

Introduction
Left ventricular hypertrophy is a common finding in clinical practice and the end result of a number of different disease processes. As such, distinguishing hypertrophy due to athletic training or chronic hypertension from more rare and potentially life-threatening genetic conditions, including hypertrophic cardiomyopathy (HCM), is of utmost clinical importance. This is true not only for the individual patient but also for the patient’s family members, who may be at risk when the cause is a heritable disease. We review how family history can be used in identifying inherited cardiac hypertrophy and in guiding ongoing management of the patient and the rest of the family. An in-depth, multi-generational family history has the potential to enhance every aspect of care: from establishing a diagnosis, to devising a genetic testing strategy, to interpreting genetic test results, to providing ongoing risk assessment for sudden cardiac death. We review that family history is not simply a static account of pre-existing deaths and diagnoses, but a dynamic ongoing process incorporating new and valuable insights from family medical records, clinical cardiology evaluations, genetic testing, and visual analysis of the patient’s family tree. Such insights enable comprehensive clinical care for families affected by HCM.
Historical Context
Scientific understanding of inherited cardiac hypertrophy dates back to 1958, when British forensic pathologist Donald Teare published an evocative series of case histories.1 Several sudden deaths in unrelated young adults had revealed, upon autopsy, asymmetric left ventricular (LV) hypertrophy accompanied by a “bizarre and disorganized” myocardium: the disease known today as HCM. In a brief addendum, Teare described a family with multiple afflicted members including a sister and brother who had each died suddenly, one while running for a bus and the other while riding a bicycle. He and colleagues would later devote an entire publication to this family, tracing its history for three generations and performing cardiology evaluations of living family members.2 The resulting family pedigree (Figure 1) was the first to reveal HCM as a hereditary disease.
Figure 1.
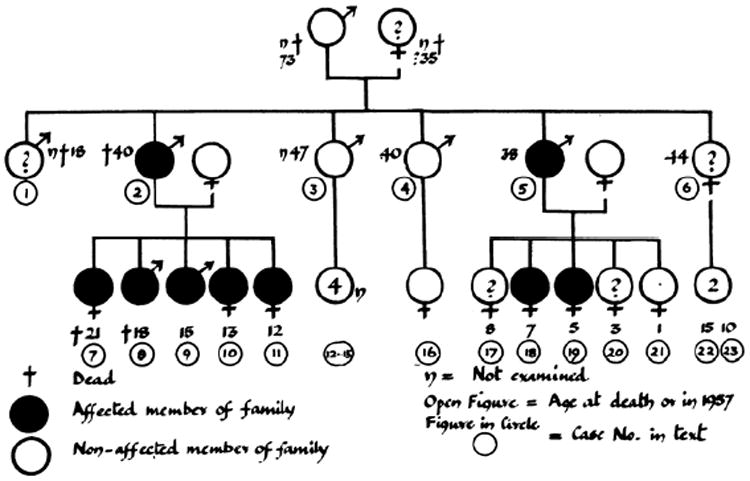
Published in 1960, this family pedigree was the first to show autosomal dominant inheritance of hypertrophic cardiomyopathy (HCM). Females are represented by the symbol for Venus (♀), males by the symbol for Mars (♂). Filled shapes indicate verified disease. Adapted with permission from Hollman et al.2
At the time, there was little understanding of heredity at a molecular level. By 1958, only five years had passed since James Watson and Francis Crick first revealed the physical structure of DNA,3 and a family pedigree remained the sole diagnostic tool for inherited disease.
Even in the current molecular era, more than two decades after the discovery that HCM is caused by genetic changes in the contractile apparatus of heart muscle cells, and with whole-genome sequencing on the clinical horizon, there are certain aspects of clinical care that are guided by a traditional family tree and cannot be accomplished by any other method. Clinical practice guidelines recommend a multi-generational family history as part of the care of all individuals with cardiomyopathy4—and, increasingly, time-saving tools make it easier for clinicians to incorporate a thorough family history into patient care. We now explore the role of family history in a clinical approach to LV hypertrophy.
Family History: Distinguishing Genetic Disease from Secondary LV Hypertrophy
Given the prevalence of hypertension in the adult population, and the popularity of competitive athletics among adolescents, it is common in cardiology practice to encounter patients with some degree of LV hypertrophy detected on electrocardiogram (ECG) or noninvasive cardiac imaging studies. Most cases of mild hypertrophy can be confidently determined to be secondary. Nonetheless, there exists a well-recognized phenotypic overlap with more rare and life-threatening disease processes including HCM. Such cases can be particularly concerning to the clinician when the patient is a young athlete, at an age when sudden cardiac death from HCM is most likely to occur.5-7
Clinical testing can offer clarity in some instances. Evidence of diastolic dysfunction, for example, can be useful in discriminating HCM from the athletic heart.8, 9 As we and others have previously described, this intrinsic feature of HCM can be present in individuals genetically predisposed to the disease even when LV wall thickness is normal.10-15 Other classic features of HCM, but not of physiological hypertrophy, include asymmetric septal hypertrophy,16, 17 small or normal LV cavity size, left atrial enlargement, anatomic abnormalities of the mitral valve or papillary muscles,18 and dynamic left ventricular outflow tract obstruction.6, 7, 19 LV hypertrophy that regresses after detraining an athlete or controlling blood pressure suggests a secondary cause rather than primary HCM.8, 9
Despite these potential clues, patients can defy easy categorization. In ambiguous cases of LV hypertrophy, insights from family history may provide important clarity.
Illustrative Cases
Patient A and Patient B were 18-year-old males referred for evaluation of mild LV hypertrophy. Both had normal mitral valve function and no evidence of LV outflow tract obstruction.
Patient A was suspected of having athlete’s heart with physiological hypertrophy, as he exercised intensely for up to seven hours per day, five days per week. His ECG was distinctly abnormal, with deep inverted T-waves in the precordial leads and high voltage throughout. However, the specificity of these findings was reduced because substantial QRS voltage and inverted T-waves are more common among African-American athletes such as Patient A.20, 21
Patient A’s maternal family history, however, was notable for two premature sudden cardiac deaths as shown in Figure 2, Panel A. His grandmother’s brother (individual II-7 in the family pedigree) had died suddenly at age 29, and his grandmother’s uncle (I-3) at age 35, raising suspicion for inherited cardiomyopathy. Echocardiograms were performed on the patient’s immediate family members to look for previously unrecognized disease. Evaluation showed both his teenage sisters (IV-2 and IV-3) to exhibit mild LV hypertrophy in the absence of any other explanation. His mother (III-2), who had hypertension, had LV hypertrophy as well. Furthermore, after a 3-month detraining period, Patient A had no regression of his cardiac hypertrophy. Given the cumulative weight of the evidence, driven by his family history, we diagnosed Patient A with HCM—revising the pedigree as shown in Figure 2, Panel B.
Figure 2.
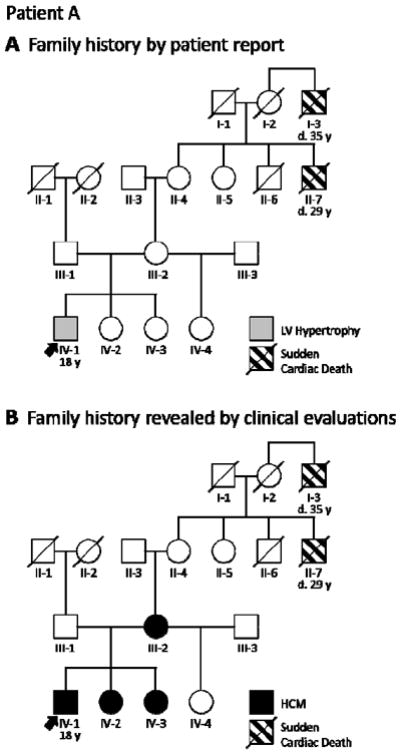
Strategic clinical assessment of a patient’s close family members aids diagnosis by adding valuable information to the family history. A, When 18-year-old Patient A (arrow) presented with mild left ventricular (LV) hypertrophy, his family history of sudden cardiac death (I-3 and II-7) raised suspicion for HCM. B, Clinical cardiology evaluation of the patient’s mother (III-2) and sisters (IV-2 and IV-3) provided the evidence needed for diagnosis. Circles indicate females; squares, males; slash, deceased. HCM = hypertrophic cardiomyopathy.
Patient B, by contrast, was not unusually athletic and had no known history of hypertension. He had recently experienced multiple episodes of syncope, one while playing basketball, raising concern that his LV hypertrophy was pathologic and a sign of HCM with exercise-induced arrhythmias.
Patient B’s family history, however, contained no suggestion of sudden death or significant cardiovascular disease (Figure 3). Accordingly, our suspicion for inherited cardiomyopathy was decreased. 24-hour ambulatory blood pressure monitoring was pursued, and revealed a substantial burden of labile hypertension. Patient B’s family history had helped to guide us toward the true cause of his hypertrophy: occult hypertension.
Figure 3.
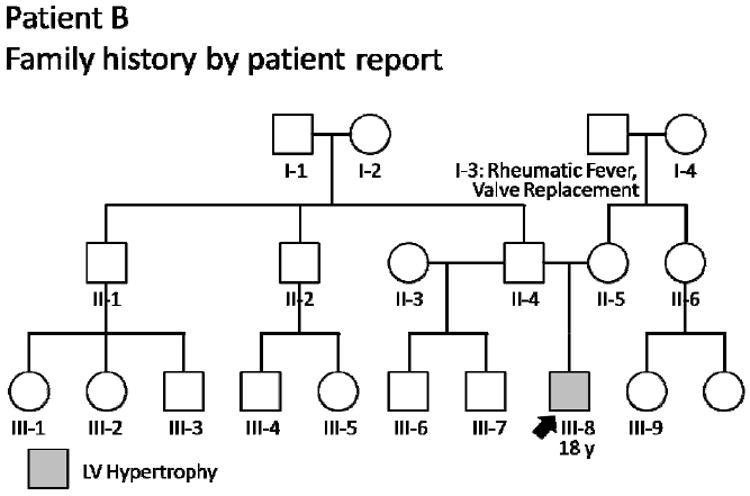
An uneventful family history lowers suspicion for HCM, prompting more thorough investigation of alternative etiologies. For 18-year-old Patient B (arrow) with mild left ventricular (LV) hypertrophy, 24-hour blood pressure monitoring revealed occult hypertension as the underlying cause. Circles indicate females; squares, males; slash, deceased. HCM = hypertrophic cardiomyopathy.
It is worth emphasizing that a comprehensive family history was needed to ascertain the informative deaths in Patient A’s family. While it is standard practice for clinicians to inquire about a patient’s first-degree relatives (parents, siblings, and children), a truly informative assessment of inherited disease risk requires delving deeper into the family tree. Even among members of the same family, the clinical presentation of HCM can vary widely,22, 23 and the diagnosis may have been missed in some relatives, particularly those who are asymptomatic with mild or even no associated health problems. What’s more, some genetically affected family members never develop LV hypertrophy—a phenomenon known as reduced penetrance. Clinical practice guidelines4 therefore recommend a careful three-generation family history that extends at least to the patient’s second-degree relatives (grandparents, aunts, uncles, nieces, and nephews).
To get the most from a family history in clinical practice, it may be necessary to go beyond tallying pre-existing deaths and diagnoses. Strategic clinical assessment of the patient’s close family members, triggered by the proband’s diagnosis with HCM or by suspicion of familial disease, can add important insights to what is already known from static history (as it did with Patient A). Such directed evaluations serve two major purposes: (1) identifying family members with unrecognized clinical disease to initiate appropriate clinical care and (2) providing key supportive evidence for a diagnosis of HCM in the original patient and, by extension, the family. For example, if an echocardiogram reveals HCM in a patient’s parent, sibling, or child, this shifts the patient’s a priori risk for HCM from 1 in 500, based on disease prevalence in the general public,24, 25 to 1 in 2, based on the likelihood of inheriting an autosomal dominant disease.
Differential Diagnosis: Other Forms of Inherited LV Hypertrophy
Informative elements of a family history for a patient with LV hypertrophy are presented in Table 1 below. It is important to ascertain whether relatives have exhibited classic symptoms of HCM such as shortness of breath, chest pain, presyncope or syncope—particularly with exertion.6, 7 Other key questions involve HCM’s more rare and serious consequences including stroke, end-stage congestive heart failure, or sudden cardiac death. Asking for details about cardiothoracic surgeries or other procedures family members have undergone can be important in distinguishing HCM from conditions such as coronary artery disease. Another essential line of inquiry involves accidental and unexpected deaths in the family, such as single-car accidents in which the family member was the driver, drownings, or Sudden Infant Death Syndrome (SIDS). These events sometimes indicate a sudden cardiac arrest that has gone unrecognized.
Table 1.
Patients with Left Ventricular Hypertrophy: Important Elements of a Family History
|
| Features relevant to differential diagnosis |
|
Cardiac hypertrophy can result, too, from a wide range of genetic conditions that affect multiple organ systems (Table 2). A careful family history may therefore detect extracardiac features that help to make a diagnosis (Table 1). Inheritance patterns can provide additional diagnostic clues: Danon disease and Fabry disease are both X-linked conditions, meaning that disease expression in carrier females may be subtle or absent.26 Father to son transmission would effectively rule out an X-linked condition.
Table 2.
Diseases Mimicking Hypertrophic Cardiomyopathy on Echocardiography
| Syndrome | Gene(s) | Gene symbol | Locus | Phenotype | Inheritance |
|---|---|---|---|---|---|
| AMP kinase disease | AMP-activated protein kinase | PRKAG2 | 7q36.1 | Cardiac hypertrophy, pre-excitation | Autosomal dominant |
| Familial amyloid disease | Transthyretin | TTR | 18q12.1 | Low voltage, severe cardiac hypertrophy, paresthesias | Autosomal dominant |
| Noonan syndrome | Protein tyrosine phosphatase, nonreceptor type 11 (aka tyrosine phosphatase SHP2) | PTPN11 | 12q24.1 | Short stature, facial dysmorphology, congenital heart defects, cardiac hypertrophy, skeletal anomalies, bleeding disorders, learning disabilities (variable) | Autosomal dominant |
| Son of sevenless homolog 1 | SOS1 | 2p22.1 | |||
| RAF proto-oncogene serine/threonine-protein kinase | RAF1 | 3p25 | |||
| GTPase KRas | KRAS | 12p12.1 | |||
| Fabry disease | Alpha-galactosidase A | GLA | Xq22 | Renal disease, paresthesias, cardiac hypertrophy | X linked |
| Females can manifest signs of disease | |||||
| Danon disease | Lysosomal-associated membrane protein 2 | LAMP2 | Xq24 | Males present in childhood with cardiac hypertrophy, skeletal myopathy, mental retardation | X linked |
| Females can manifest signs of cardiomyopathy | |||||
| Hereditary hemochromatosis | Hereditary hemochromatosis protein | HFE | 6p21.3 | Iron overload, cardiomyopathy, hypogonadotropic hypogonadism, arthropathy, hepatic fibrosis or cirrhosis, diabetes mellitis, progressive skin pigmentation/bronzing | Autosomal recessive |
| Pompe disease | Acid alpha-glucosidase (aka acid maltase) | GAA | 17q25 | Acid maltase deficiency (aka glycogen storage disease type II) | Autosomal recessive |
| Infantile and juvenile/adult forms | |||||
| Skeletal myopathy, ventilatory failure, cardiac hypertrophy |
Accurately distinguishing HCM from its mimics is important, given the significant differences in prognosis and treatment. Enzyme replacement therapy is available for Fabry disease and Pompe disease, for example.27 Anticipation of the likely need for heart transplantation may be warranted for Danon disease, which can progress rapidly to end-stage heart failure, particularly in adolescent males.28 In ambiguous cases, genetic testing can be of assistance. Genes for some of these syndromes are included on clinically-available HCM genetic testing panels—facilitating simultaneous genetic testing for primary HCM and for multi-system diseases that include LV hypertrophy.
Family History: Managing HCM
Stratifying Risk for Sudden Cardiac Death
Given that HCM is the most common form of inherited LV hypertrophy, the remainder of this review will focus on the role of family history in managing HCM. Once a diagnosis has been established, the next important contribution of family history involves assessing a patient’s risk for sudden cardiac death (SCD).
A family history of sudden death is a major consideration when assessing an individual patient’s risk to determine whether an implantable cardioverter-defibrillator (ICD) for primary prevention is appropriate.7, 29 It is one of six major risk factors considered along with prior cardiac arrest, LV thickness of 3 cm or greater, a history of unexplained syncope, nonsustained ventricular tachycardia on 48 hour Holter monitor, and abnormal blood pressure response to exercise.6, 7 Although any history of premature sudden death is concerning, deaths of greatest concern involve close family members, particularly when multiple family members have died.6, 30
Careful scrutiny is often needed to determine which events constitute premature sudden cardiac death due to HCM. Such events occur most frequently in adolescents and adults under age 35,7 suggesting that sudden death in an elderly relative may be of less clinical concern—particularly given the higher likelihood of confounding comorbidities, most prominently coronary artery disease. Yet the risk for HCM-related cardiac arrest remains elevated throughout life, and published guidelines offer no easy algorithm or well-defined age cut-off for risk stratification. Gathering medical records and autopsy reports for suspicious deaths can be highly informative, when available. If family members have been implanted with ICDs, events that previously would have resulted in sudden death may now register as appropriate shocks.
None of the risk factors for SCD is static—including family history. Family history should be updated at intervals and patients urged to contact the clinic with reports of new deaths, cardiac events, or diagnoses.
Managing family members
HCM is typically inherited in an autosomal dominant fashion, meaning that just one altered copy of a gene, inherited from just one parent, causes the disease. Although de novo genetic variants (brand new in the patient, not inherited from either parent) have been reported,31, 32 the majority of HCM seems to be familial. A patient’s diagnosis therefore implies risk to other family members even in the absence of a clear family history, and clinical screening of relatives is appropriate. Immediate family members—parents, siblings, and children—each share half of the patient’s genes, creating a 50% chance that they carry the same disease-causing variant. A de novo genetic change initiates new familial disease, placing the patient’s children, but not the patient’s siblings or parents, at risk.
Guidelines for the clinical management of these at-risk first-degree family members (Figure 4) include physical examination by a cardiologist familiar with HCM, echocardiography, and 12-lead ECG.4, 7 Cardiac magnetic resonance imaging, Holter monitoring, and exercise testing may also be beneficial in certain situations. Any family member involved in competitive sports, and any family member experiencing symptoms, also needs evaluation—even if more distantly related to the patient with HCM.
Figure 4.
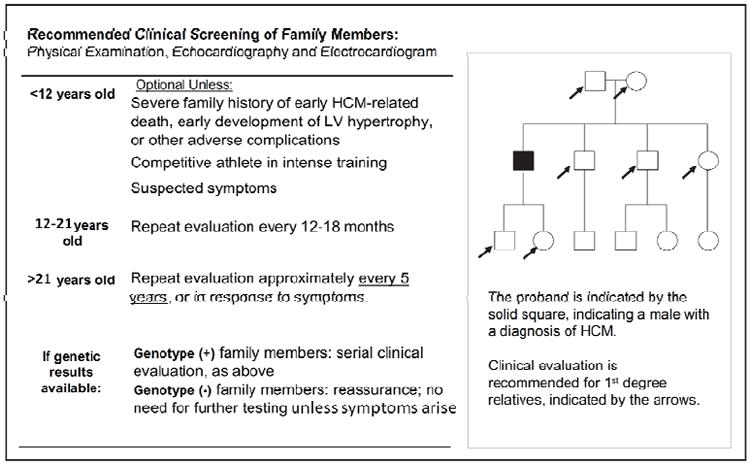
Screening recommendations for families with HCM. The frequency of screening is based on the age of the at-risk family member, due to the age-dependent penetrance of left ventricular (LV) hypertrophy. HCM = hypertrophic cardiomyopathy. Adapted with permission from Ho.70 Data from Gersh et al.7
Moreover, HCM shows age-dependent penetrance, meaning its features may emerge with time in someone previously without signs or symptoms. Cardiac evaluation should therefore be repeated at regular intervals over time. Evaluations should occur annually throughout puberty, when the first signs of HCM are most likely to appear, and every 3-5 years thereafter. The risk of developing HCM persists even past middle age,33 so unless genetic testing confirms that an at-risk relative has not inherited the family’s pathogenic variant, cardiac evaluation should be ongoing as outlined in Figure 4.
Family history can play an important role in individualizing these screening recommendations. Someone from a high-risk family with consistent development of heart failure or sudden death may warrant more frequent monitoring, since it is clear that their specific genetic milieu results in particularly grave consequences when an HCM-causing variant is present. Adult relatives who participate in competitive or high intensity athletics may also warrant more frequent screening. For a family with early-onset LV hypertrophy, childhood screening should start earlier than puberty.4, 7, 34
Family History: Pedigree Analysis
Successfully identifying at-risk individuals within a family tree requires integration of clinical history with basic laws of inheritance and probability.
Illustrative Case
The family of a 15-year-old patient with HCM is depicted in Figure 5. Panel A indicates the immediate family members at 50% risk for HCM based only on the patient’s diagnosis. In constructing a detailed, 3-generation pedigree, however, we discovered a family history of the disease that had previously been unrecognized: The patient’s mother’s cousin (II-6) had died suddenly in his thirties and was diagnosed with HCM on autopsy. Based on this new information, Panel B indicates additional at-risk family members who require ongoing cardiology screening because they are first-degree relatives of an affected patient.
Figure 5.
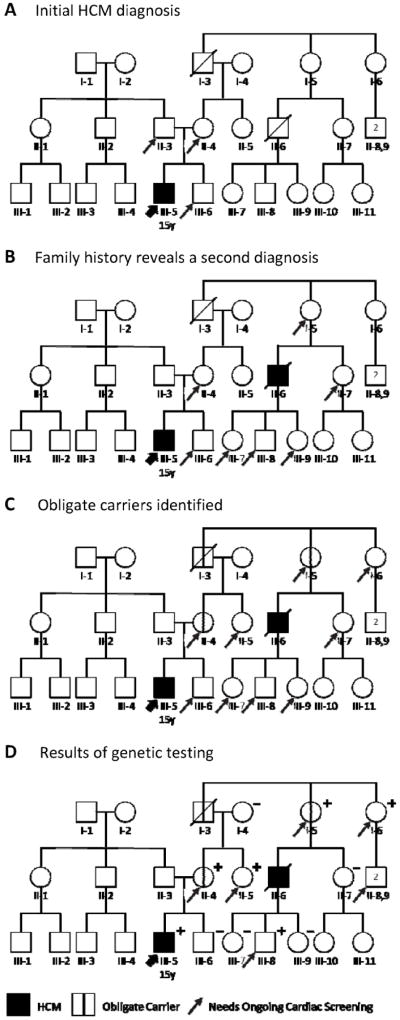
An evolving HCM family history, and careful pedigree assessment, shifts cardiology screening needs. A, Relatives needing screening based on initial diagnosis. Diagnosis of this 15-year-old patient (III-5, thick arrow) with HCM means his first-degree relatives (father II-3, mother II-4, and brother III-6; thin arrows) are each at 50% risk. B, Relatives needing screening based on initial plus a second diagnosis. When a second HCM diagnosis (II-6) is discovered in the patient’s maternal family history, this individual’s first-degree relatives (I-5, II-7, III-7, III-8, and III-9) also require cardiac screening. The patient’s father (II-3), not on the affected side of the family, is no longer considered at risk. C, Additional relatives need screening based on obligate carriers. Pedigree analysis identifies three obligate carriers (I-3, I-5, and II-4; marked with vertical line) connecting the individuals with HCM, including the patient’s mother. Each is at known risk for disease. Immediate family members of an obligate carrier are at 50% risk and also require screening (arrows added to I-6 and II-5). D, Genetic testing helps target screening to relatives definitively predisposed to HCM. In this family, several family members at 50% risk (II-7, III-6, III-7, and III-9) did not inherit the disease-causing variant; they and their descendants can be excused from further screening. By contrast, II-5 and I-6 test positive, newly revealing their children (II-8,9) to be at 50% risk. Circles indicate females; squares, males; slash, deceased; +, genetic variant present; –, genetic variant absent. HCM = hypertrophic cardiomyopathy. Adapted with permission from Ho.50
The next level of analysis involves identifying obligate carriers: individuals who logically must carry the family’s HCM-causing genetic variant in order to explain the overall disease pattern within the family. Simple visual inspection of the pedigree reveals these at-risk individuals. In this case, the chain of family members that connects our patient to his mother’s affected cousin includes three obligate carriers (I-3, I-5, and II-4), each marked with a vertical bar in Panel C. Without each of these individuals having inherited and then passing on the disease-causing variant, the two known cases of HCM could not have occurred. These carrier individuals may have undiagnosed cardiomyopathy or, if they do not, then they are each at risk to develop HCM in the future.
Immediate family members of an obligate carrier are at 50% risk to carry the predisposition to HCM, just like the brother of our initial patient (III-6); they too require ongoing cardiology screening. In Panel C, arrows indicate two such family members (I-6 and II-5) whose at-risk status we may not have recognized had we not drawn out and analyzed the pedigree.
Family History: Genetic Testing Strategy
Given the screening recommendations outlined above, a child at 50% risk of being predisposed to HCM will undergo up to twenty cardiology evaluations between the ages of 12 and 75. It would be ideal if these evaluations could be focused only on the family members who inherited a disease-causing genetic variant, and are predisposed to develop HCM, instead of screening everyone at 50% risk. Genetic testing can help to provide this focus.
Two decades ago, a key role in the pathophysiology of HCM was attributed to the sarcomere: the assembly of proteins in each cardiac muscle cell that enables contraction (Figure 6). The first disease-causing variant to be discovered was in MYH7, the beta-myosin heavy chain gene;35 since then the list of sarcomere-associated genes known to cause HCM has grown to more than a dozen, with MYH7 and MYBPC3 (myosin-binding protein C) most frequently involved.36 Multi-gene panels containing the major genes associated with HCM are clinically available, and are used when the first affected family member undergoes genetic testing.
Figure 6.
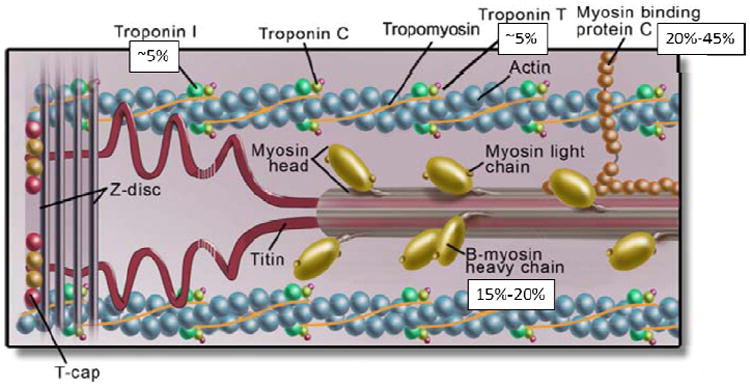
The cardiac sarcomere, highlighting protein products of genes involved in hypertrophic cardiomyopathy. Disease-causing variants in cardiac myosin-binding protein C (MYBPC3) and beta-myosin heavy chain (MYH7) are most common, accounting for 20-45% and 15-20% of the disease respectively. Cardiac troponin T type 2 (TNNT2) and troponin I type 3 (TNNI3) each account for ~5%. Variation in other sarcomere genes is less frequent. Data from Ackerman et al.36
Family history is an important guide when deciding which family member should be tested first. In general, the best candidate is the person whose HCM was diagnosed at the youngest age, or whose disease features are the most classic and severe.4 Notably, this may not be the patient who first presents to clinic. Testing the most affected family member is a well-established principle of medical genetics: It helps to minimize the chance of testing a phenocopy (someone whose LV hypertrophy is due solely to hypertension or to intense athletic activity) and to maximize the chance that the person tested actually carries the familial predisposition to HCM.
What’s more, the approach increases the likelihood of detecting all disease-causing genetic variants present in the family, as there may not be just one. Approximately 5% of patients with HCM have been reported to carry two or more sarcomere gene variants37, 38 (in the gene panels explored to date), and our appreciation of multi-genic contribution is only likely to increase as new DNA sequencing technologies reach the clinic.39 Individuals with this higher genetic “dosage” may have earlier disease onset and worse prognosis,38, 40-43 although it has been difficult to fully understand the impact of multiple variants due to phenotypic heterogeneity and the limited scale of prior studies. Of note: When a patient carries two disease-causing changes, it is possible that one was inherited from each parent. This emphasizes the importance of withholding judgment about which side of the family may be affected by HCM.4, 44 The reality is, it may be both.
Initial genetic testing with a multi-gene panel will yield one of three results: negative, positive, or uncertain. A negative result is when testing fails to locate a disease-causing variant in any of the genes sequenced. For patients with a clinical diagnosis of HCM, testing is negative approximately 40-50% of the time. This includes a range from ~70% of patients without a family history to ~30-40% of patients with a family history.7, 31, 36, 37, 45-49 This imperfect capture is a sign that our knowledge of HCM genetics remains incomplete, and a negative test result does not mean that a patient’s HCM is not hereditary. In truth, the result is simply not informative. While detecting a disease-causing genetic variant can confidently rule “in” a diagnosis of HCM, failing to find such a variant cannot rule it “out.”
A positive test result, by contrast, is highly informative. It reveals a change in the patient’s DNA that is not found in the healthy human reference genomes used for comparison. Association between a genetic variant and disease is always probabilistic, however, leaving room for the possibility for false positives—and our level of confidence that a particular variant causes disease is based on the weight of the evidence. To be confident that a variant is pathogenic requires that it has been shown to definitively segregate with disease in a sufficient number of unrelated HCM families and is absent from healthy, ethnically-matched controls. A positive result of this kind confirms the diagnosis of HCM in the patient and identifies the genetic change believed to be responsible. These test results become an important piece of family history and drive the medical management of family members.
Genetic test results of “uncertain significance”, the third possible outcome, are discussed below.
Genetic Testing Alters Family Management
Where genetics has enormous power is in identifying at-risk family members prior to the onset of disease, and in providing some relief from life-long cardiac evaluations for family members who are not at risk.4, 36, 50 Through “predictive” genetic testing, healthy family members can determine whether they carry the specific genetic variant associated with HCM in the family. Simply put, if a family member did inherit the disease-causing variant, they are very likely to develop HCM at some point in their lives and have a 50% chance to pass the predisposition on each time they have a child. If they did not inherit the variant, neither they nor their descendants should be at risk. However, given the probabilistic nature of genetic testing, family members who test negative are urged to seek cardiac evaluation if they develop symptoms of HCM. It’s worth noting that for at-risk family members with mild LV hypertrophy, predictive testing for the family’s pathogenic variant can be extremely useful in differentiating HCM from hypertensive or athlete’s heart.
Typically, predictive testing is conducted in a systematic, “cascade” fashion.36 It starts with the immediate family members of an affected patient, who are each at 50% risk. For anyone who tests positive, the offer of genetic testing (and the 50% risk) extends to all of their first-degree relatives. For an autosomal dominant disease, half of family members will carry the disease-causing variant on average. Conversely, half will test negative—meaning that cardiac screening for both them and their descendants can be dramatically reduced.50, 51 Each individual’s genetic test result, whether positive or negative, therefore adds valuable information to the family history and impacts management.
In the case of the family in Figure 5, genetic testing revealed a disease-causing variant in the MYBPC3 gene of the 15-year-old proband. As illustrated in Panel D, testing the obligate carriers—his mother (II-4) and his great-aunt (I-5)—confirmed that they did carry this familial variant, as expected. For the remaining family members, by contrast, genetic testing was truly predictive. Our patient’s brother (III-6) tested negative, releasing him from a lifetime of regular screening for HCM—as did two of his second cousins (III-7 and III-9). Genetic testing thereby substantially reduced the number of individuals in this youngest generation who require ongoing cardiac screening. By contrast, the patient’s great-aunt (I-6) tested positive, newly revealing her two sons (II-8,9) to be at 50% risk and in need of ongoing screening.
Family History: Interpreting Genetic Test Results of “Uncertain Significance”
The more we learn about the human genome, the clearer it is that we all have benign, rare alterations even within disease-causing genes,52, 53 and scientific understanding in this area is moving quickly. Thus, it is not always clear if a variant identified in an HCM-associated gene indeed contributes to disease, or if it may instead be part of the clinically inconsequential rare genetic variation present in every genome.
The fact that this normal variation exists leads to the most challenging genetic test result of all to emerge from a multi-gene panel: a “variant of uncertain clinical significance” (VUS). In these cases, the patient does have a genetic variant within an HCM-associated gene, but it is not clear that the variant is linked to disease. Hundreds of separate variants have so far been identified in patients with HCM, many of them private to a single family.36 Due to this remarkable genetic heterogeneity, HCM genetic testing frequently reveals a novel or poorly-characterized genetic variant—one whose ability to cause disease is unknown.
A VUS cannot be used for predictive testing of unaffected family members, as the power of predictive testing lies in our confidence that the genetic variant we are testing for is very likely responsible for disease. When significant uncertainty exists, the danger is that we will mistakenly excuse someone who is actually at risk from clinical surveillance, only to have them later develop HCM, undiagnosed and untreated.
Family history, however, often holds the key to clarifying a variant’s role in disease. This is accomplished through a process known as segregation analysis. A genetic variant and a disease are said to “co-segregate” if found together, without exception, in every obligate carrier and affected family member tested. This evidence associating the two suggests the variant may indeed be the underlying cause for the disease.
Testing healthy, at-risk family members for a VUS is typically not informative. Detecting a VUS in a healthy individual raises two mutually exclusive possibilities: (1) The variant is not responsible for the disease in the family or (2) The family member is genetically predisposed to develop HCM but has not yet expressed overt clinical manifestations. Therefore, understanding of the variant’s pathogenicity is not advanced. For this reason, testing for a VUS should generally be limited to HCM-affected individuals and obligate carriers to determine if it segregates with disease.54
Careful phenotyping of additional family members, through ECG and echocardiogram, can add power to this process. The weight of the evidence provided by co-segregation increases as more affected family members test positive for the variant, and particularly when distantly-related affected family members test positive. This is because it is possible to calculate the mathematical probability that all of these affected relatives would have inherited the same genetic variant simply by chance. The more distantly-related the affected relatives are, the more likely it becomes that the co-segregation of variant and disease is due to the variant’s disease-causing role rather than due to chance alone.
Illustrative Cases
Genetic testing of the patient in Figure 7 revealed a VUS in the MYH7 gene. Robust segregation analysis was possible due to the multiple affected family members and obligate carriers known. Initial testing revealed that the patient’s two siblings with HCM (III-2 and III-4) harbored the same MYH7 variant. However, the likelihood that any three siblings will have inherited the same genetic variant simply by chance is one in eight. Further testing of the patient’s affected niece (IV-3) and obligate carrier mother (II-4) revealed that they too carried the variant, strengthening the evidence for pathogenicity. If the patient’s aunt (II-9) were to test positive, this would provide more evidence still (with a less than 1% likelihood that these affected family members would all carry the variant by chance alone).
Figure 7.
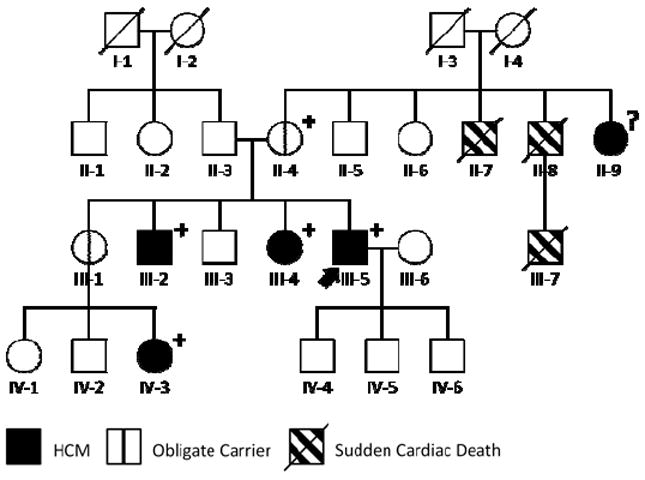
Robust segregation analysis can help to determine whether a specific genetic variant is responsible for disease. In this HCM family, genetic testing of our patient (arrow) revealed a variant of uncertain significance (VUS) in the MYH7 gene. The VUS was present in all affected family members (III-2, III-4, III-5, and IV-3) and obligate carriers (II-4) tested, increasing confidence in its pathogenicity. Circles indicate females; squares, males; slash, deceased; ?, genotype unknown; +, VUS present; −, VUS absent. HCM = hypertrophic cardiomyopathy.
Based on our segregation data, the presence of this variant in other families with HCM tested the clinical laboratory, and its absence in controls, the laboratory eventually reclassified this variant as “pathogenic.” This highlights the importance of two-way communication between clinicians and genetic testing laboratories, which can significantly move the science forward.
Segregation analysis can also be key in proving that an identified variant is not the cause of familial disease, as in the following example. A 20-year-old woman was recently diagnosed with HCM, and had a known maternal family history of disease: The patient’s mother and maternal grandmother had each been diagnosed. When genetic testing identified a VUS in the patient, we began testing other affected family members to determine whether they, too, carried the variant. The very first test result—the patient’s affected mother—was negative. We could therefore conclude that the VUS detected on the multi-gene HCM panel was not the cause of the family’s disease. The genetic etiology in this family remains undefined.
The Pedigree in Clinical Practice
Constructing a three-generation family pedigree from scratch is undeniably time-consuming and may seem prohibitively so to a busy physician. A genetic counselor typically devotes at least 20 minutes to taking a patient’s family medical history,55 with additional time spent outside the actual clinic visit on tasks such as seeking and reviewing family medical records.56
Nonetheless, there are time-efficient ways to engage patients and their family members in constructing a detailed and accurate history. For example, the Surgeon General’s Family Health History Initiative has created a web-based pedigree tool. At https://familyhistory.hhs.gov, patients can enter information about their family members and generate a printable pedigree to bring to clinic. Sending patients a family medical history questionnaire in advance of an appointment, and urging them to discuss it with knowledgeable family members before creating an online pedigree, may maximize the chances of obtaining useful information.57
Encouraging patients to begin a direct dialog about medical history with their extended family members sets in motion a highly informative process. We see family histories change dramatically, with new diagnoses and sudden deaths uncovered, once heart disease becomes the subject of conversation and detective work within the family. The physician can guide this family process by alerting patients to the relevant signs and symptoms of inherited disease and by pointing out individuals within the family pedigree whose medical histories are of greatest interest. Distributing family letters is an effective way to inform relatives and to encourage cardiology screening and/or genetic testing;58 these letters will often prompt family members to reveal diagnoses that were not previously known to the patient.
Accuracy of Family History
As clinicians know from experience, patients may possess only vague details about a family member’s medical condition or specific cause of death. Studies show that even an event as dramatic as myocardial infarction in a parent or sibling is known to and reported by the patient just 80-85% of the time, with accuracy further decreasing for more distant family members.59-62 Complicating matters, patients tend to report any life-threatening cardiac event as a “heart attack,” unfamiliar with the distinction between this and cardiac arrest.4, 61
The initial history obtained by patient report should therefore be considered a starting point for further investigation, confirmed whenever possible through medical records, death certificates, and autopsy reports. If a diagnosis of HCM is not clearly stated, it can often be inferred from the weight of the heart or from pathognomonic histological features of HCM such as cardiac myocyte hypertrophy, disarray, or increased myocardial fibrosis.6
Illustrative Case
Figure 8 illustrates how the detailed family history for the case introduced in Figure 7 was actually obtained. Like the majority of individuals newly-diagnosed with HCM, the patient did not think he had a family history of the disease at the time of his first appointment. Panel A shows the information obtained by patient report at his first genetic counseling session. However, in counseling we identified family members whose medical records might potentially reveal HCM. One brother (III-2) had experienced arrhythmias and had undergone heart surgery. A sister (III-4) had a “big heart” and required a pacemaker. His other two siblings had no heart issues, but a niece (IV-3) had been born with a “congenital heart problem.” On the patient’s mother’s side of the family, two uncles (II-7 and II-8) had died suddenly—although the patient did not know the cause.
Figure 8.
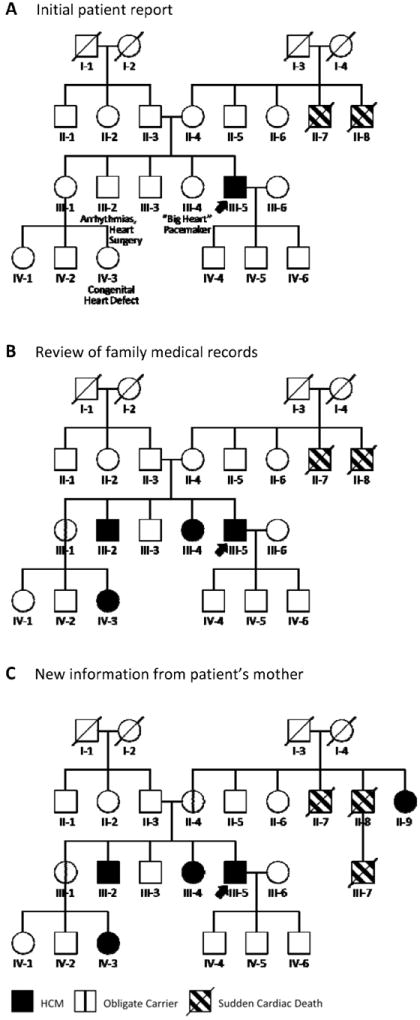
Constructing a family history is a dynamic process that unfolds over time. A, At diagnosis, this patient (arrow) had no known history of HCM but reported suspicious cardiac features in three family members (III-2, III-4, and IV-3). B, Review of family medical records dramatically altered the original history, showing those three family members to have HCM and the patient’s unaffected sister (III-1) to be an obligate carrier (vertical line). C, The patient’s mother then reported that her sister (II-9) carried a diagnosis of HCM. This definitively localized the disease to the maternal side of the family, revealing the patient’s mother (II-4) as another obligate carrier. Circles indicate females; squares, males; slash, deceased. HCM = hypertrophic cardiomyopathy.
Compare this to the family history as it looked after the patient obtained family medical records at our request (Panel B). Cardiology records for the patient’s brother and sister showed them both to have HCM. The same was true of his niece, revealing his unaffected sister (III-1) as an obligate carrier.
As conversations about heart disease continued within the family, the patient’s mother contacted us with new information (Panel C). Her sister (II-9), she had discovered, also carried a diagnosis of HCM. What’s more, the family had suffered not two but three sudden cardiac deaths: the third having occurred in the patient’s cousin (III-7). Not only did this new information definitively localize the disease to the maternal side of the family, it revealed the patient’s mother (II-4) as another obligate carrier and provided evidence that the family was at increased risk for SCD, influencing decisions about primary prevention ICD implantation. The “negative” family history was not negative at all: four of the patient’s living family members had HCM.
The Future
We have focused in this review on ways to predict and respond to HCM using family history. Even with today’s technology, however, some approaches to disease prevention are possible. Preimplantation genetic diagnosis (PGD) offers the opportunity to decrease the chance of passing a disease-causing variant to the next generation. PGD involves using embryo selection during in vitro fertilization directed by genetic testing of a single cell from each embryo considered.63 Couples attempt pregnancy using only embryos determined not to carry the disease-causing variant. At around $20,000 per cycle, however, the financial expense of IVF/PGD is considerable—and it is often not covered by insurance. It also requires identifying the family’s HCM-causing variant in advance, which for almost half of HCM patients is not currently possible.31, 36, 45-49
Looking to the future, one area of research interest involves developing new therapies that can slow or even halt development of the disease.64 As more families undergo genetic testing for HCM, a new “preclinical” population is growing. These apparently healthy individuals carry the family’s HCM-causing variant, yet currently exhibit no evidence of hypertrophy. At the moment, clinicians can only screen and wait for clinical features of HCM to appear, then assess risk for sudden death and try to palliate symptoms. To truly transform the lives of families with HCM, we will instead need to learn how to prevent preclinical cases from progressing to overt disease.
One such approach has shown promise in a mouse model of HCM, and has advanced to testing in humans. Mouse studies have shown abnormalities in intracellular calcium handling by cardiac myocytes to be among the earliest detectable manifestations of the disease.65-68 Treating “preclinical” mice with the L-type calcium channel blocker diltiazem reduced the amount of hypertrophy, disarray, and fibrosis to develop in their hearts, as compared to placebo.66 Similarly, blocking TGF-β signaling with the angiotensin II receptor antagonist losartan has been shown to prevent the emergence of hypertrophy and fibrosis.69 These findings have led to the first human placebo-controlled pilot study of a preventive approach to HCM (http://clinicaltrials.gov/ct2/show/record/NCT00319982), as well as a multi-center initiative to foster better understanding of this preclinical stage and to test new approaches for disease modification.
Conclusion
In summary, we have highlighted the power of family history in the workup of patients with cardiac hypertrophy. From clarifying diagnosis in those with unclear etiology to forming the bedrock of genetic evaluation in those with clearly demonstrated disease, the family history is more powerful now than ever before. Indeed, even as new genetic technologies usher in an unprecedented appreciation of our patients’ genomes, we will continue to rely on the family history to inform thoughtful care and rational management of patients and families with hypertrophic disease.
SUMMARY.
To engage patients in constructing their own comprehensive family medical history, suggest they:
Speak with knowledgeable family members about their relatives’ health
Contact family members with heart issues to clarify the exact diagnosis
Fill out a family history questionnaire
Diagram their family tree at https://familyhistory.hhs.gov
Gather cardiology records for family members who could potentially have HCM
Gather autopsy reports and death certificates for suspicious sudden or accidental deaths
Update you on new diagnoses or sudden deaths
Acknowledgments
We thank our patients and their families.
Sources of Funding
Dr. Ashley is supported by NIH Director’s New Innovator Award DP2 OD004613, NIH R01 award HL105993, and Stanford NIH/NCRR Clinical and Translational Science Award UL1 RR029890. Dr. Ho is supported by NIH/National Heart, Lung, and Blood Institute award 1P20HL101408-01 and NIH/National Human Genome Research Institute award 1U01 HG006500-01.
Footnotes
Disclosures
Dr. Ashley is a stockholder and consultant for Personalis, Inc.
References
- 1.Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1–8. doi: 10.1136/hrt.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hollman A, Goodwin JF, Teare D, Renwick JW. A family with obstructive cardiomyopathy (asymmetrical hypertrophy) Br Heart J. 1960;22:449–456. doi: 10.1136/hrt.22.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 4.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ. Sudden death in young athletes. N Engl J Med. 2003;349:1064–1075. doi: 10.1056/NEJMra022783. [DOI] [PubMed] [Google Scholar]
- 6.Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687–1713. doi: 10.1016/s0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- 7.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:e783–831. doi: 10.1161/CIR.0b013e318223e2bd. [DOI] [PubMed] [Google Scholar]
- 8.Pelliccia A. Athlete’s heart and hypertrophic cardiomyopathy. Curr Cardiol Rep. 2000;2:166–171. doi: 10.1007/s11886-000-0015-4. [DOI] [PubMed] [Google Scholar]
- 9.Maron BJ, Pelliccia A. The heart of trained athletes: cardiac remodeling and the risks of sports, including sudden death. Circulation. 2006;114:1633–1644. doi: 10.1161/CIRCULATIONAHA.106.613562. [DOI] [PubMed] [Google Scholar]
- 10.Cardim N, Perrot A, Ferreira T, Pereira A, Osterziel KJ, Reis RP, et al. Usefulness of Doppler myocardial imaging for identification of mutation carriers of familial hypertrophic cardiomyopathy. Am J Cardiol. 2002;90:128–132. doi: 10.1016/s0002-9149(02)02434-7. [DOI] [PubMed] [Google Scholar]
- 11.Nagueh SF, Bachinski LL, Meyer D, Hill R, Zoghbi WA, Tam JW, et al. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation. 2001;104:128–130. doi: 10.1161/01.cir.104.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagueh SF, McFalls J, Meyer D, Hill R, Zoghbi WA, Tam JW, et al. Tissue Doppler imaging predicts the development of hypertrophic cardiomyopathy in subjects with subclinical disease. Circulation. 2003;108:395–398. doi: 10.1161/01.CIR.0000084500.72232.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho CY. Hypertrophic cardiomyopathy: preclinical and early phenotype. J Cardiovasc Transl Res. 2009;2:462–470. doi: 10.1007/s12265-009-9124-7. [DOI] [PubMed] [Google Scholar]
- 14.Ho CY, Carlsen C, Thune JJ, Havndrup O, Bundgaard H, Farrohi F, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:314–321. doi: 10.1161/CIRCGENETICS.109.862128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho CY, Sweitzer NK, McDonough B, Maron BJ, Casey SA, Seidman JG, et al. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation. 2002;105:2992–2997. doi: 10.1161/01.cir.0000019070.70491.6d. [DOI] [PubMed] [Google Scholar]
- 16.Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol. 1995;26:1699–1708. doi: 10.1016/0735-1097(95)00390-8. [DOI] [PubMed] [Google Scholar]
- 17.Maron BJ. Echocardiographic assessment of left ventricular hypertrophy in patients with obstructive or nonobstructive hypertrophic cardiomyopathy. Eur Heart J. 1983;4(Suppl F):73–91. doi: 10.1093/eurheartj/4.suppl_f.73. [DOI] [PubMed] [Google Scholar]
- 18.Maron MS, Olivotto I, Harrigan C, Appelbaum E, Gibson CM, Lesser JR, et al. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation. 2011;124:40–47. doi: 10.1161/CIRCULATIONAHA.110.985812. [DOI] [PubMed] [Google Scholar]
- 19.Maron BJ, Maron MS, Wigle ED, Braunwald E. The 50-year history, controversy, and clinical implications of left ventricular outflow tract obstruction in hypertrophic cardiomyopathy from idiopathic hypertrophic subaortic stenosis to hypertrophic cardiomyopathy: from idiopathic hypertrophic subaortic stenosis to hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:191–200. doi: 10.1016/j.jacc.2008.11.069. [DOI] [PubMed] [Google Scholar]
- 20.Uberoi A, Stein R, Perez MV, Freeman J, Wheeler M, Dewey F, et al. Interpretation of the electrocardiogram of young athletes. Circulation. 2011;124:746–757. doi: 10.1161/CIRCULATIONAHA.110.013078. [DOI] [PubMed] [Google Scholar]
- 21.Baggish AL, Wood MJ. Athlete’s heart and cardiovascular care of the athlete: scientific and clinical update. Circulation. 2011;123:2723–2735. doi: 10.1161/CIRCULATIONAHA.110.981571. [DOI] [PubMed] [Google Scholar]
- 22.Arad M, Seidman JG, Seidman CE. Phenotypic diversity in hypertrophic cardiomyopathy. Hum Mol Genet. 2002;11:2499–2506. doi: 10.1093/hmg/11.20.2499. [DOI] [PubMed] [Google Scholar]
- 23.Brito D, Richard P, Isnard R, Pipa J, Komajda M, Madeira H. Familial hypertrophic cardiomyopathy: the same mutation, different prognosis. Comparison of two families with a long follow-up. Rev Port Cardiol. 2003;22:1445–1461. [PubMed] [Google Scholar]
- 24.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 25.Zou Y, Song L, Wang Z, Ma A, Liu T, Gu H, et al. Prevalence of idiopathic hypertrophic cardiomyopathy in China: a population-based echocardiographic analysis of 8080 adults. Am J Med. 2004;116:14–18. doi: 10.1016/j.amjmed.2003.05.009. [DOI] [PubMed] [Google Scholar]
- 26.Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet. 2006;43:347–352. doi: 10.1136/jmg.2005.036327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146:77–86. doi: 10.7326/0003-4819-146-2-200701160-00148. [DOI] [PubMed] [Google Scholar]
- 28.Maron BJ, Roberts WC, Arad M, Haas TS, Spirito P, Wright GB, et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA. 2009;301:1253–1259. doi: 10.1001/jama.2009.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bos JM, Maron BJ, Ackerman MJ, Haas TS, Sorajja P, Nishimura RA, et al. Role of family history of sudden death in risk stratification and prevention of sudden death with implantable defibrillators in hypertrophic cardiomyopathy. Am J Cardiol. 2010;106:1481–1486. doi: 10.1016/j.amjcard.2010.06.077. [DOI] [PubMed] [Google Scholar]
- 30.Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114:e385–484. doi: 10.1161/CIRCULATIONAHA.106.178233. [DOI] [PubMed] [Google Scholar]
- 31.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 32.Watkins H, Thierfelder L, Hwang DS, McKenna W, Seidman JG, Seidman CE. Sporadic hypertrophic cardiomyopathy due to de novo myosin mutations. J Clin Invest. 1992;90:1666–1671. doi: 10.1172/JCI116038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maron BJ, Seidman JG, Seidman CE. Proposal for contemporary screening strategies in families with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:2125–2132. doi: 10.1016/j.jacc.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 34.Ho CY, Seidman CE. A contemporary approach to hypertrophic cardiomyopathy. Circulation. 2006;113:e858–862. doi: 10.1161/CIRCULATIONAHA.105.591982. [DOI] [PubMed] [Google Scholar]
- 35.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 36.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 37.Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Sarcomeric genotyping in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:463–469. doi: 10.1016/S0025-6196(11)63196-0. [DOI] [PubMed] [Google Scholar]
- 38.Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010;55:1444–1453. doi: 10.1016/j.jacc.2009.11.062. [DOI] [PubMed] [Google Scholar]
- 39.Dewey FE, Pan S, Wheeler MT, Quake SR, Ashley EA. DNA sequencing: clinical applications of new DNA sequencing technologies. Circulation. 2012;125:931–944. doi: 10.1161/CIRCULATIONAHA.110.972828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lekanne Deprez RH, Muurling-Vlietman JJ, Hruda J, Baars MJ, Wijnaendts LC, Stolte-Dijkstra I, et al. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J Med Genet. 2006;43:829–832. doi: 10.1136/jmg.2005.040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman CE. Homozygous mutation in cardiac troponin T: implications for hypertrophic cardiomyopathy. Circulation. 2000;102:1950–1955. doi: 10.1161/01.cir.102.16.1950. [DOI] [PubMed] [Google Scholar]
- 42.Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. doi: 10.1016/j.jacc.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 43.Kelly M, Semsarian C. Multiple mutations in genetic cardiovascular disease: a marker of disease severity? Circ Cardiovasc Genet. 2009;2:182–190. doi: 10.1161/CIRCGENETICS.108.836478. [DOI] [PubMed] [Google Scholar]
- 44.Morales A, Cowan J, Dagua J, Hershberger RE. Family history: an essential tool for cardiovascular genetic medicine. Congest Heart Fail. 2008;14:37–45. doi: 10.1111/j.1751-7133.2008.08201.x. [DOI] [PubMed] [Google Scholar]
- 45.Erdmann J, Daehmlow S, Wischke S, Senyuva M, Werner U, Raible J, et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet. 2003;64:339–349. doi: 10.1034/j.1399-0004.2003.00151.x. [DOI] [PubMed] [Google Scholar]
- 46.Morner S, Richard P, Kazzam E, Hellman U, Hainque B, Schwartz K, et al. Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J Mol Cell Cardiol. 2003;35:841–849. doi: 10.1016/s0022-2828(03)00146-9. [DOI] [PubMed] [Google Scholar]
- 47.Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:739–744. doi: 10.1016/S0025-6196(11)61527-9. [DOI] [PubMed] [Google Scholar]
- 48.Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358:1899–1908. doi: 10.1056/NEJMoa075463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andersen PS, Havndrup O, Hougs L, Sorensen KM, Jensen M, Larsen LA, et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat. 2009;30:363–370. doi: 10.1002/humu.20862. [DOI] [PubMed] [Google Scholar]
- 50.Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation. 2010;122:2430–2440. doi: 10.1161/CIRCULATIONAHA.110.978924. discussion 2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201–211. doi: 10.1016/j.jacc.2009.02.075. [DOI] [PubMed] [Google Scholar]
- 52.Ashley EA, Butte AJ, Wheeler MT, Chen R, Klein TE, Dewey FE, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525–1535. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dewey FE, Chen R, Cordero SP, Ormond KE, Caleshu C, Karczewski KJ, et al. Phased whole-genome genetic risk in a family quartet using a major allele reference sequence. PLoS Genet. 2011;7:e1002280. doi: 10.1371/journal.pgen.1002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caleshu C, Day S, Rehm HL, Baxter S. Use and interpretation of genetic tests in cardiovascular genetics. Heart. 2010;96:1669–1675. doi: 10.1136/hrt.2009.190090. [DOI] [PubMed] [Google Scholar]
- 55.Cohen SA, McIlvried DE. Impact of computer-assisted data collection, evaluation and management on the cancer genetic counselor’s time providing patient care. Fam Cancer. 2011;10:381–389. doi: 10.1007/s10689-011-9417-2. [DOI] [PubMed] [Google Scholar]
- 56.McPherson E, Zaleski C, Benishek K, McCarty CA, Giampietro PF, Reynolds K, et al. Clinical genetics provider real-time workflow study. Genet Med. 2008;10:699–706. doi: 10.1097/gim.0b013e318182206f. [DOI] [PubMed] [Google Scholar]
- 57.Armel SR, McCuaig J, Finch A, Demsky R, Panzarella T, Murphy J, et al. The effectiveness of family history questionnaires in cancer genetic counseling. J Genet Couns. 2009;18:366–378. doi: 10.1007/s10897-009-9228-x. [DOI] [PubMed] [Google Scholar]
- 58.van der Roest WP, Pennings JM, Bakker M, van den Berg MP, van Tintelen JP. Family letters are an effective way to inform relatives about inherited cardiac disease. Am J Med Genet A. 2009;149A:357–363. doi: 10.1002/ajmg.a.32672. [DOI] [PubMed] [Google Scholar]
- 59.Murabito JM, Nam BH, D’Agostino RB, Sr, Lloyd-Jones DM, O’Donnell CJ, Wilson PW. Accuracy of offspring reports of parental cardiovascular disease history: the Framingham Offspring Study. Ann Intern Med. 2004;140:434–440. doi: 10.7326/0003-4819-140-6-200403160-00010. [DOI] [PubMed] [Google Scholar]
- 60.Hastrup JL, Hotchkiss AP, Johnson CA. Accuracy of knowledge of family history of cardiovascular disorders. Health Psychol. 1985;4:291–306. doi: 10.1037//0278-6133.4.4.291. [DOI] [PubMed] [Google Scholar]
- 61.Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med. 2010;12:370–375. doi: 10.1097/GIM.0b013e3181e15bd5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bensen JT, Liese AD, Rushing JT, Province M, Folsom AR, Rich SS, et al. Accuracy of proband reported family history: the NHLBI Family Heart Study (FHS) Genet Epidemiol. 1999;17:141–150. doi: 10.1002/(SICI)1098-2272(1999)17:2<141::AID-GEPI4>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 63.Iwarsson E, Malmgren H, Blennow E. Preimplantation genetic diagnosis: twenty years of practice. Semin Fetal Neonatal Med. 2011;16:74–80. doi: 10.1016/j.siny.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 64.Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, et al. Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation. 2010;122:1130–1133. doi: 10.1161/CIRCULATIONAHA.110.950089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IG, Schoen FJ, et al. An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–1359. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, et al. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–1020. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haim TE, Dowell C, Diamanti T, Scheuer J, Tardiff JC. Independent FHC-related cardiac troponin T mutations exhibit specific alterations in myocellular contractility and calcium kinetics. J Mol Cell Cardiol. 2007;42:1098–1110. doi: 10.1016/j.yjmcc.2007.03.906. [DOI] [PubMed] [Google Scholar]
- 68.Szczesna-Cordary D, Jones M, Moore JR, Watt J, Kerrick WG, Xu Y, et al. Myosin regulatory light chain E22K mutation results in decreased cardiac intracellular calcium and force transients. FASEB J. 2007;21:3974–3985. doi: 10.1096/fj.07-8630com. [DOI] [PubMed] [Google Scholar]
- 69.Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho CY. Hypertrophic cardiomyopathy. Heart Fail Clin. 2010;6:141–159. doi: 10.1016/j.hfc.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]