

Abstract
Multiple forms of plasticity are activated following reduced respiratory neural activity. For example, in ventilated rats, a central neural apnea elicits a rebound increase in phrenic and hypoglossal burst amplitude upon resumption of respiratory neural activity, forms of plasticity called inactivity-induced phrenic and hypoglossal motor facilitation (iPMF and iHMF), respectively. Here, we provide a conceptual framework for plasticity following reduced respiratory neural activity to guide future investigations. We review mechanisms giving rise to iPMF and iHMF, present new data suggesting that inactivity-induced plasticity is observed in inspiratory intercostals (iIMF) and point out gaps in our knowledge. We then survey conditions relevant to human health characterized by reduced respiratory neural activity and discuss evidence that inactivity-induced plasticity is elicited during these conditions. Understanding the physiological impact and circumstances in which inactivity-induced respiratory plasticity is elicited may yield novel insights into the treatment of disorders characterized by reductions in respiratory neural activity.
Keywords: Facilitation, Activity deprivation, Control of breathing, Respiratory Plasticity, Central apnea, Spinal injury
1. Introduction
The seemingly “simple” task of maintaining ventilation throughout life is not trivial. Motor output from multiple, diverse motor neuron pools must be coordinated to activate respiratory muscles in a precise sequence to achieve efficient and adequate ventilation. In addition, the respiratory control system must produce a stable rhythmic motor output, yet remain dynamic in order to respond to respiratory challenges (e.g., exercise, hypoxia, hypercapnia) and enable non-respiratory behaviors (e.g., airway clearance, speech). Remarkably, this task is accomplished despite ever-changing conditions that occur throughout life. The processes by which the respiratory control system maintains network stability and responsiveness in the presence of changing physiological demands and network properties are not well understood, but likely involve long-lasting adjustments in system performance (i.e., plasticity; Feldman et al., 2003; Mitchell and Johnson, 2003).
The respiratory control system exhibits a range of different mechanisms that give rise to respiratory plasticity (Bach and Mitchell, 1998; Baker et al., 2001; Devinney et al., 2013; Mahamed and Mitchell, 2008; Nichols et al., 2012; Peng et al., 2003; Strey et al., 2012; Tadjalli et al., 2010; Zhang et al., 2003). For many, a common theme is that recurrent respiratory challenges, such as hypoxia or hypercapnia, drive the expression of plasticity. Although these chemoreflex-driven forms of plasticity are associated with increases in respiratory neural activity, most are activity-independent and require neuromodulators for their induction and/or maintenance (Feldman et al., 2003; Mitchell and Johnson, 2003). By contrast, reports of activity-dependent plasticity in the control of breathing are generally rare (Johnson and Mitchell, 2002; McCrimmon et al., 1997; Zhou et al., 1997), and typically describe plasticity as a result of increased activity. However, in recent years, we have begun to appreciate that decreases in respiratory neural activity are perhaps a more potent trigger for plasticity than increases in activity (Baertsch and Baker-Herman, 2013; Castro-Moure and Goshgarian, 1996, 1997; Mahamed et al., 2011; Mantilla et al., 2007; Prakash et al., 1999; Strey et al., 2012; Tadjalli et al., 2010; Zhang et al., 2003). The apparent bias in the respiratory control network toward inactivity-(versus activity-) induced plasticity may not be surprising, since Hebbian-like plasticity may be inappropriate in a physiological system that routinely experiences recurrent increases in respiratory neural activity (e.g., during exercise) and the dire consequences that follow if the system fails to generate sufficient motor output.
One key question is: what is the physiological role of inactivity-induced plasticity? Unfortunately, the answer is not currently known. However, various conditions during health and disease are accompanied by reduced respiratory neural activity. For example, healthy individuals experience reductions in respiratory neural output during behaviors such as sleep (Javaheri and Dempsey, 2013; Uliel et al., 2004), voluntary diving (Dutschmann and Paton, 2002; Gooden, 1994) or at altitude (Berssenbrugge et al., 1984). Periods of reduced or absent respiratory motor output, may accompany a variety of other conditions, such as prematurity (Gaultier and Gallego, 2005) and aging (Ancoli-Israel et al., 1987), or may be a secondary consequence of various pathologies, including genetic disorders (Goridis et al., 2010), neurodegenerative diseases (Gaig and Iranzo, 2012) or heart failure (Yumino and Bradley, 2008). Finally, reduced respiratory neural activity is often experienced following spinal cord injury (Strakowski et al., 2007) and by patients requiring ventilatory support (Epstein, 2011; Tobin, 2001). The short- and long-term consequences of reduced neural activity in respiratory motor pools on the control of breathing in any context is not well understood.
Here, we review evidence that reduced respiratory neural activity elicits unique mechanisms of plasticity within respiratory motor circuits. To build context, we survey conditions and disorders relevant to human health that are characterized by reduced respiratory neural activity. We provide evidence that these disorders elicit endogenous mechanisms of compensatory plasticity (or whether such evidence is lacking), and if inactivity-induced plasticity may be adaptive or maladaptive in these situations. This review is not intended to be a comprehensive catalogue of conditions/disorders associated with reduced respiratory neural activity; instead we aim to identify gaps in our knowledge in basic and translational research to guide development of future lines of investigation.
2. Does-reduced respiratory neural activity elicit plasticity?
An emerging principle of neuroscience is that neural networks sense and respond to prolonged changes in activity through local homeostatic mechanisms to maintain a “set-point” level of neuronal activity (Turrigiano, 2008). As such, prolonged changes in activity that result in a deviation from the “set-point” are met with mechanisms of plasticity that attempt to restore target activity levels through a variety of negative feedback mechanisms. This homeostatic synaptic plasticity is generally bi-directional and requires hours to days of altered activity levels to induce. However, the respiratory system has unique demands that suggest a bias toward rapid induction of plasticity, particularly in response to hypoactivity, may be appropriate. In contrast to many other neural systems, it is imperative that the respiratory control system remain highly active to produce a life-sustaining, rhythmic motor output. For example, the diaphragm has a duty cycle of ~32–44% (Kong and Berger, 1986; Sieck et al., 2012), while muscles of the hind limb (e.g. soleus and extensor digitorum longus muscles) have duty cycles ranging from ~2 to 14% (Hensbergen and Kernell, 1997). Thus, the phrenic motor pool may be exquisitely tuned to a high level of activity and uniquely sensitive to a lack thereof. Here we summarize available evidence that reduced respiratory neural activity induces plasticity of inspiratory motor output and discuss the underlying cellular mechanisms. One major purpose of this review is to provide a conceptual framework in which to interpret plasticity following reduced respiratory neural activity in non-disease and disease states.
2.1. Inactivity-induced plasticity following reduced respiratory neural activity
To our knowledge, one of the first anecdotal reports of increased respiratory motor output following reduced respiratory neural activity was reported by Budzinska and colleagues (1985), who focally cooled regions of the ventral medulla to create a central apnea in anesthetized, ventilated cats. The authors remarked that upon rewarming after cold block-induced apnea, “the return of rhythmic activity after apnoea sometimes showed a ‘rebound’ with an enhanced [phrenic] peak inspiratory activity relative to the pre-block control breaths.” The authors did not quantify the frequency of occurrence nor the magnitude of phrenic facilitation following cold block-induced apnea (Budzińska et al., 1985). In a subsequent study, Castro-Moure and Goshgarian (1996) focally cooled the ventral spinal cord at C2 to block axon conduction unilaterally in descending tracts to ipsilateral phrenic motor neurons in anesthetized rats in order to mimic disruption of descending respiratory drive associated with spinal injury. As expected, C2 cold block silenced ipsilateral diaphragm EMG activity; however, to the authors’ surprise, upon reversal of the cold block and restoration of axon conduction, EMG activity in the hemidiaphragm ipsilateral (but not contralateral) to cold block was significantly increased relative to baseline and contralateral diaphragm EMG activity (Castro-Moure and Goshgarian, 1996). The duration of increased ipsilateral diaphragm EMG activity following cold block was not reported, nor could it be differentiated whether observed effects were due to central neural versus diaphragm neuromuscular junction plasticity. However, profound morphological changes were observed within the ipsilateral phrenic motor nucleus, including an increase in the number of synapses onto phrenic motor neurons (Castro-Moure and Goshgarian, 1997), consistent with the interpretation that removal of respiratory-related inputs onto phrenic motor neurons elicited local mechanisms of plasticity within the phrenic motor pool. Similar findings may have been observed by Webber and Pleschka (1984) and Martin et al. (1994). In representative traces of phrenic (Webber and Pleschka, 1984) and hypoglossal (Martin et al., 1994) motor output before, during and after inhibition of respiratory neural activity using focal cooling of the C2 spinal cord and ventral medullary surface, respectively, increased respiratory motor output was apparent following resumption of respiratory neural activity, although these authors did not remark upon nor quantify the extent of the increases in phrenic or hypoglossal burst amplitude. Nevertheless, collectively, these early reports led to the interesting suggestion that reduced respiratory-related inputs to phrenic and hypoglossal motor neurons elicits forms of plasticity that lead to enhanced respiratory motor output.
To better understand the long-lasting impact of reduced respiratory neural activity on phrenic motor output, we exposed anesthetized, ventilated rats to a prolonged (30 min) central neural apnea while monitoring phrenic neural activity (Mahamed et al., 2011). Following resumption of respiratory neural activity post-central neural apnea, a long-lasting, rebound increase in phrenic burst amplitude was apparent. Since multiple methods (with different mechanisms of action) led to similar increases in phrenic burst amplitude, we suggested that increased phrenic burst amplitude post-neural apnea was due to a common factor: reduced respiratory neural activity (Mahamed et al., 2011), and termed this form of plasticity inactivity-induced phrenic motor facilitation (iPMF; Mahamed et al., 2011). Later studies revealed that inactivity-induced plasticity is also expressed in hypoglossal motor output (Baker-Herman and Strey, 2011), albeit with a more transient pattern of expression. Fig. 1 depicts a meta-analysis of phrenic and hypoglossal nerve responses to neural apnea from multiple studies in our lab (Baertsch and Baker-Herman, 2013; Baker-Herman and Strey, 2011; Mahamed et al., 2011; Strey et al., 2012). Following a prolonged neural apnea, iPMF manifests as a long-term (>60 min) increase in phrenic amplitude (~60–80% baseline) and is associated with a proportional increase in the phrenic burst amplitude response to a hypercapnic challenge (Baertsch and Baker-Herman, 2013). By contrast, facilitation of hypoglossal nerve burst amplitude following a prolonged central neural apnea is more modest (~30–40%) and transient, returning to baseline levels within ~30 min (Baertsch and Baker-Herman, 2013; Baker-Herman and Strey, 2011). In addition to increases in burst amplitude, central neural apnea also elicits increased respiratory burst frequency (Mahamed et al., 2011; Baker-Herman and Strey, 2011; Baertsch and Baker-Herman, 2013), albeit this inactivity-induced frequency facilitation is small and has a limited time course (~15 min).
Fig. 1.

Differential expression of inactivity-induced respiratory plasticity. (A) Representative compressed and integrated phrenic (top) and hypoglossal (bottom) neurograms and inspiratory intercostal EMG activity (middle) before, during and for 60 min following a 30 min hyperventilation-induced neural apnea, illustrating a prolonged increase in phrenic burst amplitude and a transient increase in hypoglossal burst amplitude and inspiratory intercostal EMG activity following resumption of respiratory neural activity, indicating iPMF, iHMF and iIMF, respectively. (B) Average change in phrenic, hypoglossal and inspiratory intercostal EMG amplitude from baseline for 60 min following resumption of respiratory neural activity after a central neural apnea. A prolonged (>60 min) facilitation of phrenic nerve burst amplitude (diamonds) is apparent following resumption of respiratory neural activity that is significantly increased relative to phrenic time controls receiving the same surgical preparation, but no neural apnea. By contrast, hypoglossal nerve burst amplitude (squares) and intercostal EMG activity (circles) exhibit only transient (15 min) increases in inspiratory burst activity following neural apnea, relative to hypoglossal and intercostal time controls. For clarity, time controls are not shown. These data suggest that iPMF is long-lasting, whereas iHMF and iIMF are transient. * significantly increased from phrenic time controls; # significantly increased from hypoglossal time controls; Φ significantly increased from intercostal time controls (p < 0.05).
Here, we present new data from experiments testing the response of inspiratory intercostal muscles to prolonged central neural apnea using procedures similar to those described in detail elsewhere (Baertsch and Baker-Herman, 2013; Mahamed et al., 2011; Strey et al., 2012). All experiments were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin—Madison. Briefly, Harlan Sprague Dawley rats (colony 217) were urethane anesthetized, vagotomized and mechanically ventilated. EMG electrodes were placed in the external intercostal (T2) muscle. Stable baseline intercostal EMG activity was established at an ETCO2 ~45 mmHg and respiratory frequency ~45 bpm prior to a 30 min hyperventilation-induced central neural apnea (n = 10). Following reversal of neural apnea, intercostal EMG activity was monitored for 1 h. In a subset of rats, the cut left phrenic nerve was also recorded to ensure that iPMF was expressed under these conditions. In separate rats, baseline parameters were maintained for 90 min (no neural apnea) to control for any time-dependent changes in external intercostal EMG activity (time controls; n = 3). Blood samples were taken at baseline and 5, 15, 30, and 60 min following resumption of inspiratory intercostal activity to ensure PaCO2, PaO2, pH and SBEc post-neural apnea were maintained at baseline levels. Inspiratory intercostal EMG activity was rectified and integrated (PowerLab data acquisition and LabChart 7.0 software); the peak amplitude of integrated intercostal EMG activity post-neural apnea (or equivalent duration in time controls) was expressed as a percentage change from baseline (%baseline), whereas burst frequency was expressed as an absolute change from baseline (Δbaseline).
Following restoration of central respiratory neural activity, inspiratory-related external intercostal EMG amplitude was significantly increased for up to 15 min, relative to baseline and time controls (neural apnea: 64 ± 11; time controls: 5 ± 6%baseline; p < 0.05; Fig. 1), indicating inactivity-induced intercostal motor facilitation (iIMF). Although inspiratory intercostal EMG activity 30 min after restoration of respiratory neural activity was significantly increased relative to baseline (46 ± 14%baseline; p < 0.05), it was no longer significantly different than time controls (3 ± 12%baseline; p > 0.05). By 60 min following resumption of respiratory neural activity, inspiratory intercostal EMG activity was not significantly different than baseline or time controls (neural apnea: 25 ± 18, time controls: 3 ± 10%baseline; p > 0.05). Similar to previous reports (Baertsch and Baker-Herman, 2013; Baker-Herman and Strey, 2011; Mahamed et al., 2011), a transient increase in intercostal EMG burst frequency was observed for up to 15 min following neural apnea compared to baseline and time controls (neural apnea: 7 ± 1; time controls: −2 ± 0 Δbaseline; p < 0.05). At 30 min post-resumption of respiratory neural activity, intercostal EMG burst frequency was significantly increased relative to baseline (5 ± 1 Δbaseline; p < 0.05), but was not significantly different than time controls (−2 ± 2 Δbaseline; p > 0.05). At 60 min post-resumption of respiratory neural activity, EMG burst frequency was not significantly different than baseline or time controls (neural apnea: 4 ± 2; time controls: −2 ± 1 Δbaseline; p > 0.05). Collectively, these data suggest that the inspiratory intercostal response to prolonged neural apnea resembles the magnitude of phrenic amplitude facilitation observed immediately after the resumption of neural activity (5 and 15 min), but has a transient time-course similar to hypoglossal nerve output (Fig. 1), and confirms the transient frequency facilitation post-neural apnea reported in previous studies (Baertsch and Baker-Herman, 2013; Baker-Herman and Strey, 2011; Mahamed et al., 2011). The mechanisms by which inactivity-induced facilitation of inspiratory burst amplitude is transient in some motor pools and long-lasting in others is unknown; however, the “activity profile” of a motor pool may be a major determinant of the rapidity of induction and duration of plasticity elicited in response to reduced respiratory neural activity. Indeed, long-lasting periods of reduced inspiratory activity may be more common for hypoglossal and some intercostal motor pools (De Troyer et al., 2005; Pagliardini et al., 2012); as such, it may be expected that inactivity-induced plasticity has a different manifestation and/or time domain in these motor pools versus the phrenic.
Clearly, a prolonged central neural apnea lasting 30 min would rarely be encountered in any but the most artificial situation (ventilated animals). As such, this model was not intended to mimic physiological/pathophysiological situations in which a central apnea would be experienced; instead, these studies demonstrated a principle of respiratory control: Reduced central respiratory drive elicits plasticity in respiratory motor output. In order to provide a more “realistic” (albeit still somewhat artificial) context to iPMF, recent studies demonstrated that iPMF is not limited to prolonged central apnea, but is also elicited following intermittent patterns of brief central neural apnea (Baertsch and Baker-Herman, 2013). Anesthetized and ventilated rats were exposed to brief intermittent neural apnea (5, 1.5 min episodes separated by 5 min recovery); following resumption of respiratory neural activity, a sustained (>60 min) increase in phrenic burst amplitude (~60–80% baseline) was apparent, which was phenotypically similar to iPMF observed following a single prolonged neural apnea (Baertsch and Baker-Herman, 2013). In contrast, exposure to a single brief “massed” neural apnea of a similar cumulative duration (7.5 min), did not alter phrenic burst amplitude at any time point following resumption of respiratory neural activity. No changes in hypoglossal burst amplitude were observed following brief intermittent or brief massed neural apnea (Baertsch and Baker-Herman, 2013). Similarly, episodic central apneas (3, 5 min episodes separated by 5 min recovery) induced via high frequency vagal stimulation in ventilated rats elicits a phenotypically similar augmentation of phrenic burst amplitude (Zhang et al., 2003); although the authors refer to this form of plasticity as long-term facilitation, we hypothesize that it represents a variation of inactivity-induced plasticity. Collectively, these reports suggest that although a prolonged central neural apnea is sufficient to elicit iPMF (Baker-Herman and Strey, 2011; Mahamed et al., 2011; Strey et al., 2012), iPMF, but not iHMF, is more efficiently induced by recurrent (intermittent) neural apnea versus a sustained neural apnea of similar cumulative duration.
One key question is: Where in the CNS is reduced respiratory neural activity sensed and responded to? Central neural apnea results in reduced respiratory neural activity throughout the neuraxis; however, we hypothesized that local mechanisms near respiratory motor neurons sense and respond to reduced respiratory-related inputs and give rise to burst amplitude facilitation (i.e., iPMF, iHMF or iIMF in the respective motor pools), whereas mechanisms operating within brainstem respiratory rhythm generating networks give rise to neural apnea-induced increases in frequency (Baertsch and Baker-Herman, 2013; Mahamed et al., 2011; Strey et al., 2012). Supportive of this hypothesis, iPMF (but not burst frequency facilitation) is impaired by application of pharmacological inhibitors that block cellular pathways leading to iPMF to regions of the spinal cord associated with the phrenic motor nucleus (Strey et al., 2012). Further, disruption of spinal synaptic inputs to phrenic motor neurons via C2 axon conduction block (in the absence of noticeable changes in central respiratory drive) elicits iPMF (Castro-Moure and Goshgarian, 1996, 1997; Strey and Baker-Herman, 2011) and morphological plasticity within the phrenic motor pool (Castro-Moure and Goshgarian, 1997), but does not elicit frequency facilitation. Notably, a complete loss of ipsilateral phrenic motor output during axon conduction block is not required for iPMF to develop, suggesting that reductions in respiratory neural activity (versus complete inactivity) are sufficient to elicit these mechanisms (Strey and Baker-Herman, 2011).
Fig. 2 depicts our working model of cellular mechanisms giving rise to iPMF. As discussed above, we hypothesize that iPMF is the result of mechanisms operating specifically within the phrenic motor pool (Baker-Herman and Strey, 2011; Mahamed et al., 2011; Strey et al., 2012). iPMF consists of at least two mechanistically distinct phases: (1) an early, labile phase that requires activity of the atypical protein kinase C (aPKC) isoform PKCζ and/or PKCι/λ in spinal regions associated with the phrenic motor nucleus to transition to (2) a late, long-lasting increase in phrenic burst amplitude (Strey et al., 2012). Atypical PKCs include the isozymes PKCζ, PKCι/λ and PKMζ, and represent one of three subfamilies of PKCs (classical, novel and atypical) that are classified based on structure and requirement for co-activators (Reyland, 2009). Consistent with a key role for spinal PKCζ and/or PKCι/λ (referred to here as PKCζ/ι for clarity), early, but not late, iPMF is associated with an increased interaction between PKCζ/ι and the scaffolding protein ZIP (PKCζ interacting protein)/p62 in ventral spinal regions (C3–C5) associated with the phrenic motor pool; an interaction which may confer context specificity to PKCζ/ι activity. Upstream mechanisms that promote formation of the PKCζ/ι-ZIP/p62 signaling cassette are thought to include release of tumor necrosis factor alpha (TNFα) and subsequent activation of TNF receptors on phrenic motor neurons. Indeed, spinal TNFα signaling is necessary for iPMF expression, and exogenous TNFα induces an aPKC dependent increase in phrenic burst amplitude (Broytman et al., 2013). Mechanisms giving rise to inactivity-induced frequency facilitation are unknown.
Fig. 2.
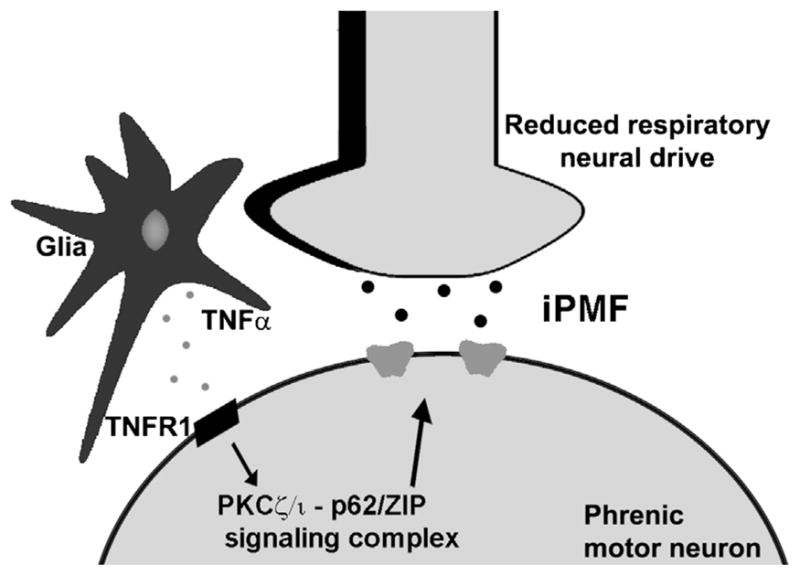
Current working model of iPMF. We hypothesize that local spinal mechanisms operating within the phrenic motor pool “sense” and “respond” to reduced bulbospinal respiratory inputs by local release of TNFα in/near the phrenic motor pool. Subsequent activation of TNFα receptors promotes the formation of the aPKCζ/ι -ZIP/p62 signalling cassette in phrenic motor neurons. This stimulus specific signaling cascade increases the synaptic strength and induces iPMF. The pathways downstream of the aPKCζ/ι -ZIP/p62 signalling cassette leading to iPMF are unknown. Similar mechanisms are proposed to occur within hypoglossal and inspiratory intercostal motor pools to give rise to iHMF and iIMF, respectively.
2.2. Gaps in our knowledge of inactivity-induced plasticity
Although we have made considerable progress in our understanding of inactivity-induced plasticity, many questions remain unanswered. For example, what cell type/s sense reduced respiratory neural activity, and what signal are these cells sensing? Are these “sensors” also the cell types that release TNFα to elicit inactivity-induced plasticity? Does inactivity-induced plasticity reflect mechanisms occurring largely within respiratory motor neurons or as part of a cellular network within or near motor nuclei? Similarly, do all phrenic motor neurons express iPMF, or are there “sub-pools” of phrenic motor neurons particularly sensitive to reduced neural activity (e.g., those involved in eupnic breathing vs. those recruited during respiratory challenges)? Is inactivity-induced plasticity in other respiratory motor pools mechanistically similar to iPMF? What mechanisms underlie differential expression of inactivity-induced facilitation among different motor pools? Do genetic or epigenetic factors influence the expression or absence of iPMF and related plasticity?
Important and challenging questions regarding the physiological role for inactivity-induced plasticity in the control of breathing also provide motivation for future studies. For example, what is the consequence of differential plasticity among inspiratory motor pools in the sculpting of a breath? In particular, what are the implications for airway stability with a prolonged facilitation of phrenic motor output in the absence of hypoglossal facilitation? What, if any, role does inactivity-induced plasticity play in physiological or pathophysiological conditions relevant to human health? How might an understanding of the mechanisms of inactivity-induced plasticity be used to manipulate physiological outcomes? Since virtually nothing is known concerning the applicability of iPMF to the control of breathing in health or disease, we will begin forming a necessary conceptual framework by discussing circumstances where reduced respiratory neural activity may be experienced.
3. Conditions/disorders associated with reduced respiratory neural activity
Although the respiratory control system is remarkably reliable for most individuals, it is susceptible to transient and recurrent or prolonged periods of reduced respiratory neural activity in a variety of physiological and pathophysiological situations. Here we briefly survey conditions and disorders of relevance to human health to outline situations in which reduced respiratory neural activity may be experienced.
3.1. Disruptions in brainstem respiratory neural activity
Central apnea may occur during normal physiological or pathophysiological processes, typically during sleep. Central sleep apnea (CSA) is characterized by recurrent episodes of absent or markedly reduced (hypopnea) respiratory neural output, whereas obstructive sleep apnea (OSA) is characterized by continued (futile) central neural output in the presence of a closed or reduced airway. In many cases, both CSA and OSA often co-exist (“mixed” apnea) in the same patient (Xie et al., 2011; Javaheri and Dempsey, 2013) or CSA may develop during treatment of OSA (“complex” sleep apnea; Dernaika et al., 2007; Lehman et al., 2007; Morgenthaler et al., 2006) for reasons that are not clearly understood.
During sleep, a number of physiological changes increase the propensity for ventilatory instability, even in otherwise healthy individuals (Eckert et al., 2007; Javaheri, 2010; Javaheri and Dempsey, 2013; Malhotra and Owens, 2010). For example, removal of the so-called wakefulness drive to breathe reveals a sensitive CO2-dependent apneic threshold (Dempsey et al., 2012; Skatrud and Dempsey, 1983). Thus, periodic cessation of inspiratory efforts will occur when PaCO2 drops below a critical level (Dempsey et al., 2012; Pack, 2011). Indeed, during sleep, even small decreases in PaCO2 (~2–5 mmHg) can result in apnea (Henke et al., 1988; Meza et al., 1998; Skatrud and Dempsey, 1983). During sleep, PaCO2 rises ~5 mmHg above wakefulness levels (Malhotra and Owens, 2010; Pack, 2011). During the transition from wakefulness to sleep in individuals with a CO2 apneic threshold close to eupnic PaCO2, a central apnea may result if the wakefulness drive to breathe is lost rapidly at sleep onset before sleep-induced reductions in ventilation occur and the establishment of the sleep PaCO2 setpoint (Javaheri and Dempsey, 2013; Leung et al., 2012). Further, a central apnea may result following a transient arousal from sleep (Trinder et al., 1992), which represents a temporary return to wakefulness. The sleeping PaCO2 represents a relative hypercapnia to the aroused brain, which then stimulates breathing to lower PaCO2 to awake eupnic levels (Eckert et al., 2007; Malhotra and Owens, 2010); if hyperventilation results in a drop of PaCO2 below the apneic threshold, then upon the resumption of sleep, the relative hypocapnia to the now sleeping brain results in a central apnea. The frequency of central apnea during sleep in an otherwise healthy individual is generally minimal; indeed, a frequency of <5 CSA events/hr is considered to be within a clinically normal range (Javaheri, 2010).
The prevalence of central apnea during sleep in the general population is not entirely clear, particularly when considering the clinically normal range. Breathing pattern instability during sleep with central apneas of short duration is characteristic of the normal, healthy infant breathing pattern (Kahn et al., 1982; Hoppenbrouwers et al., 1977; Ng and Chan, 2013). Indeed, periodic breathing is apparent in 78% of infants between 0 and 2 weeks of age and declines to 29% by 39–52 weeks of age, although the apneic events are typically <10 s in duration and occupy <1% of the sleep time (Kelly et al., 1985). However, premature infants are particularly susceptible to central apnea and periodic breathing (Martin et al., 2004), with nearly 100% of preterm infants exhibiting episodes of periodic breathing that are longer in duration and more frequent than full-term infants (Henderson-Smart, 1981; Glotzbach et al., 1989). Although the frequency of central apnea decreases with increasing gestational age, central apnea is also relatively common in children (Scholle et al., 2011), with 30–40% of children experiencing a total of 1 to 7 central apneic events lasting >10 s during sleep (Marcus et al., 1992; Uliel et al., 2004). The frequency of such events appears to diminish in adolescence (Tapia et al., 2008). As an individual ages, the frequency of central apnea in sleep increases (Chowdhuri and Badr, 2010; Bixler et al., 1998; Carskadon and Dement, 1981). For example, in a large study of men aged 20–100 yr., Bixler and colleagues report that 12.1% of subjects over the age of 65 had a central apnea index ≥2.5, whereas only 1.7% of middle-aged subjects and no subjects in the young age group had a central apnea index ≥2.5 (Bixler et al., 1998).
Although central apnea may occur during sleep in healthy individuals, the frequency of such events is typically minimal and not considered to be clinically relevant (Javaheri, 2010). However, the prevalence and frequency of CSA increases during certain conditions. For example, many individuals will experience periodic breathing and frequent central apneas upon ascent to high altitude. At altitude, the ventilatory response to hypoxia (HVR) lowers PaCO2 (Pack, 2011), thereby narrowing the eupnic PaCO2—apneic threshold difference (i.e., “CO2 reserve”) and creating breathing instability during sleep (Bloch et al., 2010; Kohler et al., 2008; Berssenbrugge et al., 1984). Typically, individuals develop a periodic breathing pattern characterized by short crescendo–decrescendo cycles (15–30 s) of hyperventilation alternating with periods of apnea/hypopnea (Pack, 2011). At altitudes >4500 m, an apnea/hypopnea index of ~60 h−1 during sleep is not uncommon (Burgess et al., 2004; Nussbaumer-Ochsner et al., 2012), suggesting considerable cumulative time spent with low (or zero) respiratory neural activity. Over several days at altitude, the magnitude of CSA is reduced in many individuals (Berssenbrugge et al., 1984).
An increase in the prevalence or frequency of CSA may occur during some pathological conditions. For example, approximately ~30–40% of patients with heart failure (HF) develop frequent episodes of CSA (Javaheri et al., 1998; MacDonald et al., 2008; Sin et al., 1999). Indeed, HF is the most common cause of clinically diagnosed CSA in the general population (Javaheri, 2010; Javaheri and Dempsey, 2013). CSA in HF patients is characterized by a long (~60–90 s) crescendo-decrescendo pattern of breathing interspersed with central apneas lasting ~10–40 s, a form of periodic breathing known as Cheyne–Stokes Respiration (CSR; Hall et al., 1996; Pack, 2011). Enhanced chemosensitivity and chronic hyperventilation during sleep in HF patients reduces sleeping PaCO2 levels (Javaheri, 2010; Tkacova et al., 1997; Yumino and Bradley, 2008; Xie et al., 2002), bringing eupnic PaCO2 closer to the apneic threshold and predisposing HF patients to central apnea. Although reports vary, patients with HF and CSR on average have an AHI ≥15 h−1 with more than 50% of central origin (Wang et al., 2007). Other conditions that may be associated with an increased incidence of central apnea include patients with idiopathic CSA (Bradley et al., 1986; Bradley and Phillipson, 1992), endocrine disorders (Grunstein et al., 1991; Millman et al., 1983; Rosenow et al., 1998), neuromuscular disorders (Chokroverty et al., 1978, 1984; Ferguson et al., 1996; Gaig and Iranzo, 2012; Glass et al., 2006; Labanowski et al., 1996; Santos et al., 2003), congenital central hypoventilation syndrome (CCHS; Goridis et al., 2010; Fleming et al., 1980; Weese-Mayer et al., 2010) and Rett syndrome (Weese-Mayer et al., 2008; Katz et al., 2009).
3.2. Disruption in inputs to spinal respiratory motor neurons
Reduced respiratory neural activity may also be caused by disruption of central neural drive in transit to respiratory motor neurons as result of injuries to the spinal cord. More than half (52% since 2010) of all spinal cord injuries (SCIs) occur in the cervical spinal region while the remaining injuries are localized to thoracic, lumbar or sacral regions (NSCISC, 2013). In contrast to central neural apnea, SCIs reduce respiratory neural activity in spinal respiratory motor neurons caudal to injury, while brainstem respiratory centers continue to generate normal (or even elevated) neural drive to breathe. In addition to disrupting bulbospinal respiratory axons in white matter tracts, spinal cord injury typically disrupts gray matter continuity and damages propriospinal interneurons and/or respiratory motor neurons in the cervical spinal cord (Lane et al., 2009). Depending on the location and severity of the injury, the level of reduced respiratory neural activity varies. For example, high cervical SCIs interrupt the descending excitatory drive to phrenic motor neurons innervating the diaphragm (Golder et al., 2011; Lane et al., 2012), while injuries below cervical regions impair breathing by damaging axons innervating accessory inspiratory muscles or muscles mainly involved in expiration (Gorini et al., 2000).
3.3. Artificial reductions in respiratory neural activity
Mechanical ventilation is a life-saving treatment for hundreds of thousands of critically ill patients each year. Most modern modes of mechanical ventilation attempt to synchronize the quantity, timing and pattern of a patient’s neural drive with the ventilator, thereby avoiding deleterious effects associated with removing patient respiratory efforts altogether (Epstein, 2011). Unfortunately, attempts to achieve synchronization are often unsuccessful (de Wit et al., 2009a, b; Epstein, 2011; Thille et al., 2006; Tobin, 2001), often due to ineffective ventilator triggering, auto-triggering or poor correspondence in the flow or timing of the patients neural breath and the ventilator breath (de Wit et al., 2009a, b; Kondili et al., 2007; Mellott et al., 2009; Pierson, 2011). In addition, many ventilated patients are at risk for over-assistance, which suppresses or diminishes the patient’s neural drive (Colombo et al., 2008; Delisle et al., 2011; Meza et al., 1998; Parthasarathy and Tobin, 2002). Thus, many patients receiving ventilatory support experience reduced central respiratory neural activity while on the ventilator (de Wit et al., 2009a, b; Epstein, 2011; Kondili et al., 2007; Younes, 2006).
In most patients, mechanical ventilation may be discontinued abruptly (Esteban et al., 1995, 2000). However, up to 30% of mechanically ventilated patients have difficulty resuming breathing on their own, even after their underlying disorder has been resolved (Epstein, 2009). Indeed, up to 40% of the time spent on the ventilator is associated with the weaning process (Esteban et al., 2008). Ventilation strategies that suppress spontaneous breathing are associated with longer duration of mechanical ventilation (Chao et al., 1997; de Wit et al., 2009b; Putensen et al., 2006; Thille et al., 2006); however, even patients that maintain spontaneous breathing for the most part, but have a high level of over-assistance or ventilator asynchrony have a longer duration of weaning from mechanical ventilation than their counterparts that do not (Chao et al., 1997; de Wit et al., 2009b; Thille et al., 2006). The pathophysiology underlying weaning failure is multifactorial and likely vary from patient to patient. Much research has been focused on the impact of reduced diaphragm muscle activity during mechanical ventilation on subsequent attempts to breathe spontaneously since many weaning failure patients exhibit a reduced ability to convert central respiratory drive into an effective breath (Liu et al., 2012), in large part due to muscle weakness (Anzueto et al., 1997; McClung et al., 2007; Powers et al., 2009; Shanely et al., 2002). However, the long-lasting impact of reduced respiratory neural activity during mechanical ventilation on a patient’s subsequent attempts to resume spontaneous respiratory efforts are completely unknown.
4. Is there evidence for plasticity in conditions and disorders associated with reduced respiratory neural activity?
Although recurrent or prolonged disruptions in respiratory neural activity are experienced in many conditions, little is known regarding the consequences of this reduced respiratory neural activity. Other than in reduced animal models, little direct evidence is available concerning links between repetitive central neural apneas or prolonged reduced respiratory activity with respiratory neuroplasticity. We suggest that this lack of evidence stems from a lack of systematic investigation since the first description of inactivity-induced respiratory motor neuroplasticity was only in 2011 (Mahamed et al., 2011).
Review of the literature reveals suggestive evidence that mechanically ventilated patients exhibit respiratory behaviors consistent with inactivity-induced plasticity. For example, central respiratory drive is higher than normal in many mechanically ventilated patients, which is apparent almost immediately after being disconnected from the ventilator (Laghi, 2005; Nemer et al., 2009; Tobin et al., 2009). Central respiratory “drive” is often approximated by airway occlusion pressure during the first 0.1 s of a breath (P0.1); since it is measured at zero flow, airway occlusion pressure is independent of respiratory system compliance and resistance (Whitelaw et al., 1975). Although both weaning failure and weaning success patients exhibit increased P0.1 relative to normal values, P0.1 is often higher at the onset and increases progressively in patients that cannot resume spontaneous, independent breathing (Herrera et al., 1985; Hilbert et al., 1998; Nemer et al., 2009; Perrigault et al., 1999; Sassoon and Mahutte, 1993; Tobin et al., 2009). Although the progressive increase in P0.1 in weaning failure patients may be due to deteriorating gas exchange secondary to worsening respiratory mechanics (Jubran and Tobin, 1997a; Tobin, 2001), the initial increase in P0.1 that is apparent immediately upon the discontinuation of mechanical ventilation is often apparent prior to any measurable deterioration in respiratory mechanics (Jubran and Tobin, 1997b; Tobin, 2001). Consistent with increased central respiratory output, many mechanically ventilated patients exhibit elevated EMG activity in the diaphragm and accessory inspiratory muscles within minutes of being disconnected from the ventilator, particularly in patients that ultimately fail the weaning trial (Dres et al., 2012; Liu et al., 2012). Although a multitude of factors may contribute to increased respiratory neuromuscular drive in mechanically ventilated patients, the potential role of central neural plasticity in reconfiguring respiratory motor output in response to prolonged reductions in respiratory neural activity experienced while on the ventilator should be considered.
To date, the best evidence that reduced respiratory neural activity elicits neuroplasticity is following SCI. Multiple reports suggest that following cervical SCI, diaphragmatic function spontaneously improves over time in humans (Axen et al., 1985; McKinley, 1996; Oo et al., 1999; Strakowski et al., 2007) and rodents (Baussart et al., 2006; El-Bohy et al., 1998; Fuller et al., 2003, 2006, 2008; Golder et al., 2011; Golder and Mitchell, 2005; Lane et al., 2009; Vinit et al., 2007). The recovery of phrenic output and diaphragm activity is associated with a functional recovery of breathing (Strakowski et al., 2007). Since regrowth of damaged axons across the spinal lesion is limited (Sharma et al., 2012), return of phrenic activity following SCI is likely due to endogenous mechanisms of compensatory plasticity (Goshgarian, 2003). Remodeling of spinal circuits post-injury may restore ipsilateral phrenic motor output by recruiting latent contralateral pathways (the “crossed phrenic phenomenon”; Goshgarian, 2009; Lane et al., 2009; Darlot et al., 2012) or strengthening spared ipsilateral pathways (Vinit et al., 2008; Vinit and Kastner, 2009). However, the cellular mechanisms giving rise to spontaneous functional recovery after SCI are unknown. Although spinal injury causes many changes in the spinal microenvironment, including local tissue damage, inflammation and ischemia (Hausmann, 2003), we propose that reduced synaptic inputs to the phrenic motor pool have the potential to play a prominent role in inducing spontaneous functional recovery by strengthening spared pathways via mechanisms similar to iPMF. Indeed, minutes after spinal injury, TNFα is increased caudal to the site of injury (Pineau and Lacroix, 2007; Wang et al., 1996), triggering a rapid increase in synaptic AMPA receptor expression in motor neurons caudal to injury (Ferguson et al., 2008; Yin et al., 2012). In addition, aPKC activity within the ipsilateral phrenic motor pool is increased shortly after cervical spinal hemisection, an effect that is still observed 28 days post-SCI (Guenther et al., 2012). Consistent with a key role for withdrawal of neural inputs inducing spontaneous recovery, a 4 h disruption in descending inputs to phrenic motor neurons via unilateral cold block causes profound morphological changes within the phrenic motor pool (Castro-Moure and Goshgarian, 1997), similar to those observed 2 h after SCI (Sperry and Goshgarian, 1993). These inactivity-induced morphological changes within the phrenic motor pool are associated with enhanced ipsilateral diaphragm EMG activity following removal of cold block and restoration of axon conduction (Castro-Moure and Goshgarian, 1996). Understanding the stimulus and mechanisms for spontaneous recovery after SCI is vital to further enhance these pathways and improve ventilation following SCI.
5. Is inactivity-induced respiratory plasticity adaptive or maladaptive?
Without a clear understanding of the role for inactivity-induced plasticity in the control of breathing, it is difficult to address whether it is adaptive or maladaptive. However, we speculate that the nature of reduced respiratory neural activity determines the functional consequences of inactivity-induced plasticity in the creation of a stable breathing pattern. For example, as discussed above, even otherwise healthy individuals experience central apnea, particularly during sleep-onset or arousals, although the frequency and duration of these apneas is generally minimal (Eckert et al., 2007; Gaultier and Gallego, 2005; Javaheri, 2010; Javaheri and Dempsey, 2013). However, these infrequent and minor disruptions in respiratory neural activity may induce mechanisms of inactivity-induced plasticity that reconfigure network properties to augment respiratory motor output, thereby preventing future episodes. In preliminary studies, we find evidence that inactivity-induced plasticity is associated with a decrease in the CO2 apneic threshold (Baertsch and Baker-Herman,2012), which may stabilize breathing and further protect against future apneas by increasing the CO2 reserve. In this sense, inactivity-induced respiratory plasticity may be an important endogenous mechanism to stabilize respiratory motor output throughout life.
On the other hand, too much enhancement and ventilatory instability may result; increased inspiratory efforts may cause large swings in PaCO2, perpetuating the cycle of apnea/hypopnea and predisposing an individual to periodic breathing. Such may be the case in pathological conditions associated with an increased frequency of CSA, such as patients with heart failure (although at this early stage we cannot rule out that these conditions represent a failure of inactivity-induced plasticity). Further, an imbalance between the duration and magnitude of inactivity-induced plasticity in different respiratory muscle groups may alter the recruitment patterns of inspiratory/expiratory muscles (Feroah et al., 2001), potentially impairing the coordination of a breath. Of interest, our findings in experimental models suggest that inactivity-induced plasticity may preferentially enhance inspiratory pump muscles versus those stabilizing the upper airway, which may increase the propensity for airway obstruction during breathing. Thus, in some cases, it may be desirable to reduce the magnitude of inactivity-induced plasticity to stabilize breathing.
Inactivity-induced plasticity may be beneficial in situations in which reduced respiratory neural activity is prolonged. For example, following a cervical spinal injury, induction of inactivity-induced plasticity may partially restore respiratory motor output. Thus, enhancing inactivity-induced plasticity in patients with compromised breathing following SCI may optimize respiratory motor function and ventilation. In this case, a clear understanding of when and in which motor pool inactivity-induced plasticity should be enhanced following SCI will be critical to maximize therapeutic benefit. However, it is important to keep in mind that this restoration of activity may be double-edged since too much plasticity may result in hyperexcitability of respiratory motor neurons and contribute to respiratory discoordination and muscle spasticity (Bos et al., 2013; Boulenguez et al., 2010). Indeed, respiratory muscle spasticity compromises breathing in many SCI patients, contributing to their ventilator dependence (Britton et al., 2005; Silver and Lehr, 1981).
By contrast, induction of inactivity-induced plasticity may be inappropriate in situations where respiratory neural activity is reduced artificially, such as during mechanical ventilation. In this context, inactivity-induced plasticity may be imposed on the system, and functional adjustments may inappropriately alter system performance, such that when artificial ventilation ends, unstable breathing may result. Accordingly, preventing induction of inactivity-induced plasticity by maintaining respiratory neural activity during mechanical ventilation may be critical for the resumption of a stable breathing pattern and ventilator weaning. On the other hand, induction of inactivity-induced plasticity may serve some benefit in facilitating resumption of spontaneous breathing after mechanical ventilation by strengthening inspiratory motor output to offset weakened respiratory muscles. Clearly, the consequence of inactivity-induced plasticity in shaping future performance of the respiratory control system in any clinical context is poorly understood, and further investigation of the role for inactivity-induced plasticity in the control of breathing is warranted.
6. How do distinct forms of respiratory plasticity interact?
Reduced respiratory neural activity rarely occurs in the absence of additional stimuli that are also capable of eliciting plasticity. Indeed, during central neural apneas, intermittent reductions in neural activity are associated with intermittent hypoxia, hypocapnia, hypercapnia and diminished vagal feedback. As summarized in Fig. 3, each of these stimuli in isolation has been shown to elicit respiratory plasticity via distinct cellular mechanisms, and each differentially affects motor output in specific motor pools (Bach and Mitchell, 1996; Baertsch and Baker-Herman, 2013; Baker et al., 2001; Dale-Nagle et al., 2010; Devinney et al., 2013; Fregosi and Mitchell, 1994; Mahamed and Mitchell, 2008; Mahamed et al., 2011; Peng et al., 2003; Strey et al., 2012; Tadjalli et al., 2010; Zhang et al., 2003). For example, acute intermittent ventilator apneas in paralyzed rats (3 or 6, 25 s apneas, separated by 5 min; without disruption in central neural drive) induces a long-lasting hypoglossal and phrenic motor facilitation (Mahamed and Mitchell, 2008); similarly, hypoglossal and phrenic motor facilitation are observed following acute isocapnic intermittent hypoxia alone (Bach and Mitchell, 1996). On the other hand, diminished vagal feedback does not elicit phrenic motor facilitation, but preferentially elicits genioglossus (and presumably, hypoglossal) facilitation (Tadjalli et al., 2010). Interestingly, intermittent hypoxia in the presence of diminished vagal feedback does not appear to elicit phrenic motor facilitation (Tadjalli et al., 2010), suggesting that simultaneous induction of multiple forms of plasticity may confer unique responses.
Fig. 3.
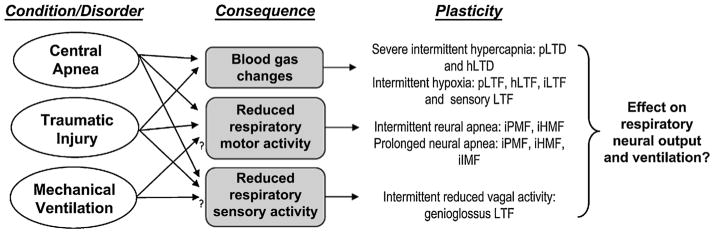
Reduced respiratory neural activity has the potential to elicit multiple forms of plasticity. In non-ventilated animals, central apnea results in reduced respiratory neural activity, profound changes in arterial blood gases and diminished sensory feedback. Similar effects may be observed following disruption of descending inputs to spinal motor neurons (depending on extent and location of disruption). Mechanical ventilation may be associated with reduced respiratory neural activity and/or altered sensory feedback in some patients. Animal models suggest that each of these stimuli independently elicit unique and possibly overlapping forms of plasticity. For example, acute intermittent hypoxia elicits long-term facilitation (LTF) in phrenic, hypoglossal and intercostal nerves (pLTF, hLTF and iLTF, respectively; Bach and Mitchell, 1996; Fregosi and Mitchell, 1994). Additional forms of plasticity are elicited during chronic exposures to intermittent hypoxia, specifically facilitation of carotid body afferent feedback (sensory LTF; Peng et al., 2003). Severe hypercapnia elicits long-term depression of phrenic and hypoglossal nerve activity (pLTD and hLTD; Bach and Mitchell, 1998; Baker et al., 2001). Reduced respiratory neural (motor) activity elicits long-lasting iPMF, and transient iHMF and iIMF (Baertsch and Baker-Herman, 2013; Baker-Herman and Strey, 2011; Mahamed et al., 2011), whereas reduced intermittent sensory (vagal) feedback elicits genioglossus facilitation (Tadjalli et al., 2010). To date, we lack a clear understanding regarding how these multiple forms of plasticity interact to shape necessary long-term adaptations in the respiratory control system.
An important question for future studies is: How do these multiple forms of plasticity interact to shape necessary long-term adaptations in the respiratory control system? Certainly, information is gained through investigations of each form of plasticity in isolation (e.g., intermittent hypoxia without reduced central drive or airway obstruction and vice versa); however, distinct mechanisms of plasticity may interact in complex ways to give rise to the final motor output (Devinney et al., 2013; Nichols et al., 2012). Indeed, cross-talk inhibition between different forms of respiratory plasticity has been demonstrated in response to intermittent hypoxia (Dale-Nagle et al., 2010; Devinney et al., 2013; Nichols et al., 2012). A clear understanding of these mechanisms and how interactions among various forms of plasticity lend context specificity to unique respiratory challenges in order to shape ventilatory adaptations is necessary to grasp the significance of plasticity within the control of breathing.
7. Conclusions
We are beginning to grasp basic mechanisms of inactivity-induced respiratory plasticity. It is tempting to speculate that this form of plasticity provides a “boost”, ensuring that the respiratory control system produces adequate motor output at all times; however, little is known regarding its functional consequences during physiological and/or pathophysiological situations. Since many conditions and disorders of relevance to human health are associated with reduced respiratory neural activity, an understanding of inactivity-induced plasticity and how (and if) it applies in these conditions is important. However, it’s imperative to consider the potentially complex interactions among multiple forms of plasticity likely induced during the same event. Since our appreciation of inactivity-induced respiratory plasticity is new (Mahamed et al., 2011), we anticipate rapid increases in our understanding from here forward.
Footnotes
Sources of Support: Supported by NIH NL105511.
Contributor Information
K.A. Strey, Email: strey@wisc.edu.
T.L. Baker-Herman, Email: bakert@svm.vetmed.wisc.edu.
References
- Ancoli-Israel S, Kripke DF, Mason W. Characteristics of obstructive and central sleep apnea in the elderly: an interim report. Biol Psychiatry. 1987;22 (6):741–750. doi: 10.1016/0006-3223(87)90206-x. [DOI] [PubMed] [Google Scholar]
- Anzueto A, Peters JI, Tobin MJ, de los Santos R, Seidenfeld JJ, Moore G, Cox WJ, Coalson JJ. Effects of prolonged controlled mechanical ventilation on diaphragmatic function in healthy adult baboons. Crit Care Med. 1997;25 (7):1187–1190. doi: 10.1097/00003246-199707000-00021. [DOI] [PubMed] [Google Scholar]
- Axen K, Pineda H, Shunfenthal I, Haas F. Diaphragmatic function following cervical cord injury: neurally mediated improvement. Arch Phys Med Rehabil. 1985;66 (4):219–222. doi: 10.1016/0003-9993(85)90146-7. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent? Respir Physiol. 1996;104 (2–3):251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypercapnia-induced long-term depression of respiratory activity requires alpha2-adrenergic receptors. J Appl Physiol. 1998;84 (6):2099–2105. doi: 10.1152/jappl.1998.84.6.2099. [DOI] [PubMed] [Google Scholar]
- Baker TL, Fuller DD, Zabka AG, Mitchell GS. Respiratory plasticity: differential actions of continuous and episodic hypoxia and hypercapnia? Respir Physiol. 2001;129 (1–2):25–35. doi: 10.1016/s0034-5687(01)00280-8. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Strey KA. Similarities and differences in mechanisms of phrenic and hypoglossal motor facilitation. Respir Physiol Neurobiol. 2011;179:48–56. doi: 10.1016/j.resp.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baertsch NA, Baker-Herman TL. Inactivity-induced phrenic motor facilitation (iPMF) is pattern sensitive and associated with a decrease in the CO2 apneic threshold. Oxford Abstract 2012 [Google Scholar]
- Baertsch NA, Baker-Herman TL. Inactivity-induced phrenic and hypoglossal motor facilitation are differentially expressed following intermittent versus sustained neural apnea. J Appl Physiol. 2013;114 (10):1388–1395. doi: 10.1152/japplphysiol.00018.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baussart B, Stamegna JC, Polentes J, Tadié M, Gauthier P. A new model of upper cervical spinal contusion inducing a persistent unilateral diaphragmatic deficit in the adult rat. Brain. 2006;130 (Pt 2):469–475. doi: 10.1016/j.nbd.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Berssenbrugge AD, Dempsey JA, Skatrud JB. Effects of sleep state on ventilatory acclimatization to hypoxia in humans. J Appl Physiol. 1984;57 (4):1089–1096. doi: 10.1152/jappl.1984.57.4.1089. [DOI] [PubMed] [Google Scholar]
- Bixler EO, Vgontzas AN, Ten Have T, Tyson K, Kales A. Effects of age on sleep apnea in men: I. Prevalence and severity. Am J Respir Crit Care Med. 1998;157 (1):144–148. doi: 10.1164/ajrccm.157.1.9706079. [DOI] [PubMed] [Google Scholar]
- Bloch KE, Latshang TD, Turk AJ, Hess T, Hefti U, Merz TM, Bosch MM, Barthelmes D, Hefti JP, Maggiorini M, Schoch OD. Nocturnal periodic breathing during acclimatization at very high altitude at Mount Muztagh Ata (7546 m) Am J Respir Crit Care Med. 2010;182 (4):562–568. doi: 10.1164/rccm.200911-1694OC. [DOI] [PubMed] [Google Scholar]
- Bos R, Sadlaoud K, Boulenguez P, Buttigieg D, Liabeuf S, Brocard C, Haase G, Bras H, Vinay L. Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. PNAS. 2013;110 (1):348–353. doi: 10.1073/pnas.1213680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med. 2010;16 (3):302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- Bradley TD, McNicholas WT, Rutherford R, Popkin J, Zamel N, Phillipson EA. Clinical and physiologic heterogeneity of the central sleep apnea syndrome. Am Rev Respir Dis. 1986;34 (2):217–221. doi: 10.1164/arrd.1986.134.2.217. [DOI] [PubMed] [Google Scholar]
- Bradley TD, Phillipson EA. Central sleep apnea. Clin Chest Med. 1992;13 (3):493–505. [PubMed] [Google Scholar]
- Britton D, Goldstein B, Jones-Redmond J, Esselman P. Baclofen pump intervention for spasticity affecting pulmonary function. J Spinal Cord Med. 2005;28 (4):343–347. doi: 10.1080/10790268.2005.11753832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broytman O, Baertsch NA, Baker-Herman TL. Spinal TNFa is necessary for inactivity-induced phrenic motor facilitation. J Physiol. 2013 doi: 10.1113/jphysiol.2013.256644. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess KR, Johnson PL, Edwards N. Central and obstructive sleep apnoea during ascent to high altitude. Respirology. 2004;9 (2):222–229. doi: 10.1111/j.1440-1843.2004.00576.x. [DOI] [PubMed] [Google Scholar]
- Budzińska K, von Euler C, Kao FF, Pantaleo T, Yamamoto Y. Effects of graded focal cold block in rostral areas of the medulla. Acta Physiol Scand. 1985;124 (3):329–340. doi: 10.1111/j.1748-1716.1985.tb07668.x. [DOI] [PubMed] [Google Scholar]
- Carskadon MA, Dement WC. Respiration during sleep in the aged human. J Gerontol. 1981;36 (4):420–423. doi: 10.1093/geronj/36.4.420. [DOI] [PubMed] [Google Scholar]
- Castro-Moure F, Goshgarian HG. Reversible cervical hemispinalization of the rat spinal cord by a cooling device. Exp Neurol. 1996;141:102–112. doi: 10.1006/exnr.1996.0143. [DOI] [PubMed] [Google Scholar]
- Castro-Moure F, Goshgarian HG. Morphological plasticity induced in the phrenic nucleus following cervical cold block of descending respiratory drive. Exp Neurol. 1997;147 (2):299–310. doi: 10.1006/exnr.1997.6615. [DOI] [PubMed] [Google Scholar]
- Chao DC, Scheinhorn DJ, Stearn-Hassenpflug M. Patient–ventilator trigger asynchrony in prolonged mechanical ventilation. Chest. 1997;112 (6):1592–1599. doi: 10.1378/chest.112.6.1592. [DOI] [PubMed] [Google Scholar]
- Chokroverty S, Sachdeo R, Masdeu J. Autonomic dysfunction and sleep apnea in olivopontocerebellar degeneration. Arch Neurol. 1984;41 (9):926–931. doi: 10.1001/archneur.1984.04050200032014. [DOI] [PubMed] [Google Scholar]
- Chokroverty S, Sharp JT, Barron KD. Periodic respiration in erect posture in Shy-Drager syndrome. J Neurol Neurosurg Psychiatry. 1978;41 (11):980–986. doi: 10.1136/jnnp.41.11.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhuri S, Badr MS. Central sleep apnoea. Indian J Med Res. 2010;131:150–164. [PubMed] [Google Scholar]
- Colombo D, Cammarota G, Bergamaschi V, De Lucia M, Corte FD, Navalesi P. Physiologic response to varying levels of pressure support and neurally adjusted ventilatory assist in patients with acute respiratory failure. Intensive Care Med. 2008;34 (11):2010–2018. doi: 10.1007/s00134-008-1208-3. [DOI] [PubMed] [Google Scholar]
- Dale-Nagle EA, Hoffman MS, MacFarlane PM, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv Exp Med Biol. 2010;669:225–230. doi: 10.1007/978-1-4419-5692-7_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlot F, Cayetanot F, Gauthier P, Matarazzo V, Kastner A. Extensive respiratory plasticity after cervical spinal cord injury in rats: axonal sprouting and rerouting of ventrolateral bulbospinal pathways. Exp Neurol. 2012;236 (1):88–102. doi: 10.1016/j.expneurol.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Delisle S, Ouellet P, Bellemare P, Tétrault JP, Arsenault P. Sleep quality in mechanically ventilated patients: comparison between NAVA and PSV modes. Annu Intensive Care. 2011;1 (1):42. doi: 10.1186/2110-5820-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey JA, Smith CA, Blain GM, Xie A, Gong Y, Teodorescu M. Role of central/peripheral chemoreceptors and their interdependence in the pathophysiology of sleep apnea. Adv Exp Med Biol. 2012;758:343–349. doi: 10.1007/978-94-007-4584-1_46. [DOI] [PubMed] [Google Scholar]
- Dernaika T, Tawk M, Nazir S, Younis W, Kinasewitz GT. The significance and outcome of continuous positive airway pressure-related central sleep apnea during split-night sleep studies. Chest. 2007;132 (1):81–87. doi: 10.1378/chest.06-2562. [DOI] [PubMed] [Google Scholar]
- De Troyer A, Kirkwood PA, Wilson TA. Respiratory action of the intercostal muscles. Physiol Rev. 2005;85 (2):717–756. doi: 10.1152/physrev.00007.2004. [DOI] [PubMed] [Google Scholar]
- Devinney MJ, Huxtable AG, Nichols NL, Mitchell GS. Hypoxia-induced phrenic long-term facilitation: emergent properties. Ann NY Acad Sci. 2013;1279 (1):143–153. doi: 10.1111/nyas.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit M, Pedram S, Best AM, Epstein SK. Observational study of patient–ventilator asynchrony and relationship to sedation level. J Crit Care. 2009a;24(1):74–80. doi: 10.1016/j.jcrc.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit M, Miller KB, Green DA, Ostman HE, Gennings C, Epstein SK. Ineffective triggering predicts increased duration of mechanical ventilation. Crit Care Med. 2009b;37 (10):2740–2745. doi: 10.1097/ccm.0b013e3181a98a05. [DOI] [PubMed] [Google Scholar]
- Dres M, Schmidt M, Ferre A, Mayaux J, Similowski T, Demoule A. Diaphragm electromyographic activity as a predictor of weaning failure. Intensive Care Med. 2012;38 (12):2017–2025. doi: 10.1007/s00134-012-2700-3. [DOI] [PubMed] [Google Scholar]
- Dutschmann M, Paton JF. Influence of nasotrigeminal afferents on medullary respiratory neurones and upper airway patency in the rat? Pflugers Arch. 2002;444 (1–2):227–235. doi: 10.1007/s00424-002-0797-x. [DOI] [PubMed] [Google Scholar]
- Eckert DJ, Jordan AS, Merchia P, Malhotra A. Central sleep apnea: pathophysiology and treatment. Chest. 2007;131 (2):595–607. doi: 10.1378/chest.06.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Bohy AA, Schrimsher GW, Reier PJ, Goshgarian HG. Quantitative assessment of respiratory function following contusion injury of the cervical spinal cord. Exp Neurol. 1998;150 (1):143–152. doi: 10.1006/exnr.1997.6757. [DOI] [PubMed] [Google Scholar]
- Epstein SK. How often does patient–ventilator asynchrony occur and what are the consequences? Respir Care. 2011;56 (1):25–38. doi: 10.4187/respcare.01009. [DOI] [PubMed] [Google Scholar]
- Epstein SK. Weaning from ventilatory support. Curr Opin Crit Care. 2009;15:36–43. doi: 10.1097/MCC.0b013e3283220e07. [DOI] [PubMed] [Google Scholar]
- Esteban A, Anzueto A, Alía I, Gordo F, Apezteguía C, Pálizas F, Cide D, Gold-waser R, Soto L, Bugedo G, Rodrigo C, Pimentel J, Raimondi G, Tobin MJ. How is mechanical ventilation employed in the intensive care unit? An international utilization review. Am J Respir Crit Care Med. 2000;161 (5):1450–1458. doi: 10.1164/ajrccm.161.5.9902018. [DOI] [PubMed] [Google Scholar]
- Esteban A, Ferguson ND, Meade MO, Frutos-Vivar F, Apezteguia C, Brochard L, Raymondos K, Nin N, Hurtado J, Tomicic V, González M, Elizalde J, Nightingale P, Abroug F, Pelosi P, Arabi Y, Moreno R, Jibaja M, D‘Empaire G, Sandi F, Matamis D, Montañez AM, Anzueto A VENTILA Group. Evolution of mechanical ventilation in response to clinical research. Am J Respir Crit Care Med. 2008;177 (2):170–177. doi: 10.1164/rccm.200706-893OC. [DOI] [PubMed] [Google Scholar]
- Esteban A, Frutos F, Tobin MJ, Alía I, Solsona JF, Valverdú I, Fernández R, de la Cal MA, Benito S, Tomás R, Carriedo D, Macías A, Blanco J Spanish Lung Failure Collaborative Group. A comparison of four methods of weaning patients from mechanical ventilation. N Engl J Med. 1995;332 (6):345–350. doi: 10.1056/NEJM199502093320601. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Bresnahan JC, Beattie MS. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci. 2008;28 (44):11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KA, Strong MJ, Ahmad D, George CF. Sleep-disordered breathing in amyotrophic lateral sclerosis. Chest. 1996;110 (3):664–669. doi: 10.1378/chest.110.3.664. [DOI] [PubMed] [Google Scholar]
- Feroah TR, Forster HV, Pan L, Wenninger J, Martino P, Rice T. Effect of slow wave and REM sleep on thyropharyngeus and stylopharyngeus activity during induced central apneas. Respir Physiol. 2001;124 (2):129–140. doi: 10.1016/s0034-5687(00)00192-4. [DOI] [PubMed] [Google Scholar]
- Fleming PJ, Cade D, Bryan MH, Bryan AC. Congenital central hypoventilation and sleep state. Pediatrics. 1980;66:425–428. [PubMed] [Google Scholar]
- Fregosi RF, Mitchell GS. Long-term facilitation of inspiratory intercostal nerve activity following carotid sinus nerve stimulation in cats. J Physiol. 1994;477:469–479. doi: 10.1113/jphysiol.1994.sp020208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Johnson SM, Olson EB, Jr, Mitchell GS. Synaptic pathways to phrenic motoneurons are enhanced by chronic intermittent hypoxia after cervical spinal cord injury. J Neurosci. 2003;23:2993–3000. doi: 10.1523/JNEUROSCI.23-07-02993.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Golder FJ, Olson EB, Jr, Mitchell GS. Recovery of phrenic activity and ventilation after cervical spinal hemisection in rats. J Appl Physiol. 2006;100 (3):800–806. doi: 10.1152/japplphysiol.00960.2005. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Doperalski NJ, Dougherty BJ, Sandhu MS, Bolser DC, Reier PJ. Modest spontaneous recovery of ventilation following chronic high cervical hemisection in rats. Exp Neurol. 2008;211 (1):97–106. doi: 10.1016/j.expneurol.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaig C, Iranzo A. Sleep-disordered breathing in neurodegenerative diseases. Curr Neurol Neurosci Rep. 2012;12 (2):205–217. doi: 10.1007/s11910-011-0248-1. [DOI] [PubMed] [Google Scholar]
- Gaultier C, Gallego J. Development of respiratory control: evolving concepts and perspectives? Respir Physiol Neurobiol. 2005;149 (1–3):3–15. doi: 10.1016/j.resp.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Glass GA, Josephs KA, Ahlskog JE. Respiratory insufficiency as the primary presenting symptom of multiple-system atrophy. Arch Neurol. 2006;63 (7):978–981. doi: 10.1001/archneur.63.7.978. [DOI] [PubMed] [Google Scholar]
- Glotzbach SF, Baldwin RB, Lederer NE, Tansey PA, Ariagno RL. Periodic breathing in preterm infants: incidence and characteristics. Pediatrics. 1989;84 (5):785–792. [PubMed] [Google Scholar]
- Golder FJ, Mitchell GS. Spinal synaptic enhancement with acute intermittent hypoxia improves respiratory function after chronic cervical spinal cord injury. J Neurosci. 2005;25 (11):2925–2932. doi: 10.1523/JNEUROSCI.0148-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Fuller DD, Lovett-Barr MR, Vinit S, Resnick DK, Mitchell GS. Breathing patterns after mid-cervical spinal contusion in rats. Exp Neurol. 2011;231 (1):97–103. doi: 10.1016/j.expneurol.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooden BA. Mechanism of the human diving response. Integr Physiol Behav Sci. 1994;29 (1):6–16. doi: 10.1007/BF02691277. [DOI] [PubMed] [Google Scholar]
- Goridis C, Dubreuil V, Thoby-Brisson M, Fortin G, Brunet JF. Phox2b, congenital central hypoventilation syndrome and the control of respiration. Semin Cell Dev Biol. 2010;21 (8):814–822. doi: 10.1016/j.semcdb.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Gorini M, Corrado A, Aito S, Ginanni R, Villella G, Lucchesi G, De Paola E. Ventilatory and respiratory muscle responses to hypercapnia in patients with paraplegia. Am J Respir Crit Care Med. 2000;162:203–208. doi: 10.1164/ajrccm.162.1.9906029. [DOI] [PubMed] [Google Scholar]
- Goshgarian HG. The crossed phrenic phenomenon: a model for plasticity in the respiratory pathways following spinal cord injury. J Appl Physiol. 2003;94 (2):795–810. doi: 10.1152/japplphysiol.00847.2002. [DOI] [PubMed] [Google Scholar]
- Goshgarian HG. The crossed phrenic phenomenon and recovery of function following spinal cord injury. Respir Physiol Neurobiol. 2009;169 (2):85–93. doi: 10.1016/j.resp.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein RR, Ho KY, Sullivan CE. Sleep apnea in acromegaly. Ann Intern Med 1. 1991;115 (7):527–532. doi: 10.7326/0003-4819-115-7-527. [DOI] [PubMed] [Google Scholar]
- Guenther CH, Windelborn JA, Tubon TC, Yin JC, Mitchell GS. Increased atypical PKC expression and activity in the phrenic motor nucleus following cervical spinal injury. Exp Neurol. 2012;234 (2):513–520. doi: 10.1016/j.expneurol.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MJ, Xie A, Rutherford R, Ando S, Floras JS, Bradley TD. Cycle length of periodic breathing in patients with and without heart failure. Am J Respir Crit Care Med. 1996;154 (2 Pt 1):376–381. doi: 10.1164/ajrccm.154.2.8756809. [DOI] [PubMed] [Google Scholar]
- Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41 (7):369–378. doi: 10.1038/sj.sc.3101483. [DOI] [PubMed] [Google Scholar]
- Henderson-Smart DJ. The effect of gestational age on the incidence and duration of recurrent apnoea in newborn babies. Aust Paediatr J. 1981;17 (4):273–276. doi: 10.1111/j.1440-1754.1981.tb01957.x. [DOI] [PubMed] [Google Scholar]
- Henke KG, Arias A, Skatrud JB, Dempsey JA. Inhibition of inspiratory muscle activity during sleep: chemical and nonchemical influences. Am Rev Respir Dis. 1988;138 (1):8–15. doi: 10.1164/ajrccm/138.1.8. [DOI] [PubMed] [Google Scholar]
- Hensbergen E, Kernell D. Daily durations of spontaneous activity in cat’s ankle muscles. Exp Brain Res. 1997;115 (2):325–332. doi: 10.1007/pl00005701. [DOI] [PubMed] [Google Scholar]
- Herrera M, Blasco J, Venegas J, Barba R, Doblas A, Marquez E. Mouth occlusion pressure (P0.1) in acute respiratory failure. Intensive Care Med. 1985;(3):134–139. doi: 10.1007/BF00258538. [DOI] [PubMed] [Google Scholar]
- Hilbert G, Gruson D, Portel L, Vargas F, Gbikpi-Benissan G, Cardinaud JP. Airway occlusion pressure at 0.1 s (P0.1) after extubation: an early indicator of postextubation hypercapnic respiratory insufficiency. Intensive Care Med. 1998;24 (12):1277–1282. doi: 10.1007/s001340050762. [DOI] [PubMed] [Google Scholar]
- Hoppenbrouwers T, Hodgman JE, Harper RM, Hofmann E, Sterman MB, McGinty DJ. Polygraphic studies of normal infants during the first six months of life: III. Incidence of apnea and periodic breathing. Pediatrics. 1977;60 (4):418–425. [PubMed] [Google Scholar]
- Javaheri S. Central sleep apnea. Clin Chest Med. 2010;31 (2):235–248. doi: 10.1016/j.ccm.2010.02.013. [DOI] [PubMed] [Google Scholar]
- Javaheri S, Dempsey JA. Central sleep apnea. Compr Physiol. 2013;3 (1):141–163. doi: 10.1002/cphy.c110057. [DOI] [PubMed] [Google Scholar]
- Javaheri S, Parker TJ, Liming JD, Corbett WS, Nishiyama H, Wexler L, Roselle GA. Sleep apnea in 81 ambulatory male patients with stable heart failure: types and their prevalences, consequences, and presentations. Circulation. 1998;97 (21):2154–2159. doi: 10.1161/01.cir.97.21.2154. [DOI] [PubMed] [Google Scholar]
- Johnson SM, Mitchell GS. Activity-dependent plasticity in descending synaptic inputs to respiratory spinal motoneurons? Respir Physiol Neurobiol. 2002;131 (1–2):79–90. doi: 10.1016/s1569-9048(02)00039-3. [DOI] [PubMed] [Google Scholar]
- Jubran A, Tobin MJ. Pathophysiologic basis of acute respiratory distress in patients who fail a trial of weaning from mechanical ventilation. Am J Respir Crit Care Med. 1997a;155 (3):906–915. doi: 10.1164/ajrccm.155.3.9117025. [DOI] [PubMed] [Google Scholar]
- Jubran A, Tobin MJ. Passive mechanics of lung and chest wall in patients who failed or succeeded in trials of weaning. Am J Respir Crit Care Med. 1997b;155 (3):916–921. doi: 10.1164/ajrccm.155.3.9117026. [DOI] [PubMed] [Google Scholar]
- Kahn A, Blum D, Engelman E, Waterschoot P. Effects of central apneas on transcutaneous PO2 in control subjects, siblings of victims of sudden infant death syndrome, and near miss infants. Pediatrics. 1982;69 (4):413–418. [PubMed] [Google Scholar]
- Katz DM, Dutschmann M, Ramirez JM, Hilaire G. Breathing disorders in Rett syndrome: progressive neurochemical dysfunction in the respiratory network after birth? Respir Physiol Neurobiol. 2009;168 (1–2):101–108. doi: 10.1016/j.resp.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DH, Stellwagen LM, Kaitz E, Shannon DC. Apnea and periodic breathing in normal full-term infants during the first twelve months. Pediatr Pulmonol. 1985;1 (4):215–219. doi: 10.1002/ppul.1950010409. [DOI] [PubMed] [Google Scholar]
- Kohler M, Kriemler S, Wilhelm EM, Brunner-LaRocca H, Zehnder M, Bloch KE. Children at high altitude have less nocturnal periodic breathing than adults. Eur Respir J. 2008;32 (1):189–197. doi: 10.1183/09031936.00119807. [DOI] [PubMed] [Google Scholar]
- Kondili E, Xirouchaki N, Georgopoulos D. Modulation and treatment of patient–ventilator dyssynchrony. Curr Opin Crit Care. 2007;13 (1):84–89. doi: 10.1097/MCC.0b013e328011278d. [DOI] [PubMed] [Google Scholar]
- Kong FJ, Berger AJ. Firing properties and hypercapnic responses of single phrenic motor axons in the rat. J Appl Physiol. 1986;61:1999–2004. doi: 10.1152/jappl.1986.61.6.1999. [DOI] [PubMed] [Google Scholar]
- Labanowski M, Schmidt-Nowara W, Guilleminault C. Sleep and neuromuscular disease: frequency of sleep-disordered breathing in a neuromuscular disease clinic population. Neurology. 1996;47 (5):1173–1180. doi: 10.1212/wnl.47.5.1173. [DOI] [PubMed] [Google Scholar]
- Laghi F. Assessment of respiratory output in mechanically ventilated patients. Respir Care Clin N Am. 2005;11 (2):173–199. doi: 10.1016/j.rcc.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Lane MA, Lee KZ, Fuller DD, Reier PJ. Spinal circuitry and respiratory recovery following spinal cord injury. Respir Physiol Neurobiol. 2009;169:123–132. doi: 10.1016/j.resp.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA, Lee KZ, Salazar K, O’Steen BE, Bloom DC, Fuller DD, Reier PJ. Respiratory function following bilateral mid-cervical contusion injury in the adult rat. Exp Neurol. 2012;235 (1):197–210. doi: 10.1016/j.expneurol.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman S, Antic NA, Thompson C, Catcheside PG, Mercer J, McEvoy RD. Central sleep apnea on commencement of continuous positive airway pressure in patients with a primary diagnosis of obstructive sleep apnea–hypopnea. J Clin Sleep Med. 2007;3 (5):462–466. [PMC free article] [PubMed] [Google Scholar]
- Leung RS, Comondore VR, Ryan CM, Stevens D. Mechanisms of sleep-disordered breathing: causes and consequences. Pflugers Arch. 2012;463 (1):213–230. doi: 10.1007/s00424-011-1055-x. [DOI] [PubMed] [Google Scholar]
- Liu L, Liu H, Yang Y, Huang Y, Liu S, Beck J, Slutsky AS, Sinderby C, Qiu H. Neuroventilatory efficiency and extubation readiness in critically ill patients. Crit Care. 2012;16 (4):R143. doi: 10.1186/cc11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald M, Fang J, Pittman SD, White DP, Malhotra A. The current prevalence of sleep disordered breathing in congestive heart failure patients treated with beta-blockers. J Clin Sleep Med. 2008;4 (1):38–42. [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Simulated apnoeas induce serotonin-dependent respiratory long-term facilitation in rats. J Physiol. 2008;586 (8):2171–2181. doi: 10.1113/jphysiol.2007.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Strey KA, Mitchell GS, Baker-Herman TL. Reduced respiratory neural activity elicits phrenic motor facilitation. Respir Physiol Neurobiol. 2011;175 (3):303–309. doi: 10.1016/j.resp.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra A, Owens RL. What is central sleep apnea? Respir Care. 2010;55 (9):1168–1178. [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Rowley KL, Zhan WZ, Fahim MA, Sieck GC. Synaptic vesicle pools at diaphragm neuromuscular junctions vary with motoneuron soma, not axon terminal, inactivity. Neuroscience. 2007;146:178–189. doi: 10.1016/j.neuroscience.2007.01.048. [DOI] [PubMed] [Google Scholar]
- Marcus CL, Omlin KJ, Basinki DJ, Bailey SL, Rachal AB, Von Pechmann WS, Keens TG, Ward SL. Normal polysomnographic values for children and adolescents. Am Rev Respir Dis. 1992;146 (5 Pt 1):1235–1239. doi: 10.1164/ajrccm/146.5_Pt_1.1235. [DOI] [PubMed] [Google Scholar]
- Martin RJ, Abu-Shaweesh JM, Baird TM. Apnoea of prematurity. Paediatr Respir Rev. 2004;5 (Suppl A):S377–S382. doi: 10.1016/s1526-0542(04)90067-x. [DOI] [PubMed] [Google Scholar]
- Martin RJ, Dreshaj IA, Miller MJ, Haxhiu MA. Hypoglossal and phrenic responses to central respiratory inhibition in piglets. Respir Physiol Neurobiol. 1994;97:93–100. doi: 10.1016/0034-5687(94)90014-0. [DOI] [PubMed] [Google Scholar]
- McClung JM, Kavazis AN, DeRuisseau KC, Falk DJ, Deering MA, Lee Y, Sugiura T, Powers SK. Caspase-3 regulation of diaphragm myonuclear domain during mechanical ventilation-induced atrophy. Am J Respir Crit Care Med. 2007;175 (2):150–159. doi: 10.1164/rccm.200601-142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrimmon DR, Zuperku EJ, Hayashi F, Dogas Z, Hinrichsen CF, Stuth EA, Tonkovic-Capin M, Krolo M, Hopp FA. Modulation of the synaptic drive to respiratory premotor and motor neurons? Respir Physiol. 1997;110 (2–3):161–176. doi: 10.1016/s0034-5687(97)00081-9. [DOI] [PubMed] [Google Scholar]
- McKinley WO. Late return of diaphragm function in a ventilator dependent patient with a high cervical tetraplegia: a case report and interactive review. Spinal Cord. 1996;34:626–629. doi: 10.1038/sc.1996.112. [DOI] [PubMed] [Google Scholar]
- Mellott KG, Grap MJ, Munro CL, Sessler CN, Wetzel PA. Patient-ventilator dyssynchrony: clinical significance and implications for practice. Crit Care Nurse. 2009;29 (6):41–55. doi: 10.4037/ccn2009612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza S, Giannouli E, Younes M. Control of breathing during sleep assessed by proportional assist ventilation. J Appl Physiol. 1998;84 (1):3–12. doi: 10.1152/jappl.1998.84.1.3. [DOI] [PubMed] [Google Scholar]
- Millman RP, Bevilacqua J, Peterson DD, Pack AI. Central sleep apnea in hypothyroidism. Am Rev Respir Dis. 1983;127 (4):504–507. doi: 10.1164/arrd.1983.127.4.504. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol. 2003;94 (1):358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Morgenthaler TI, Kagramanov V, Hanak V, Decker PA. Complex sleep apnea syndrome: is it a unique clinical syndrome? Sleep. 2006;29 (9):1203–1209. doi: 10.1093/sleep/29.9.1203. [DOI] [PubMed] [Google Scholar]
- National Spinal Cord Injury Statistical Center Spinal cord injury. Facts and figures at a glance. J Spinal Cord Med. 2013;36 (1):1–2. doi: 10.1179/1079026813Z.000000000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemer SN, Barbas CS, Caldeira JB, Cárias TC, Santos RG, Almeida LC, Azeredo LM, Noé RA, Guimarães BS, Souza PC. A new integrative weaning index of discontinuation from mechanical ventilation. Crit Care. 2009;13 (5):R152. doi: 10.1186/cc8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng DK, Chan CH. A review of normal values of infant sleep polysomnography. Pediatr Neonatol. 2013;54 (2):82–87. doi: 10.1016/j.pedneo.2012.11.011. [DOI] [PubMed] [Google Scholar]
- Nichols NL, Dale EA, Mitchell GS. Severe acute intermittent hypoxia elicits phrenic long-term facilitation by a novel adenosine-dependent mechanism. J Appl Physiol. 2012;112 (10):1678–1688. doi: 10.1152/japplphysiol.00060.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaumer-Ochsner Y, Ursprung J, Siebenmann C, Maggiorini M, Bloch KE. Effect of short-term acclimatization to high altitude on sleep and nocturnal breathing. Sleep. 2012;35 (3):419–423. doi: 10.5665/sleep.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo T, Watt JWH, Soni BM, Sett PK. Delayed diaphragm recovery in 12 patients after high cervical spinal cord injury: a retrospective review of the diaphragm status of 107 patients ventilated after acute spinal cord injury. Spinal Cord. 1999;37:117–122. doi: 10.1038/sj.sc.3100775. [DOI] [PubMed] [Google Scholar]
- Pack AI. Central sleep apnea. Handb Clin Neurol. 2011;98:411–9. doi: 10.1016/B978-0-444-52006-7.00027-7. [DOI] [PubMed] [Google Scholar]
- Pagliardini S, Greer JJ, Funk GD, Dickson CT. State-dependent modulation of breathing in urethane-anesthetized rats. J Neurosci. 2012;32 (33):11259–11270. doi: 10.1523/JNEUROSCI.0948-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy S, Tobin MJ. Effect of ventilator mode on sleep quality in critically ill patients. Am J Respir Crit Care Med. 2002;166 (11):1423–1429. doi: 10.1164/rccm.200209-999OC. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. PNAS. 2003;100 (17):10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrigault PF, Pouzeratte YH, Jaber S, Capdevila XJ, Hayot M, Boccara G, Ramonatxo M, Colson P. Changes in occlusion pressure (P0. 1) and breathing pattern during pressure support ventilation. Thorax. 1999;54 (2):119–123. doi: 10.1136/thx.54.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson DJ. Patient–ventilator interaction. Respir Care. 2011;56 (2):214–228. doi: 10.4187/respcare.01115. [DOI] [PubMed] [Google Scholar]
- Pineau I, Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol. 2007;500:267–285. doi: 10.1002/cne.21149. [DOI] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, Levine S. Prolonged mechanical ventilation alters diaphragmatic structure and function. Crit Care Med. 2009;37 (10 Suppl):S347–S353. doi: 10.1097/CCM.0b013e3181b6e760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash YS, Miyata H, Zhan W, Sieck GC. Inactivity-induced remodeling of neuromuscular junctions in rat diaphragmatic muscle. Muscle Nerve. 1999;22:307–319. doi: 10.1002/(sici)1097-4598(199903)22:3<307::aid-mus3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Putensen C, Muders T, Varelmann D, Wrigge H. The impact of spontaneous breathing during mechanical ventilation. Curr Opin Crit Care. 2006;12 (1):13–18. doi: 10.1097/01.ccx.0000198994.37319.60. [DOI] [PubMed] [Google Scholar]
- Reyland ME. Protein kinase C isoforms: multi-functional regulators of cell life and death. Front Biosci. 2009;14:2386–2399. doi: 10.2741/3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenow F, McCarthy V, Caruso AC. Sleep apnoea in endocrine diseases. J Sleep Res. 1998;7 (1):3–11. doi: 10.1046/j.1365-2869.1998.00086.x. [DOI] [PubMed] [Google Scholar]
- Santos C, Braghiroli A, Mazzini L, Pratesi R, Oliveira LV, Mora G. Sleep-related breathing disorders in amyotrophic lateral sclerosis. Monaldi Arch Chest Dis. 2003;59 (2):160–165. [PubMed] [Google Scholar]
- Sassoon CS, Mahutte CK. Airway occlusion pressure and breathing pattern as predictors of weaning outcome. Am Rev Respir Dis. 1993;148:860–866. doi: 10.1164/ajrccm/148.4_Pt_1.860. [DOI] [PubMed] [Google Scholar]
- Scholle S, Wiater A, Scholle HC. Normative values of polysomnographic parameters in childhood and adolescence: cardiorespiratory parameters. Sleep Med. 2011;12 (10):988–996. doi: 10.1016/j.sleep.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, Belcastro A, Powers SK. Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med. 2002;166 (10):1369–1374. doi: 10.1164/rccm.200202-088OC. [DOI] [PubMed] [Google Scholar]
- Sharma H, Alilain W, Sadhu A, Silver J. Treatments to restore respiratory function after spinal cord injury and their implications for regeneration, plasticity and adaptation. Exp Neurol. 2012;235:18–25. doi: 10.1016/j.expneurol.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieck DC, Zhan WZ, Fang YH, Ermilov LG, Sieck GC, Mantilla CB. Structure-activity relationships in rodent diaphragm muscle fibers vs. neuromuscular junctions. Respir Physiol Neurobiol. 2012;180 (1):88–96. doi: 10.1016/j.resp.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver JR, Lehr RP. Dyspnoea during generalised spasms in tetraplegic patients. J Neurol Neurosurg Psychiatry. 1981;44 (9):842–845. doi: 10.1136/jnnp.44.9.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin DD, Fitzgerald F, Parker JD, Newton G, Floras JS, Bradley TD. Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. Am J Respir Crit Care Med. 1999;160 (4):1101–1106. doi: 10.1164/ajrccm.160.4.9903020. [DOI] [PubMed] [Google Scholar]
- Skatrud JB, Dempsey JA. Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. J Appl Physiol. 1983;55 (3):813–822. doi: 10.1152/jappl.1983.55.3.813. [DOI] [PubMed] [Google Scholar]
- Sperry MA, Goshgarian HG. Ultrastructural changes in the rat phrenic nucleus developing within 2 h after cervical spinal cord hemisection. Exp Neurol. 1993;120 (2):233–244. doi: 10.1006/exnr.1993.1058. [DOI] [PubMed] [Google Scholar]
- Strakowski JA, Pease WS, Johnson EW. Phrenic nerve stimulation in the evaluation of ventilator-dependent individuals with C4- and C5-level spinal cord injury. Am J Phys Med Rehabil. 2007;86 (2):153–157. doi: 10.1097/PHM.0b013e31802edce9. [DOI] [PubMed] [Google Scholar]
- Strey KA, Baker-Herman TL. Local reductions in synaptic inputs to the phrenic motor pool elicits inactivity-induced phrenic motor facilitation (iPMF). 5th SFN Satellite Symposium on Motor Systems Abstract. National Institute of Health; Washington DC. 2011. p. Abstr. 58. [Google Scholar]
- Strey KA, Nichols NL, Baertsch NA, Broytman O, Baker-Herman TL. Spinal atypical protein kinase C activity is necessary to stabilize inactivity-induced phrenic motor facilitation. J Neurosci. 2012;32 (46):16510–16520. doi: 10.1523/JNEUROSCI.2631-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadjalli A, Duffin J, Peever J. Identification of a novel form of noradrenergic-dependent respiratory motor plasticity triggered by vagal feedback. J Neurosci. 2010;30 (50):16886–16895. doi: 10.1523/JNEUROSCI.3394-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia IE, Karamessinis L, Bandla P, Huang J, Kelly A, Pepe M, Schultz B, Gallagher P, Brooks LJ, Marcus CL. Polysomnographic values in children undergoing puberty: pediatric vs. adult respiratory rules in adolescents. Sleep. 2008;31 (12):1737–1744. doi: 10.1093/sleep/31.12.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thille AW, Rodriguez P, Cabello B, Lellouche F, Brochard L. Patient-ventilator asynchrony during assisted mechanical ventilation. Intensive Care Med. 2006;32 (10):1515–1522. doi: 10.1007/s00134-006-0301-8. [DOI] [PubMed] [Google Scholar]
- Tkacova R, Hall MJ, Liu PP, Fitzgerald FS, Bradley TD. Left ventricular volume in patients with heart failure and Cheyne–Stokes respiration during sleep. Am J Respir Crit Care Med. 1997;156 (5):1549–1555. doi: 10.1164/ajrccm.156.5.9612101. [DOI] [PubMed] [Google Scholar]
- Tobin MJ. Advances in mechanical ventilation. N Engl J Med. 2001;344 (26):1986–1996. doi: 10.1056/NEJM200106283442606. [DOI] [PubMed] [Google Scholar]
- Tobin MJ, Laghi F, Brochard L. Role of the respiratory muscles in acute respiratory failure of COPD: lessons from weaning failure. J Appl Physiol. 2009;107 (3):962–970. doi: 10.1152/japplphysiol.00165.2009. [DOI] [PubMed] [Google Scholar]
- Trinder J, Whitworth F, Kay A, Wilkin P. Respiratory instability during sleep onset. J Appl Physiol. 1992;73 (6):2462–2469. doi: 10.1152/jappl.1992.73.6.2462. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135 (3):422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uliel S, Tauman R, Greenfeld M, Sivan Y. Normal polysomnographic respiratory values in children and adolescents. Chest. 2004;125 (3):872–878. doi: 10.1378/chest.125.3.872. [DOI] [PubMed] [Google Scholar]
- Vinit S, Kastner A. Descending bulbospinal pathways and recovery of respiratory motor function following spinal cord injury. Respir Physiol Neurobiol. 2009;169:115–122. doi: 10.1016/j.resp.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Vinit S, Darlot F, Stamegna JC, Sanchez P, Gauthier P, Kastner A. Long-term reorganization of respiratory pathways after partial cervical spinal cord injury. Eur J Neurosci. 2008;27 (4):897–908. doi: 10.1111/j.1460-9568.2008.06072.x. [DOI] [PubMed] [Google Scholar]
- Vinit S, Stamegna JC, Boulenguez P, Gauthier P, Kastner A. Restorative respiratory pathways after partial cervical spinal cord injury: role of ipsilateral phrenic afferents. Eur J Neurosci. 2007;25 (12):3551–3560. doi: 10.1111/j.1460-9568.2007.05619.x. [DOI] [PubMed] [Google Scholar]
- Wang CX, Nuttin B, Heremans H, Dom R, Gybels J. Production of tumor necrosis factor in spinal cord following traumatic injury in rats. J Neuroimmunol. 1996;69:151–156. doi: 10.1016/0165-5728(96)00080-x. [DOI] [PubMed] [Google Scholar]
- Wang H, Parker JD, Newton GE, Floras JS, Mak S, Chiu KL, Ruttanaumpawan P, Tomlinson G, Bradley TD. Influence of obstructive sleep apnea on mortality in patients with heart failure. J Am Coll Cardiol. 2007;49 (15):1625–1631. doi: 10.1016/j.jacc.2006.12.046. [DOI] [PubMed] [Google Scholar]
- Webber CL, Pleschka K. Respiratory effects of high cervical cord cold blockade on efferent vagal and phrenic discharges in the rabbit. Pflügers Arch. 1984;402:10–17. doi: 10.1007/BF00584825. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, Ramirez JM. Autonomic dysregulation in young girls with Rett Syndrome during nighttime in-home recordings. Pediatr Pulmonol. 2008;43 (11):1045–1060. doi: 10.1002/ppul.20866. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Trang H. An official ATS clinical policy statement: congenital central hypoventilation syndrome genetic basis, diagnosis and management. Am J Respir Crit Care Med. 2010;181:626–44. doi: 10.1164/rccm.200807-1069ST. [DOI] [PubMed] [Google Scholar]
- Whitelaw WA, Derenne JP, Milic-Emili J. Occlusion pressure as a measure of respiratory center output in conscious man. Respir Physiol. 1975;23 (2):181–199. doi: 10.1016/0034-5687(75)90059-6. [DOI] [PubMed] [Google Scholar]
- Xie A, Bedekar A, Skatrud JB, Teodorescu M, Gong Y, Dempsey JA. The heterogeneity of obstructive sleep apnea (predominant obstructive vs pure obstructive apnea) Sleep. 2011;34 (6):745–750. doi: 10.5665/SLEEP.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Puleo DS, Rahko PS, Dempsey JA. Apnea-hypopnea threshold for CO2 in patients with congestive heart failure. Am J Respir Crit Care Med. 2002;165 (9):1245–1250. doi: 10.1164/rccm.200110-022OC. [DOI] [PubMed] [Google Scholar]
- Yin HZ, Hsu CI, Yu S, Rao SD, Sorkin LS, Weiss JH. TNF-α triggers rapid membrane insertion of Ca(2+) permeable AMPA receptors into adult motor neurons and enhances their susceptibility to slow excitotoxic injury. Exp Neurol. 2012;238 (2):93–102. doi: 10.1016/j.expneurol.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes M. Control of breathing during mechanical ventilation. In: Slutsky AS, Brochard L, editors. Mechanical Ventilation. Springer; Berlin, Heidelberg: 2006. pp. 63–82. [Google Scholar]
- Yumino D, Bradley TD. Central sleep apnea and Cheyne–Stokes respiration. Proc Am Thorac Soc. 2008;5 (2):226–236. doi: 10.1513/pats.200708-129MG. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McGuire M, White DP, Ling L. Episodic phrenic-inhibitory vagus nerve stimulation paradoxically induces phrenic long-term facilitation in rats. J Physiol. 2003;551 (Pt 3):981–991. doi: 10.1113/jphysiol.2003.048157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Champagnat J, Poon CS. Phasic and long-term depression in brainstem nucleus tractus solitarius neurons: differing roles of AMPA receptor desensitization. J Neurosci. 1997;17 (14):5349–5356. doi: 10.1523/JNEUROSCI.17-14-05349.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]