase-α manifest nephromegaly [8]. However, mice expressing higher hepatic G6Pase-α activity had lower kidney weights, suggesting good metabolic control reduced nephromegaly [8]. In normal kidneys, the G6PC gene is expressed primarily in the proximal tubular epithelial cells [19], suggesting that the systemic delivery is probably a suboptimal route for renaltranduction. A successful renal tubular epithelial cell transduction has been obtained with an AAV2 vector using a catheter-based renal artery delivery system [20], and this approach warrants further investigation. More study of kidney transduction and renal tissue targeting through capsid modifications are also warranted.
Minimizing the induction of host immune responses is a critical consideration for any human gene therapy trial. In general, the AAV capsid proteins are poor activators of the innate and adaptive immune responses, but the encoded transgene product could represent a more substantial immune challenge [21]. While our studies showed minimal immune reactivity to our vectors [7,8], mouse and canine models are not always predictive of the immune reactivity in primates. [22-24]. Healthy human subjects do carry AAV capsid-specific CD8+ T cell populations that can expand upon rAAV-mediated gene transfer. There are a variety of strategies to overcome humoral immunity to the AAV capsids that can be considered [21], and many clinical trials using a new therapy may pre-screen participants for existing AAV capsid serotype reactivity and will opt for some level of immunosuppression therapy during primary virus exposure, for increased safety. In conclusion, the rAAV8-GPE vector that shows high efficacy in treating murine GSD-Ia, averts fasting hypoglycemia, prevents HCA development is a promising candidate for clinical trials in human GSD-Ia.
Highlights.
Identify the best rAAV vector for clinical translation in human glycogen storage disease type Ia.
The additional control elements in the G6PC promoter/enhancer are critical for optimal hepatic transgene expression
Acknowledgement
This research was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, and The Children’s Fund for Glycogen Storage Disease Research.
Abbreviations
- GSD-Ia
glycogen storage disease type Ia
- G6Pase
glucose-6-phosphatase
- G6P
glucose-6-phosphate
- HCA
hepatocellular adenoma
- AAV
adeno-associated virus
- GPE
G6PC promoter/enhancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement The authors reported no potential conflicts of interest.
References
- 1.Chou JY, Matern D, Mansfield BC, Chen YT. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr. Mol. Med. 2002;2:121–143. doi: 10.2174/1566524024605798. [DOI] [PubMed] [Google Scholar]
- 2.Chou JY, Jun HS, Mansfield BC. Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nat. Rev. Endocrinol. 2010;6:676–688. doi: 10.1038/nrendo.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greene HL, Slonim AE, O’Neill JA, Jr., Burr IM. Continuous nocturnal intragastric feeding for management of type 1 glycogen-storage disease. N. Engl. J. Med. 1976;294:423–425. doi: 10.1056/NEJM197602192940805. [DOI] [PubMed] [Google Scholar]
- 4.Chen YT, Cornblath M, Sidbury JB. Cornstarch therapy in type I glycogen storage disease. N. Engl. J. Med. 1984;310:171–175. doi: 10.1056/NEJM198401193100306. [DOI] [PubMed] [Google Scholar]
- 5.Chou JY, Mansfield BC. Recombinant AAV-directed gene therapy for type I glycogen storage diseases. Expert Opin. Biol. Ther. 2011;11:1011–1024. doi: 10.1517/14712598.2011.578067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koeberl DD, Pinto C, Sun B, Li S, Kozink DM, Benjamin DK, Jr., Demaster AK, Kruse MA, Vaughn V, Hillman S, Bird A, Jackson M, Brown T, Kishnani PS, Chen YT. AAV vector-mediated reversal of hypoglycemia in canine and murine glycogen storage disease type Ia. Mol. Ther. 2008;16:665–672. doi: 10.1038/mt.2008.15. [DOI] [PubMed] [Google Scholar]
- 7.Yiu WH, Lee YM, Peng WT, Pan CJ, Mead PA, Mansfield BC, Chou JY. Complete normalization of hepatic G6PC deficiency in murine glycogen storage disease type Ia using gene therapy. Mol. Ther. 2010;18:1076–1084. doi: 10.1038/mt.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee YM, Jun HS, Pan CJ, Lin SR, Wilson LH, Mansfield BC, Chou JY. Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy. Hepatology. 2012;56:1719–1729. doi: 10.1002/hep.25717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarty DM. Self-complementary AAV vectors; advances and applications. Mol. Ther. 2008;16:1648–1656. doi: 10.1038/mt.2008.171. [DOI] [PubMed] [Google Scholar]
- 10.Fisher KJ, Gao GP, Weitzman MD, DeMatteo R, Burda JF, Wilson JM. Transduction with recombinant adeno-associated virus for gene therapy is limited by leading-strand synthesis. J. Virol. 1996;70:520–532. doi: 10.1128/jvi.70.1.520-532.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lei KJ, Chen H, Pan CJ, Ward JM, Mosinger B, Lee EJ, Westphal H, Mansfield BC, Chou JY. Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type 1a mouse. Nat. Genet. 1996;13:203–209. doi: 10.1038/ng0696-203. [DOI] [PubMed] [Google Scholar]
- 12.Cunningham SC, Dane AP, Spinoulas A, Logan GJ, Alexander IE. Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol. Ther. 2008;16:1081–1088. doi: 10.1038/mt.2008.72. [DOI] [PubMed] [Google Scholar]
- 13.Labrune P, Trioche P, Duvaltier I, Chevalier P, Odievre M. Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J. Pediatr. Gastroenterol. Nutr. 1997;24:276–279. doi: 10.1097/00005176-199703000-00008. [DOI] [PubMed] [Google Scholar]
- 14.Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I) Eur. J. Pediatr. 2002;161(Suppl 1):S20–S34. doi: 10.1007/s00431-002-0999-4. [DOI] [PubMed] [Google Scholar]
- 15.Franco LM, Krishnamurthy V, Bali D, Weinstein DA, Arn P, Clary B, Boney A, Sullivan J, Frush DP, Chen YT, Kishnani PS. Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J. Inherit. Metab. Dis. 2005;28:153–162. doi: 10.1007/s10545-005-7500-2. [DOI] [PubMed] [Google Scholar]
- 16.Michelfelder S, Trepel M. Adeno-associated viral vectors and their redirection to cell-type specific receptors. Adv. Genet. 2009;67:29–60. doi: 10.1016/S0065-2660(09)67002-4. [DOI] [PubMed] [Google Scholar]
- 17.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 18.Luo X, Hall G, Li S, Bird A, Lavin PJ, Winn MP, Kemper AR, Brown TT, Koeberl DD. Hepatorenal correction in murine glycogen storage disease type I with a double-stranded adeno-associated virus vector. Mol. Ther. 2011;19:1961–1970. doi: 10.1038/mt.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan CJ, Lei KJ, Chen H, Ward JM, Chou JY. Ontogeny of the murine glucose-6-phosphatase system. Arch. Biochem. Biophys. 1998;358:17–24. doi: 10.1006/abbi.1998.0849. [DOI] [PubMed] [Google Scholar]
- 20.Takeda S, Takahashi M, Mizukami H. Successful gene transfer using adeno-associated virus vectors into the kidney: comparison among adeno-associated virus serotype 1-5 vectors in vitro and in vivo. Nephron Exp. Nephrol. 2004;96:e119–e126. doi: 10.1159/000077378. [DOI] [PubMed] [Google Scholar]
- 21.Mays LE, Wilson JM. The Complex and Evolving Story of T cell Activation to AAV Vector-encoded Transgene Products. Mol. Ther. 2011;19:16–27. doi: 10.1038/mt.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, Ozelo MC, Hoots K, Blatt P, Konkle B, Dake M, Kaye R, Razavi M, Zajko A, Zehnder J, Rustagi PK, Nakai H, Chew A, Leonard D, Wright JF, Lessard RR, Sommer JM, Tigges M, Sabatino D, Luk A, Jiang H, Mingozzi F, Couto L, Ertl HC, High KA, Kay MA. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 23.Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr. Gene Ther. 2007;7:316–324. doi: 10.2174/156652307782151425. [DOI] [PubMed] [Google Scholar]
- 24.Hurlbut GD, Ziegler RJ, Nietupski JB, Foley JW, Woodworth LA, Meyers E, Bercury SD, Pande NN, Souza DW, Bree MP, Lukason MJ, Marshall J, Cheng SH, Scheule RK. Preexisting immunity and low expression in primates highlight translational challenges for liver-directed AAV8-mediated gene therapy. Mol. Ther. 2010;18:1983–1994. doi: 10.1038/mt.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]