

Abstract
MLL (mixed‐lineage leukemia)‐fusion genes induce the development of leukemia through deregulation of normal MLL target genes, such as HOXA9 and MEIS1. Both HOXA9 and MEIS1 are required for MLL‐fusion gene‐induced leukemogenesis. Co‐expression of HOXA9 and MEIS1 induces acute myeloid leukemia (AML) similar to that seen in mice in which MLL‐fusion genes are over‐expressed. p27kip1 (p27 hereafter), a negative regulator of the cell cycle, has also been defined as an MLL target, the expression of which is up‐regulated in MLL leukemic cells (LCs). To investigate whether p27 plays a role in the pathogenesis of MLL‐leukemia, we examined the effects of p27 deletion (p27−/−) on MLL‐AF9 (MA9)‐induced murine AML development. HOXA9/MEIS1 (H/M)‐induced, p27 wild‐type (p27+/+) and p27−/− AML were studied in parallel as controls. We found that LCs from both MA9‐AML and H/M‐AML can be separated into three fractions, a CD117‐CD11bhi differentiated fraction as well as CD117+CD11bhi and CD117+CD11blo, two less differentiated fractions. The CD117+CD11blo fraction, comprising only 1–3% of total LCs, expresses higher levels of early hematopoietic progenitor markers but lower levels of mature myeloid cell markers compared to other populations of LCs. p27 is expressed and is required for maintaining the quiescent and drug‐resistant states of the CD117+CD11blo fraction of MA9‐LCs but not of H/M‐LCs. p27 deletion significantly compromises the leukemogenic capacity of CD117+CD11blo MA9‐LCs by reducing the frequency of leukemic stem cells (LSCs) but does not do so in H/M‐LCs. In addition, we found that p27 is highly expressed and required for cell cycle arrest in the CD117‐CD11bhi fraction in both types of LCs. Furthermore, we found that c‐Myc expression is required for maintaining LCs in an undifferentiated state independently of proliferation. We concluded that p27 represses the proliferation of LCs, which is specifically required for maintaining the quiescent and drug‐resistant states of a small subset of MA9‐LSCs in collaboration with the differentiation blockage function of c‐Myc.
Keywords: MLL-leukemia, p27kip1, Leukemia stem cell, Quiescence
Highlights
p27 is specifically expressed in a small subset of MLL‐AF9 LSC but not in HOXA9/MEIS1 LSC.
The niche‐generated factors Flt3L and SCF induce p27 expression in MLL‐AF9 LSC but not in HOXA9/MEIS1 LSC.
p27 restricts proliferation and maintains resistance to chemotherapeutic agents in MLL‐AF9 LSC.
Over‐expression of p27 in HOXA9/MEIS1 LSC leads to quiescence and resistance to chemotherapeutic agents.
1. Introduction
MLL protein, a large histone methyltransferase, maintains the expression of a subset of genes by specifically methylating histone H3 at lysine 4 (H3K4) in the promoter regions of its target genes (Milne et al., 2002). Most MLL‐target genes, such as those within the Hox loci and many other genes, are master regulators of development and are also key regulators of hematopoiesis (Abramovich and Humphries, 2005). Gene knockout studies suggested that Mll is dispensable for the initiation of stage‐specific expression of its target Hox genes but is absolutely required for the maintenance of expression of such genes during early development (Yu et al., 1995). Mice with homozygous Mll mutation suffer early embryonic lethality with significant defects in both yolk sac primary and AGM definitive hematopoiesis (Yagi et al., 2004, 2004, 1997, 1998). The self‐renewal and proliferation of hematopoietic stem cells (HSCs) are significantly compromised by Mll inactivation (McMahon et al., 2007; Jude et al., 2007). The hematopoietic defects in Mll‐knockout mice (Mll −/−) might be mainly due to a reduction in the expression of Hox genes because the proliferative defects of hematopoietic stem and progenitor cells (HSPCs) derived from Mll −/− embryonic stem cells can be largely rescued by the over‐expression of Hox‐cluster genes (Ernst et al., 2004, 2004).
MLL‐rearrangements are found in >70% of infant acute lymphoblastic leukemias (ALL), approximately 10% adult acute myeloid leukemias (AMLs) and >30% of therapy‐related leukemias (Liu et al., 2009). MLL‐fusion proteins, the products of MLL rearrangements, induce the development of leukemia through deregulating MLL target genes, such as HOXA9, HOXA7, MEIS1 and PBX1. (Marschalek, 2011; Muntean and Hess, 2011; Faber et al., 2009; Dou and Hess, 2008; Zeisig et al., 2004; Kroon et al., 1998; Li et al., 2009; Andreeff et al., 2008). Although they lack histone methyltransferase activity, MLL‐fusion proteins contain Menin/LEDGF binding sites, a nuclear localization sequence and a CxxC motif, all of which are features essential for the correct targeting of the MLL complex to the promoter regions of its target genes (Milne et al., 2010; Milne et al., 2005; Zeleznik‐Le et al., 1994; Ayton et al., 2004). Interrupting the DNA binding ability of MLL‐fusion proteins by disrupting these DNA binding motifs will significantly compromise their leukemogenic capacity due to their consequent failure to up‐regulate MLL target genes (Slany et al., 1998; Cierpicki et al., 2010; Yokoyama et al., 2005; Caslini et al., 2007; Yokoyama and Cleary, 2008; Bach et al., 2009). The requirement for the wild‐type (WT) allele of MLL for MLL‐fusion protein‐induced leukemic transformation suggested that both MLL‐fusion protein and WT‐MLL might be recruited to the regulatory regions of target genes. An MLL‐fusion protein might be dependent upon WT‐MLL's methyltransferase activity and hijack the normal transcription/elongation machinery to induce target gene expression (Milne et al., 2010; Muntean et al., 2010; Bernt et al., 2011; Chang et al., 2010; Thiel et al., 2011). Many MLL target genes are consistently up‐regulated in human MLL cells (Slany, 2009; Imamura et al., 2002; Lawrence et al., 1999; Armstrong et al., 2002). Several of these genes, including HOXA9, HOXA7, MEIS1 and PBX1/2, have been shown to be required for MLL‐fusion protein‐induced leukemia development as shown by genetic studies (Faber et al., 2009; Wong et al., 2007; Kumar et al., 2009; Ayton and Cleary, 2003). In addition, co‐expression of HOXA9 and MEIS1 induces leukemic transformation in HSCs by promoting self‐renewal/proliferation and blocking differentiation, which mimic most of the functions of MLL‐fusion proteins (Zeisig et al., 2004; Kroon et al., 1998). These studies demonstrated the key role of Hox‐cluster genes in the pathogenesis of MLL‐fusion protein‐induced leukemia.
p27kip1 (p27 hereafter), a negative regulator of the cell cycle, has been identified as a direct target of MLL (Milne et al., 2005; Xia et al., 2005). Menin, the product of the tumor suppressor gene Multiple endocrine neoplasia, represses cell growth by inducing p27 expression (Horiguchi et al., 2009). It was found that Menin functions as an adaptor for the interface of MLL and LEDGF (lens epithelium‐derived growth factor), which mediates the specific binding of MLL to chromatin and DNA (Milne et al., 2005; Caslini et al., 2007; Yokoyama and Cleary, 2008). Both Menin and LEDGF are required for the correct localization of MLL to its target genes and inducing target gene expression. Genetic disruption of the MLL/Menin/LEDGF interaction leads to down‐regulation of MLL target gene expression in MLL‐fusion gene‐transduced cells and blocks the development of leukemia (Yokoyama et al., 2005; Caslini et al., 2007; Yokoyama and Cleary, 2008). In MLL‐fusion gene‐transduced LCs, Menin recruits both WT MLL and MLL‐fusion proteins to the p27 promoter, which might cooperatively up‐regulate p27 expression (Milne et al., 2005; Xia et al., 2005). In contrast to other leukemogenic fusion protein‐related LCs, Flt3‐L (Flt3‐Ligand) specifically induces p27 expression in MLL‐fusion‐related LCs and represses their proliferation (Furuichi et al., 2007). This specific up‐regulation of p27 in MLL LCs suggested that it might play a distinct role in the pathogenesis of MLL‐fusion gene‐induced leukemia.
To investigate the role of p27 in the development of MLL‐fusion gene‐induced leukemia, we evaluated the effects of deletion of the p27 gene on the pathogenesis of MLL leukemia by comparative study of the in vitro growth behaviors and in vivo leukemogenic activity of p27 +/+ (p27 WT) and p27 −/− (p27‐knockout), MA9 (MLL‐AF9) murine LCs, as well as p27 +/+ and p27 −/− H/M (HOXA9/MEIS1) murine LCs. We found that in both types of leukemia, p27 is highly expressed in CD117‐ differentiated LCs, and this factor is responsible for the cell cycle arrest state of these cells. However, in CD117+ undifferentiated LCs, p27 is expressed only in the CD11blo population of MA9‐LCs but not in the same population of H/M‐LCs. The CD117+CD11blo population represents a small subset of LCs (1–3%) in both types of leukemia that express several markers specific for HSPCs. Interestingly, p27 expression can be induced by Flt3‐L and SCF in CD117+ MA9‐LCs but not in CD117+ H/M‐LCs. Moreover, we found that p27 is responsible for maintaining quiescence, leukemogenic ability and drug‐resistance in the CD117+CD11blo population of MA9‐LCs but not in H/M LCs.
2. Materials and methods
2.1. Mice
All experiments were performed in strict accordance with the provisions of Loyola University Chicago IACUC protocol #06‐013. p27 −/− mice (C57Bl6J background, which are CD45.2+, Stock Number: 010834) and Ptprc recipient mice (C57Bl6J background, which are CD45.1+, Stock Number: 002725) were purchased from the Jackson Laboratory and maintained in the Department of Comparative Medicine, Loyola University Medical Center. c‐Myc fx/fx conditional c‐Myc knockout mice (de Alboran et al., 2001) were kindly provided by Dr. Frederick W. Alt of the Howard Hughes Medical Institute (C57Bl6J background, CD45.2+). c‐Myc fx/fx p27 −/− compound‐mutant mice were generated by crossing p27 −/− mice with c‐Myc fx/fx mice. Genotyping for p27 −/− and c‐Myc fx/fx mice was performed by tail biopsy using PCR assay with the following primers: p27 forward: GAACTAACCCGGGACTTGG AGAAGC; p27 reverse: TAACCCAGCCTGATTGTCTGACGAG; c‐Myc forward: GCCCCTGAATTGCTAGGAAGACTG; c‐Myc reverse: CCGACCGGGTCCGAGTCCCTATT.
2.2. CD117+ HSPC isolation, infection and in vitro culture
HoxA9‐GFP (Cat. No. 20977) and Meis1‐YFP (Cat. No. 21013) plasmids were purchased from Addgene. MLL‐AF9 (MA9), HoxA9 or Meis1‐expressing retroviruses were produced by co‐transfecting 293T‐HEK cells with packaging plasmids and the appropriate retroviral vector (MLL‐AF9 (MA9)‐neo, HoxA9‐GFP or Meis1‐YFP) using Lipofectamine 2000 ® reagent (Invitrogen). Bone marrow (BM) CD117+ HSPCs from p27 +/+ and p27 −/− mice were enriched using EasySep ® Mouse CD117 Positive Selection Kit (StemCell Technologies) and cultured in 12‐well plates in 4‐cytokine stem cell culture medium (RPMI 1640 medium containing 10% FBS, 100 ng/ml SCF, 50 ng/ml IL‐6, 10 ng/ml IL‐3, and 20 ng/ml GM‐CSF) overnight to induce cell proliferation (pre‐stimulation) in order to improve the efficiency of infection. Pre‐stimulated HSPCs were infected with MA9‐expressing retrovirus or co‐infected with HoxA9 or Meis1‐expressing retrovirus by spinoculation in the presence of 4 μg/mL polybrene. Twenty‐four hours after the spinoculation, MA9‐infected cells were selected with 1.2 mg/ml G418 for 2 weeks with daily medium change, whereas HoxA9 and Meis1 co‐infected cells were purified by sorting for GFP+/YFP+ cells using FACS and continuously culturing for two weeks. HoxA9 and Meis1 co‐infected cells were analyzed by flow cytometry to confirm their purity. Samples with >99% GFP+/YFP+ cells were used for further studies. All cytokines used in this study were purchased from eBioscience.
2.3. In vitro colony‐forming assay
Indicated numbers of pre‐LCs or LCs were seeded into MethoCult ® GF M3434 medium (StemCell Technologies) and incubated at 37 °C., 100% humidity, and 5% CO2. Numbers of colonies were counted on day 7 of culturing.
2.4. In vivo transplantation and leukemogenic assay
Recipient mice were lethally irradiated (10.5 Gy) in a Nordion Gammacell 40 irradiator. Indicated numbers of pre‐LCs or LCs (CD45.2+) were mixed with normal support BM mononuclear cells (MNCs, CD45.1+) and transplanted into recipient mice (CD45.1+). Mice were monitored for signs of leukemia. Leukemia was diagnosed by the detection of leukemic blasts in PB and BM, as well as by observing liver infiltration. The donor origin of the leukemic cells was confirmed by CD45.2 staining for MA9 leukemia or GFP+/YFP+ for H/M leukemia. Immunophenotypes of LCs were examined by staining the cells with fluorescein‐conjugated antibodies followed by flow cytometric analysis. Antibodies used in this study included: rat anti‐mouse CD117, CD11b, CD34, CD123, CD135, CD48, CD150, CD19, Ter119, CD11c, F4/80 and Gr1, all of which were purchased from eBioscience. After mice were sacrificed, their livers were immediately collected. After 2–3 days fixation in zinc formalin, liver tissues were embedded in paraffin. Sections were cut for H & E staining.
2.5. BrdU incorporation
To examine the proliferation of CD117‐CD11bhi, CD117+CD11bhi and CD117+CD11blo LCs, 4 h prior to being sacrificed, mice were injected with BrdU at 50 μg/g body weight. MNCs were collected from leukemic mice. The percentages of BrdU+ cells within different subsets of LCs were assessed by CD117 and CD11b cell‐surface marker staining followed by cell permeabilization and BrdU antibody staining, as previously reported (Zhang et al., 2003). To examine the proliferation of pre‐LCs and LCs cultured in vitro, cells were treated with BrdU at a concentration of 10 ng/ml commencing 1–2 h prior to collection. The percentages of BrdU+ cells were detected by cell permeabilization and BrdU antibody staining.
2.6. Cell cycle analysis
CD117−CD11bhi, CD117+CD11bhi and CD117+CD11blo LCs were sorted from the spleens of leukemic mice by FACS and fixed in cold 70% ethanol for 3 h. Cells were then washed twice with PBS containing 0.5 μg/ml pyronin‐Y and 2 μg/ml Hoechst 33342 and stained in the same solution for 10 min. Data were collected using a FACSAria ® cytometer (BD Bioscience) and analyzed using FlowJo ® software.
2.7. Annexin‐V staining to assess apoptosis
Cells were stained with allophycocyanin‐conjugated Annexin‐V in binding buffer following the manufacturer's instructions (BD Biosciences). Death of cells was determined by examination of the percentage of Annexin‐V+ cells by flow cytometry.
2.8. qRT‐PCR
RNA was isolated using RNeasy Plus ® Mini kit (Qiagen). cDNA was prepared using SuperScript ® First‐Strand Synthesis System (Invitrogen). Quantitative PCR for detecting p27, p21 and p57 mRNA expression levels was conducted using SYBR Green PCR Master ® (Applied Biosystems). Information concerning the primers used can be found in Supplementary Table 1. Triplicate RT‐PCRs were performed.
2.9. Statistical analyses
The serial dilution and competitive transplantation data were analyzed by L‐Calc™ Software (StemCell Technologies). All other data were analyzed using the 2‐tailed Student t Test with GraphPad Prism ® software (Version 5.04) to identify significant differences between groups. Values are expressed as means ± SEM. Differences were regarded as significant at p < 0.05.
3. Results
3.1. p27 deletion does not affect the immunophenotype of MA9‐AML and H/M‐AML LCs in mice
To study whether p27 is required for the pathogenesis of MA9‐AML, we generated both p27 +/+ and p27 −/− murine MA9‐AMLs by transplanting MA9‐transduced p27 +/+ and p27 −/− HSPCs (CD45.2+) into lethally‐irradiated congenic mice (CD45.1+) with the support of CD45.1+ normal BM‐MNCs. We also generated p27 +/+ and p27 −/− murine H/M‐AMLs by transplanting H/M‐transduced p27 +/+ and p27 −/− HSPCs into lethally‐irradiated congenic mice. The recipient mice were monitored for signs of leukemia by examining their peripheral blood (PB) for the percentage of CD45.2+ LCs (for MA9 transduction) and the percentage of GFP+YFP+ LCs (for H/M transduction). After leukemia developed (indicated by LCs > 60% in PB), mice were sacrificed and LCs from the BM and spleens of the mice were collected for phenotype analysis. We found that p27 deletion delayed MA9‐leukemia development but had no significant influence on H/M leukemia development as shown by the survival curves of the recipient mice (Supplementary Figure 1). In addition, we found that almost all LCs derived from both MA9‐ and H/M‐transduced HSPCs were CD11b+Gr1+ myeloid blasts which were not affected by the deletion of p27 (Figure 1A). Consistent with previous studies (Somervaille and Cleary, 2006), we found that 35–60% (average 43%) of MA9‐LCs and 21–46% (average 32%) of H/M‐LCs also express moderate levels of CD117 (CD117+). Again here, these percentages were not affected by p27 deletion (Figure 1B). In addition, we found that CD117+ LCs in both types of leukemia can be further separated into CD11bhi and CD11blo populations. The percentage of CD117+CD11blo cells is 1–3% in both MA9‐LCs and H/M‐LCs, which was not affected by p27 deletion (Figure 1B). Furthermore, no significant differences in LCs in PB (Figure 1C) or liver infiltrates (Figure 1D) were observed between mice receiving MA9‐transduced p27 +/+ and p27 −/− HPSCs, nor between mice receiving H/M‐transduced p27 +/+ and p27 −/− HSPCs. These data indicated that p27 is not required for the leukemic transformation of either MA9‐transduced HSPCs or H/M‐transduced HSPCs.
Figure 1.
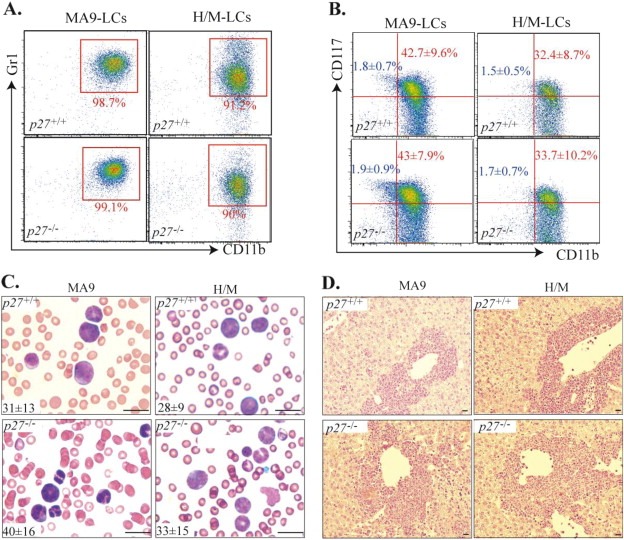
p27 deletion does not affect the phenotype of either MA9‐ or H/M‐induced leukemia. Mice which had received MA9‐transduced p27+/+ or p27−/− HPSCs as well as mice which had received H/M‐transduced p27+/+ or p27−/− HPSCs were monitored for leukemia development. After leukemia developed, mice were sacrificed. LCs were collected from the spleens of leukemic mice and stained with Gr1, CD11b and CD117 antibodies. Immunophenotype was compared between p27+/+ and p27−/− MA9‐LCs as well as between p27+/+ and p27−/− H/M‐LCs (A & B). Percentages of CD117+ and CD117+CD11blo cells are shown in red and blue, respectively (B). Leukemia was verified by peripheral blood smear (C) and liver histologic section studies (D). Average numbers ( × 106/ml) of white blood cell counts (WBC) are shown at the lower left corner of each picture (C). n = 6. Bars represent 50 μm in C and 100 μm in D.
3.2. p27 is required for maintaining the quiescent state of the CD117+CD11blo population of MA9‐LCs but not of H/M‐LCs
It has been reported that the p27 gene is a direct target of MLL and MLL‐fusion proteins, the expression of which is up‐regulated in MLL LCs but not in other types of LCs (Milne et al., 2005; Xia et al., 2005; Furuichi et al., 2007). However, we found that levels of p27 were comparable between MA9‐LCs and H/M‐LCs when an unsorted population of cells was examined (Supplementary Figure 2). Interestingly, we found that in MA9‐LCs, p27 is expressed in the CD117+CD11blo population and is down‐regulated in the CD117+CD11bhi population, and is significantly up‐regulated in CD117‐CD11bhi population, while in H/M‐LCs, the expression levels of p27 are low in both the CD117+CD11blo and CD117+CD11bhi populations, and is also up‐regulated in the CD117‐CD11bhi population (Figure 2A–C). However, the expression of p21cip1 (p21 hereafter) and p57kip2 (p57 hereafter), which are other cell cycle inhibitory proteins in the p27 family, as well as the expression of p18ink4c, are comparable between CD117+CD11blo and CD117+CD11bhi populations in both types of LCs, while p15ink4b and p16 ink4b cannot be detected in all of those types of LCs. In addition, the expression of p21, p57 and p18 is not altered by p27 deletion. (Supplementary data 3). These data suggested that p27 might play a unique role in the pathogenesis of MA9‐AML. Consistent with its function, we found that expression levels of p27 are well correlated to the proliferative and cell cycle states of LCs (Figure 2D and E). In MA9‐LCs, the CD117+CD11blo population is relatively quiescent compared to the CD117+CD11bhi population, as shown by both BrdU pulse‐labeling (Figure 2D) and pyronin‐Y/Hoechst staining (Supplementary data 4C), whereas in H/M‐LCs, proliferation of the CD117+CD11blo population is comparable to that of the CD117+CD11bhi population (Figure 2E). Proliferation of the CD117‐CD11bhi population in both types of LCs is significantly reduced (Figure 2D and E). Furthermore, we found that p27 deletion significantly promotes the proliferation of the CD117+CD11blo population of MA9‐LCs (Figure 2D) and the CD117‐CD11bhi population of both types of LCs (Figure 2D and E), but does not affect the proliferation of the CD117+CD11bhi population in either type of LC (Figure 2D and E). It was known that the CD117‐CD11bhi population consists of differentiated LCs whereas LSCs are enriched in CD117+ LCs. We found that the CD117‐CD11bhi population of both types of LCs contains differentiated granulocytes as shown by Wright's Giemsa staining (Figure 2F and G). The differentiated state of CD117‐CD11bhi populations in both types of LCs is not affected by p27 deletion (Figure 2F and G). These data suggested that p27 inhibits the proliferation of both MA9 and H/M LCs but is not required for the differentiation of either type. The difference in p27 expression and cell cycle state of the CD117+CD11blo population between MA9‐LCs and H/M‐LCs indicated that p27 might be required for maintaining the quiescent state in a small subset of MA9‐LSCs but not of H/M‐LSCs.
Figure 2.
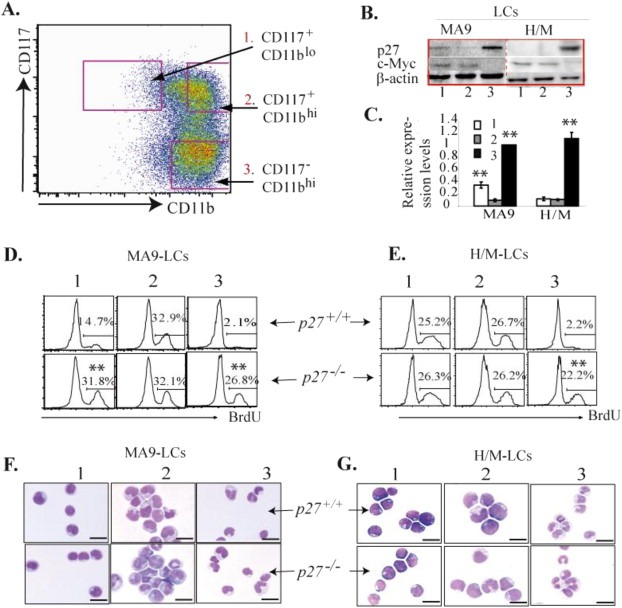
p27 is required for maintaining the quiescent state of the CD117+CD11blo population of MA9‐LCs but not of H/M‐LCs. A–C. LCs were collected from the spleens of p27+/+ MA9 (CD45.2+) and p27+/+ H/M (GFP+/YFP+) leukemic mice and were separated into CD117+CD11blo, CD117+CD11bhi and CD117‐CD11bhi populations by FACS (A). p27 and c‐Myc expression in these populations of cells was examined by Western Blotting (B) and p27 expression by qRT‐PCR assays (C). The relative levels of mRNA expression in C were first normalized to the expression levels of β‐actin in each sample and then further normalized by relative expression levels of p27 in the CD117‐CD11bhi population of MA9‐LCs. ** indicates significant increase compared to CD117+CD11bhi groups (p < 0.01). D & E. Leukemic mice were injected with 1 mg BrdU 2 h prior to being sacrificed. Proliferation of CD117+CD11blo, CD117+CD11bhi and CD117‐CD11bhi populations of LCs was compared between p27+/+ and p27−/− MA9‐LCs (D) as well as between p27+/+ and p27−/− H/M‐LCs (E). ** indicates significant increase compared to corresponding population in p27+/+ LCs (p < 0.01). Comparison of the morphology of the three populations in both p27+/+ and p27−/− MA9‐LCs (F) as well as between p27+/+ and p27−/− H/M‐LCs (G). Bars represent 50 μm.
3.3. Proliferation and differentiation of LCs are uncoupled and controlled by p27 and c‐Myc respectively
c‐Myc has been shown to play a critical role in the pathogenesis of most acute leukemias by promoting the proliferation and blocking the differentiation of HSPCs (Hoffman et al., 2002; Weng et al., 2006; Fang et al., 2009; Schreiner et al., 2001). We found that c‐Myc is expressed in both CD117+CD11blo and CD117+CD11bhi LC populations but is significantly down‐regulated in the CD117‐CD11bhi population of both MA9‐ and H/M‐LCs (Figure 2A–B). c‐Myc expression correlates well with the differentiation state but not the proliferation rate of LCs (Figure 2). Moreover, we found that knockout of c‐Myc (c‐Myc −/−) promotes MA9‐LC differentiation followed by cell cycle arrest (Figure 3A and B). Inactivation of p27 can largely prevent cell cycle arrest in LCs without significantly affecting differentiation induced by c‐Myc gene deletion as shown in Figure 3A–C. In colony‐forming assays, we found that c‐Myc ‐/‐ LCs can only divide 1–3 times and form 2‐ to 8‐cell clusters (data not shown), whereas many of the c‐Myc −/− p27 −/− LCs were able to divide for 4–5 times and generate clusters of 10–30 cells. Cells from these clusters are mature granulocytes (Figure 3D). However, despite the fact that they were proliferating, due to their complete maturation after day 3 of culturing (Figure 3A), c‐Myc ‐/‐ p27 −/− LCs fail to form large colonies.
Figure 3.
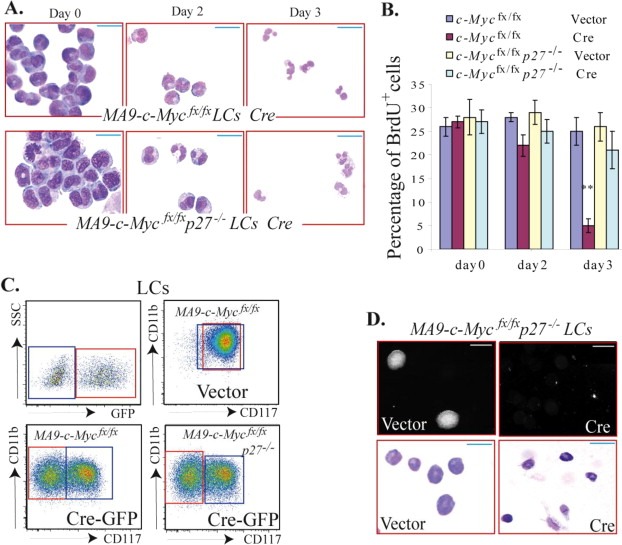
p27‐deficiency prevents cell cycle arrest but not differentiation in MA9‐LCs induced by c‐Myc deletion. CD117+ HSPCs from c‐Mycfx/fx (conditional c‐Myc knockout) mice and c‐Mycfx/fxp27−/− mice were infected with MA9‐expressing retrovirus. The infected cells were transplanted into mice for leukemia development. c‐Mycfx/fx MA9‐LCs and c‐Mycfx/fxp27−/− MA9‐LCs were collected from the spleens of leukemic mice and infected with Cre‐GFP‐expressing retrovirus to induce c‐Myc deletion. Vector‐only virus infection was studied in parallel as a control. A & B. Infected cells were purified by FACS of GFP+ cells on the 2nd day after infection and cultured in 4‐cytokine medium. Differentiation and proliferation of infected cells were examined on days 0, 2 and 3 after sorting. The differentiation of infected cells was evaluated by morphology (A) and proliferation was examined after 2 h of BrdU pulse‐labeling (B). C. The mixture of infected and uninfected cells was cultured in 4‐cytokine medium for 4 days. Differentiation of the infected cells (GFP+ in red gate) was examined by detecting CD117 expression using flow cytometry. c‐Myc‐deleted cells (Cre‐infected) became CD117‐CD11b+ differentiated cells despite their p27 status, whereas c‐Myc intact‐LCs (GFP‐ uninfected cells in blue gate and vector‐only control cells) retained their CD117+CD11b+ undifferentiated status. D. Infected cells were purified by FACS and seeded into MethoCult® GF M3434 medium for 5 days for colony‐forming analysis (C, upper panel; photomicrographs were obtained using the same magnification). The morphology of the cells from the colonies was examined by Wright's Giemsa staining (C, lower panel). ** in B indicates significant reduction compared to other groups (p < 0.01). Bars represent 50 μm in A and lower panel of D; bar is 0.1 μm in upper panel of D.
3.4. CD117 + CD11b lo populations of MA9‐LCs and H/M‐LCs express early hematopoietic progenitor markers regardless of their p27 status
We found that populations of both MA9‐LCs and H/M‐LCs consist of 1–3% of CD117+CD11blo cells (Figure 4A and B). This percentage in both types of LCs is not affected by p27 deletion (Figure 4B). Morphologically, for both types of LCs, the CD117+CD11blo cells are relatively smaller with higher nucleus‐to‐cytoplasm ratios compared to those in the CD117+CD11bhi population. Again, this type of morphologic distinction is not altered by p27 deletion (Figure 2F and G).
Figure 4.
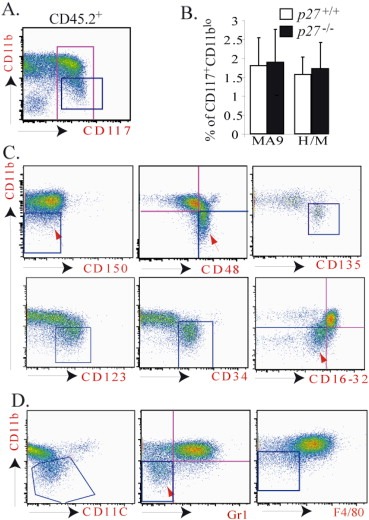
CD117+CD11blo LCs express early hematopoietic progenitor markers and lower levels of myeloid cell markers. LCs were collected from the spleens of leukemic mice and stained with cell surface markers as indicated. The phenotypes of LCs were analyzed by flow cytometry. To distinguish LCs from normal hematopoietic cells (both recipient and support BM cells are CD45.1+), cells were first gated on CD45.2+ (for MLL‐AF9 leukemia) or GFP+/YFP+ (for HoxA9/Meis1 leukemia) and then gated on CD117+ for further analysis (A). The percentages of CD117+CD11blo populations in p27+/+ and p27−/− MA9‐LCs as well as in p27+/+ and p27−/− H/M‐LCs were summarized (B). CD117+ LCs (the cells inside the red gate of figure A) were analyzed for expression of HSPC markers (C) and mature myeloid or B lymphocyte markers (D). Cells in the blue gate are CD117+CD11blo populations in all figures. Data shown in C and D are from p27+/+ MA9‐LCs. Similar data were obtained from p27−/− MA9‐LCs, p27+/+ H/M‐LCs and p27−/− H/M‐LCs (data not shown).
To study whether the CD117+CD11blo population of LCs is phenotypically different from the CD117+CD11bhi population, we compared the expression of hematopoietic cell markers between these two populations in both MA9‐LCs and H/M‐LCs. We found that in both types of LCs, compared to the CD117+CD11bhi population, the CD117+CD11blo populations express higher levels of several HSPC markers, including CD48, CD123 (IL3‐receptor), and CD135 (Flt3) (Figure 4C) but lower levels of mature myeloid cell markers, including Gr1, CD11C, F4/80 and CD16/32 (Figure 4D). In addition, in contrast to the CD117+CD11bhi population, which represents a mixture of CD34+ and CD34‐ cells (Supplementary data 4A), almost all CD117+CD11blo LCs are CD34+ (Figure 4C and Supplementary data 4A). Within the CD117+CD11bhi population, CD34+ and CD34‐ cells behaved similarly in terms of their proliferation and colony‐forming ability (Supplementary data 4C and D). These data indicated that CD117+CD11blo LCs might be relatively undifferentiated compared to CD117+CD11bhi LCs.
3.5. CD117+CD11blo MA9‐LCs contain a higher frequency of leukemogenic LSCs than CD117+CD11bhi MA9‐LCs, which can be normalized by p27 deletion
To study whether the CD117+CD11blo population of LCs is functionally different from the CD117+CD11bhi population, we compared the in vitro colony‐forming ability and in vivo leukemogenic capacity between these two populations in both MA9‐LCs and H/M‐LCs. We found that CD117+CD11blo MA9‐LCs generated significantly more large colonies (2–3 times more cells in each colony) (Figure 5A, B and Supplementary data 4D) and contained a significantly higher frequency of leukemogenic LSCs than did CD117+CD11bhi MA9‐LCs (Figure 5C and Supplementary data 5). Interestingly, p27 deletion significantly compromised the leukemogenic capacity of the CD117+CD11blo population, which makes the CD117+CD11blo population equivalent to the CD117+CD11bhi population in terms of their in vitro colony growth and in vivo leukemogenic capacity (Figure 5A and C). However, CD117+CD11blo H/M‐LCs generated comparable numbers/sizes of colonies and contained a comparable frequency of leukemogenic LSCs as CD117+CD11bhi H/M‐LCs. The clonogenic ability and leukemogenic capacity of H/M‐LCs are independent of their p27 status (Figure 5C and D). Furthermore, mice receiving CD117+CD11blo and CD117+CD11bhi LCs developed AML of exactly the same phenotype as the donor mice from which the cells had been harvested, being composed of similar percentages of CD117+CD11blo and CD117+CD11bhi cells (Figure 5E and F). These data suggested that p27 is required to maintain the leukemogenic capacity of CD117+CD11blo MA9‐LCs but not H/M‐LCs. The CD117+CD11blo and CD117+CD11bhi populations are, in effect, phenotypically inter‐convertible in vivo, with each population being able to give rise to the other.
Figure 5.
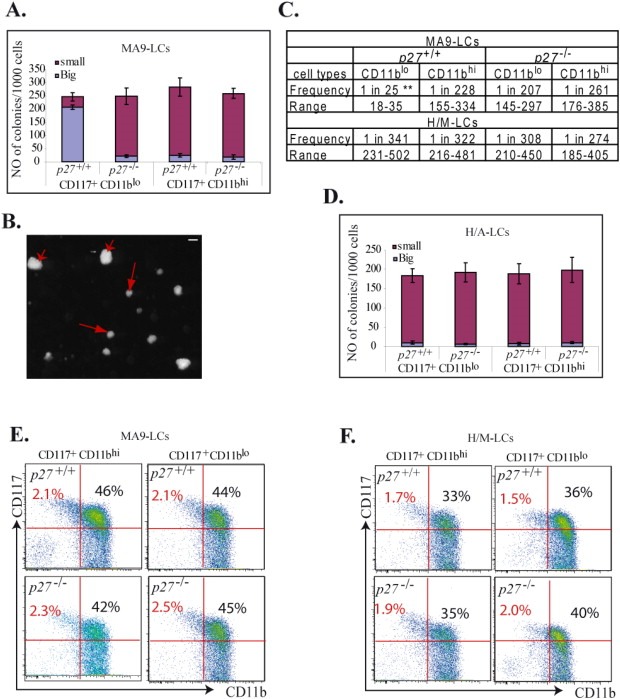
CD117+CD11blo MA9‐LCs contain a higher frequency of leukemogenic LSCs than do CD117+CD11bhi MA9‐LCs. A‐D. LCs were collected from the spleens of p27+/+ MA9 (CD45.2+) and p27+/+ H/M (GFP+/YFP+) leukemic mice and were separated into CD117+CD11blo, CD117+CD11bhi and CD117‐CD11bhi populations by FACS. Cells were seeded into MethoCult® GF M3434 medium for colony‐forming analysis. Clonogenic ability of CD117+CD11blo, CD117+CD11bhi and CD117‐CD11bhi populations was compared between p27+/+ MA9‐LCs and p27−/− MA9‐LCs (A), as well as between p27+/+ H/M‐LCs and p27−/− H/M‐LCs (D). Large and small colonies were counted in each sample. The size of large colonies (arrowhead) is 2–3 times larger than the size of small colonies (arrow) as shown in B. The frequency of LSCs in the CD117+CD11blo and CD117+CD11bhi populations in p27+/+ MA9‐LCs, p27−/− MA9‐LCs, p27+/+ H/M‐LCs and p27−/− H/M‐LCs was examined by serial dilution and competitive repopulation assays (C). ** indicates significant increase compared to other groups (p < 0.01). The percentages of CD117+CD11blo and CD117+CD11bhi populations of cells were examined in leukemic mice which had received CD117+CD11blo and CD117+CD11bhi LCs (E & F). Bar in B represents 0.1 μm.
3.6. p27 can be induced by Flt3‐ligand and SCF, and its expression is required to maintain drug resistance in CD117+CD11blo MA9‐LCs
To study whether p27 expression in MA9‐LCs can be stimulated by certain cytokines, we isolated p27 +/+ CD117+ MA9‐LCs from the spleens of leukemic mice and treated them with the indicated concentration of cytokines. p27 +/+ CD117+ H/M‐LCs were studied in parallel as controls. Interestingly, we found that, in MA9‐LCs, p27 expression was significantly induced by Flt3 Ligand (Flt3L) and SCF stimulation but not by IL‐3 nor GM‐CSF, as shown by both Western Blotting (Figure 6A) and qRT‐PCR (Figure 6B). In contrast, in H/M‐LCs, none of these cytokines increased p27 expression.
Figure 6.
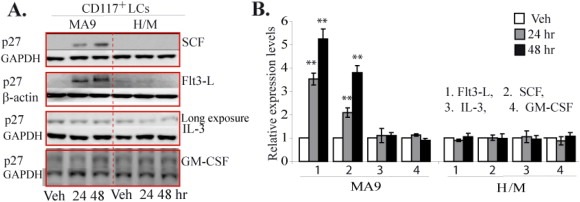
p27 expression can be induced by Flt3 and SCF in CD117+ MA9‐LCs but not in CD117+H/M‐LCs. CD117+ LCs were isolated from spleens of MA9 and H/M leukemic mice by FACS and incubated in RPMI 1640 medium supplemented with 10% FBS. Cells were treated with 100 ng/ml Flt3, 100 ng/ml SCF, 20 ng/ml IL‐3 or 25 ng/ml GM‐CSF for the indicated times. p27 expression was examined by Western Blotting (A) and qRT‐PCR (B). Vehicle (Veh) treated cells were used as controls. In B, the relative levels of mRNA expression were first normalized to the expression levels of β‐actin in each sample and then further normalized to the levels in control samples of each group. ** indicates significant increase compared to other groups (p < 0.01).
Because the CD117+CD11blo population of MA9‐LCs is relatively quiescent compared to the CD117+CD11bhi population, we predicted that the former cells might be more resistant to chemotherapeutic drug treatment. To test this hypothesis, we treated CD117+CD11blo and CD117+CD11bhi LCs with different dosages of cytosine arabinoside (Ara‐C) or daunorubicin (DNR) for 6 h. The drug sensitivity of the cells was evaluated by both Annexin‐V staining and colony‐forming assay. We found that the CD117+CD11blo population of MA9‐LCs is relatively more resistant to both Ara‐C and DNR treatment compared to the CD117+CD11bhi population (Figure 7A and C). We believe that this drug resistance in the CD117+CD11blo population of MA9‐LCs is p27‐dependent, as the cells can be re‐sensitized to the drugs' effects by p27 deletion (Figure 7A and C). However, the sensitivity of CD117+CD11blo H/M‐LCs to Ara‐C is comparable to that of the CD117+CD11bhi population (Figure 7B and D).
Figure 7.
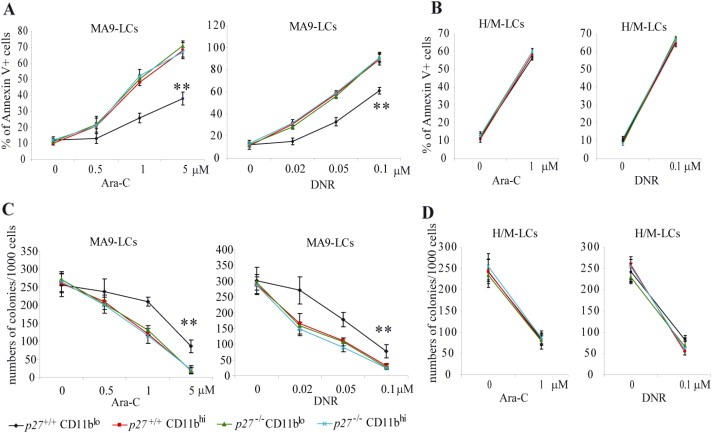
CD117+CD11blo LCs are relatively more drug resistant than are CD117+CD11bhi LCs. Populations of CD117+CD11blo and CD117+CD11bhi LCs were isolated from the spleens of p27+/+ MA9, p27−/− MA9, p27+/+ H/M and p27−/− H/M leukemic mice by FACS. Cells were incubated in 96‐well plates in 4‐cytokine culture medium with indicated dosages of cytosine arabinoside (Ara‐C) or doxorubicin (DNR). Cells were seeded at a density of 2 × 104 or 1 × 103 per well for cell death or colony‐forming analysis, respectively. Cell death was determined after 16 h of treatment by Annexin‐V staining and analyzed by flow‐cytometry (A & B). The remaining colony‐forming cells in each well after 6 h of Ara‐C or doxorubicin treatment were examined by suspension culture in MethoCult® GF M3434 (C & D). ** indicates significant difference compared to other groups (p < 0.01).
3.7. Forced p27 expression inhibits proliferation and induces drug resistance in the CD117+CD11blo population of H/M‐LCs
To further investigate whether the functional difference of the CD117+CD11blo population in MA9‐LCs compared to H/M‐LCs is due to p27, we forced the expression of p27 in the CD117+CD11blo population isolated from H/M‐LCs. We found that p27 expression inhibits proliferation in such cells as shown by BrdU pulse‐labeling (Figure 8A). We also found that p27‐transduced CD117+CD11blo H/M‐LCs are relatively more resistant to chemotherapeutic drug treatment than vector‐only‐transduced CD117+CD11blo H/M‐LCs (Figure 8B and C). These data further support the key role of p27 in inducing the quiescent/drug‐resistant feature of CD117+CD11blo LCs.
Figure 8.
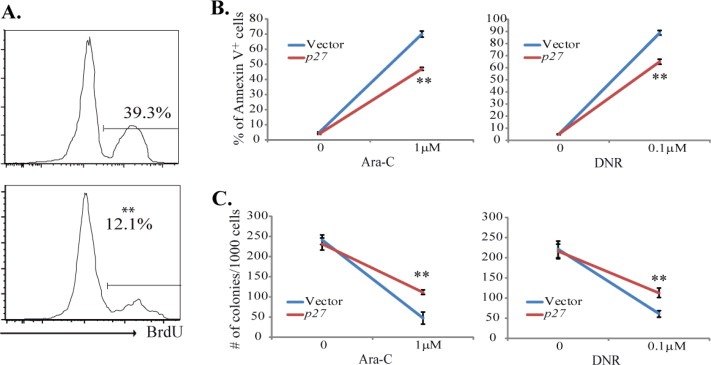
p27 over‐expression represses proliferation and induces drug resistance in CD117+CD11blo H/M‐LCs. CD117+CD11blo cells were sorted from H/M‐LCs and infected with p27‐mcherry‐expressing virus. Infected cells were purified one day after infection by sorting mcherry+ cells and were incubated in 4‐growth factor medium for 24 h mCherry vector‐only infected cells were studied in parallel as a control. A. Cells were treated with 10 ng/ml BrdU 2 h prior to collection. Cell proliferation was examined by detecting the BrdU+ cell percentage. B. Cells were seeded at a density of 2 × 104 per well and treated with indicated dosages of Ara‐C or DNR. Cell death was determined 16 h after the initiation of treatment by Annexin‐V staining and analyzed by flow‐cytometry. C. Cells were seeded at a density of 1 × 103 per well and treated with indicated dosages of Ara‐C or DNR. The remaining colony‐forming cells in each well after 6 h of Ara‐C or DNR treatment were examined by suspension culture in MethoCult® GF M3434. **indicates significant difference compared to other groups (p < 0.01).
4. Discussion
LSCs have been implicated as central to the processes involved in the progression and relapse of leukemia. The complete elimination of all LSCs from patients' tissues, therefore, has been proposed to be the ultimate and necessary goal in curing this fatal disease (Konopleva and Jordan, 2011; Becker and Jordan, 2011). However, accumulated evidence suggests that LSCs representing a special subset of LCs are able to survive intensive chemotherapeutic interventions because of the influence of niche microenvironmental factors (Saito et al., 2010, 2010). Maintaining quiescence has been proposed to be one of the mechanisms by which LSCs evade the cytotoxic effects of chemotherapeutic agents because most currently‐available chemotherapeutic drugs used to treat leukemia typically target cycling cells (Weissman, 2005; Dick, 2005; Jamieson et al., 2004; Reya et al., 2001; Guzman and Jordan, 2004). In‐depth studies of the molecular mechanisms by which LSCs maintain their state of replicative quiescence should help point the way toward the development of novel strategies to induce such cells into the cell cycle and hence become susceptible to chemotherapeutic killing. We found that, in spite of the phenotypic similarity between MA9‐LCs and H/M‐LCs, within the population of CD117+ undifferentiated LCs, p27 is specifically expressed in only a small subset of CD11blo MA9‐LCs but not in the corresponding population in H/M‐LCs. CD117+CD11blo MA9‐LCs are relatively quiescent and consist of a greater percentage of LSCs than other populations. p27 expression can be induced by Flt3‐L and SCF in MA9‐LCs, which is required for maintaining the quiescent state, drug resistance and leukemogenic capacity of CD117+CD11blo MA9‐LCs. We propose that Flt3‐L and SCF might stimulate niche signals to specifically protect these populations of LSCs from cytotoxic stress by inducing p27 expression and restricting their proliferation. This model might explain the poor prognosis associated with MLL‐fusion gene‐related leukemias compared to non‐MLL leukemias (He et al., 2011; Krivtsov and Armstrong, 2007) and in general the more favorable prognosis of leukemias with low levels of p27 expression compared to leukemias in which p27 is expressed at high levels (Haferlach et al., 2011). The molecular mechanism by which Flt3‐L and SCF specifically induce p27 in MA9‐LCs but not H/M‐LCs need to be addressed. In addition, clinical studies have suggested that a high level HoxA9 is also key marker of poor prognosis in leukemia (Li et al., 2013). Future studies need to determine whether the prognosis of leukemia with high levels of both p27 and HoxA9 is even poorer than leukemia with high levels of either p27 or HoxA9.
p27 is a member of the cip/kip cyclin‐dependent kinase inhibitor (CDKi) family of proteins which suppress cell cycle progression by repressing the activities of cyclinE‐CDK2 complexes in late G1 phase and cyclin A‐CDK2 in early S phase. Members of this family of proteins have been shown to play critical roles in normal hematopoiesis by restricting the proliferation of both HSCs and hematopoietic progenitors. p57 is highly expressed in HSCs and is down‐regulated during differentiation (Umemoto et al., 2005). It has been demonstrated that p57 expression can be induced by BM niche signals such as TPO and TGF1β (Yamazaki et al., 2006; Yamazaki et al., 2009; Yoshihara et al., 2007; Scandura et al., 2004), which are required to maintain the quiescent state and self‐renewal capacity of primitive long‐term HSCs (Matsumoto et al., 2011; Zou et al., 2011). The expression of p21 is low in HSCs and is increased in multipotent progenitors (MPPs). Studies have demonstrated that p21 is a key effector of p53‐mediated G1 cell cycle arrest and apoptosis, whose expression can be induced by treatment with various genotoxic agents or by oncogene‐induced DNA damage (Seoane et al., 2002; Viale et al., 2009). p21 deficiency has fewer effects on normal hematopoiesis during homeostasis but sensitizes HSCs to DNA damage‐induced depletion stimulated by myelotoxic injury (Cheng et al., 2000, 2000, 2007). p27 is consistently expressed in HSCs and MPPs at a relatively low level. Due to the compensation of p57, p27 deficiency has no effect on the self‐renewal and proliferation of HSCs but significantly induces the expansion of MPPs (Cheng et al., 2000, 2000). Interestingly, despite evidence of their growth inhibitory function on LCs, as shown by in vitro culture studies, there have not yet been any reports of mutations within this family of genes in human leukemias. The role of this family of CDKi's in the pathogenesis of leukemia remains elusive.
Leukemia cell line studies have indicated that p27 expression is normally repressed in undifferentiated LCs and is markedly up‐regulated during differentiation, which is commonly correlated with c‐Myc down‐regulation. p27 over‐expression induces LC differentiation and cell cycle arrest (Munoz‐Alonso et al., 2005). These studies suggested that differentiation is always coupled with arrest of the cell cycle. However, in our current studies, we found that in both MA9‐LCs and H/M‐LCs, p27 deletion promotes the proliferation of CD117‐ differentiated LCs without affecting their differentiation state, as shown by cell marker staining, morphology and colony‐formation assays. Our studies indicate that differentiation and proliferation of LCs are controlled by c‐Myc and p27 respectively, which is consistent with a previous report. In this report, Acosta et al. found that c‐Myc over‐expression can prevent differentiation but fails to rescue the inhibition of proliferation of K652 cells induced by p27 over‐expression (Acosta et al., 2008). Interestingly, in CD117+CD11blo MA9‐LCs, which are enriched in quiescent LSCs, both c‐Myc and p27 are expressed. We predict that c‐Myc prevents differentiation and collaborates with p27 to maintain LSCs in quiescence in murine MA9‐leukemia. Whether this co‐expression of c‐Myc and p27 is also required to maintain the quiescence and drug resistance of LSCs in human MLL‐leukemias needs additional study.
p27 has both tumor suppressive and tumor promoting activities, thus playing a dual role in tumorigenesis (Besson et al., 2007). The tumor suppressive activity of p27 is accomplished in the nucleus through inhibition of CDK‐dependent cell cycle progression, whereas its tumor promoting activity is cytoplasmic, through the regulation of RhoA activation or possibly a currently‐unknown mechanism which is CDK‐independent (Besson et al., 2004). We speculate that the role p27 plays in murine MA9 and H/M leukemias is accomplished through a CDK‐dependent mechanism because deletion of the p27 gene mainly alters the proliferation of both types of LCs while having minor effects on the leukemogenic ability of MA9‐LCs (only within the CD117+CD11blo population). Whether this CDK‐independent pro‐oncogenic function of p27 is involved in the pathogenesis of other types of leukemia needs to be addressed.
Quiescence is one of the key features of self‐renewable HSCs (Wilson et al., 2008). It was demonstrated that long‐term reconstitutive HSCs are largely enriched in quiescent cell populations. Inducing cell cycle entry of HSCs will significantly compromise their function (Baldridge et al., 2010). This quiescent state of HSCs is essentially maintained by p57, with additional support from p27 (Matsumoto et al., 2011). Studies of clinical samples have indicated that LSCs are also quiescent (Saito et al., 2010a; Zou et al., 2011). A recent study suggested that in murine PML‐RARα and AML1–ETO leukemia models, p21 is indispensable for maintaining self‐renewal in LSCs by restricting cellular proliferation and limiting the accumulation of DNA damage. LSCs with p21 deficiency will be exhausted through apoptosis and senescence as a consequence of the marked accumulation of DNA damage (Viale et al., 2009). Whether p21 is required for LSC self‐renewal in MLL‐related leukemias has not yet been studied. We found that LSCs are enriched in both CD117+CD11bhi and CD117+CD11blo populations of MA9‐LCs. p27 deletion affects only the proliferation and leukemogenic ability of CD117+CD11blo LSCs, having no influence on the CD117+CD11bhi population. We are currently in the process of investigating whether LSCs in the CD117+CD11bhi population are also quiescent. If so, we will subsequently determine which CDKi, p21 or p57, restricts the proliferation of these LSCs, and also which niche signal(s) induce(s) cell cycle arrest in these LSCs.
Contributions
Jun Zhang, Christopher Seet, Clear Sun, Jing Li, Dewen You, Yechen Xiao, Andrew Volk, Peter Breslin, Shubin Zhang and Wei Wei performed the research and analyzed the data. Nancy J. Zeleznik‐Le, Manual Diaz, Zhijiang Qian and Jiwang Zhang designed the experiments. Peter Breslin, Zhou Zhang and Jiwang Zhang analyzed the data and wrote the paper.
Conflict‐of‐interest disclosure
The authors declare no competing financial interests.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
We appreciate the excellent animal care services provided by the staff of the Department of Comparative Medicine, Loyola University Medical Center. This work was supported by NIH – R01 HL95896, NSFC Project 81071774, 31201104 and 31270883, National Basic Research Program of China 2013CB966800, the Program for Professor of Special Appointment (Eastern Scholar) at the Shanghai Institutions of Higher Learning, the Food Safety and Nutrition Program of Shanghai Normal University DXL123, the Innovation Program of Shanghai Municipal Education Commission 13ZZ103 and 13YZ051, as well as by a grant from the Jimmy Burns Foundation.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.07.011.
Zhang Jun, Seet Christopher S., Sun Clare, Li Jing, You Dewen, Volk Andrew, Breslin Peter, Li Xingyu, Wei Wei, Qian Zhijian, Zeleznik-Le Nancy J., Zhang Zhou, Zhang Jiwang, (2013), p27kip1 maintains a subset of leukemia stem cells in the quiescent state in murine MLL‐leukemia, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.07.011.
References
- Abramovich, C. , Humphries, R.K. , 2005. Hox regulation of normal and leukemic hematopoietic stem cells. Curr. Opin. Hematol.. 12, 210–216. [DOI] [PubMed] [Google Scholar]
- Acosta, J.C. , 2008. Myc inhibits p27-induced erythroid differentiation of leukemia cells by repressing erythroid master genes without reversing p27-mediated cell cycle arrest. Mol. Cell Biol.. 28, 7286–7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeff, M. , 2008. HOX expression patterns identify a common signature for favorable AML. Leukemia. 22, 2041–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, S.A. , 2002. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet.. 30, 41–47. [DOI] [PubMed] [Google Scholar]
- Ayton, P.M. , Cleary, M.L. , 2003. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev.. 17, 2298–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton, P.M. , Chen, E.H. , Cleary, M.L. , 2004. Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol. Cell Biol.. 24, 10470–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach, C. , Mueller, D. , Buhl, S. , Garcia-Cuellar, M.P. , Slany, R.K. , 2009. Alterations of the CxxC domain preclude oncogenic activation of mixed-lineage leukemia 2. Oncogene. 28, 815–823. [DOI] [PubMed] [Google Scholar]
- Baldridge, M.T. , King, K.Y. , Boles, N.C. , Weksberg, D.C. , Goodell, M.A. , 2010. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 465, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, M.W. , Jordan, C.T. , 2011. Leukemia stem cells in 2010: current understanding and future directions. Blood Rev.. 25, 75–81. [DOI] [PubMed] [Google Scholar]
- Bernt, K.M. , 2011. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 20, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson, A. , 2007. Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor phenotype. Genes Dev.. 21, 1731–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson, A. , Gurian-West, M. , Schmidt, A. , Hall, A. , Roberts, J.M. , 2004. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev.. 18, 862–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caslini, C. , 2007. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res.. 67, 7275–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, M.J. , 2010. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res.. 70, 10234–10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, T. , 2000. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 287, 1804–1808. [DOI] [PubMed] [Google Scholar]
- Cheng, T. , Rodrigues, N. , Dombkowski, D. , Stier, S. , Scadden, D.T. , 2000. Stem cell repopulation efficiency but not pool size is governed by p27(kip1). Nat. Med.. 6, 1235–1240. [DOI] [PubMed] [Google Scholar]
- Cierpicki, T. , 2010. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct. Mol. Biol.. 17, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alboran, I.M. , 2001. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity. 14, 45–55. [DOI] [PubMed] [Google Scholar]
- Dick, J.E. , 2005. Acute myeloid leukemia stem cells. Ann. N. Y. Acad. Sci.. 1044, 1–5. [DOI] [PubMed] [Google Scholar]
- Dou, Y. , Hess, J.L. , 2008. Mechanisms of transcriptional regulation by MLL and its disruption in acute leukemia. Int. J. Hematol.. 87, 10–18. [DOI] [PubMed] [Google Scholar]
- Ernst, P. , 2004. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev. Cell. 6, 437–443. [DOI] [PubMed] [Google Scholar]
- Ernst, P. , Mabon, M. , Davidson, A.J. , Zon, L.I. , Korsmeyer, S.J. , 2004. An Mll-dependent Hox program drives hematopoietic progenitor expansion. Curr. Biol.. 14, 2063–2069. [DOI] [PubMed] [Google Scholar]
- Faber, J. , 2009. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood. 113, 2375–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, Z.H. , 2009. Transcriptional regulation of survivin by c-Myc in BCR/ABL-transformed cells: implications in antileukemic strategy. J. Cell Mol. Med.. 13, 2039–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi, Y. , 2007. Fms-like tyrosine kinase 3 ligand stimulation induces MLL-rearranged leukemia cells into quiescence resistant to antileukemic agents. Cancer Res.. 67, 9852–9861. [DOI] [PubMed] [Google Scholar]
- Guzman, M.L. , Jordan, C.T. , 2004. Considerations for targeting malignant stem cells in leukemia. Cancer Control. 11, 97–104. [DOI] [PubMed] [Google Scholar]
- Haferlach, C. , 2011. CDKN1B, encoding the cyclin-dependent kinase inhibitor 1B (p27), is located in the minimally deleted region of 12p abnormalities in myeloid malignancies and its low expression is a favorable prognostic marker in acute myeloid leukemia. Haematologica. 96, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, N. , 2011. Human polymerase-associated factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc. Natl. Acad. Sci. U. S. A.. 108, E636–E645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, J.L. , Yu, B.D. , Li, B. , Hanson, R. , Korsmeyer, S.J. , 1997. Defects in yolk sac hematopoiesis in Mll-null embryos. Blood. 90, 1799–1806. [PubMed] [Google Scholar]
- Hoffman, B. , Amanullah, A. , Shafarenko, M. , Liebermann, D.A. , 2002. The proto-oncogene c-myc in hematopoietic development and leukemogenesis. Oncogene. 21, 3414–3421. [DOI] [PubMed] [Google Scholar]
- Horiguchi, K. , 2009. Transcriptional activation of the mixed lineage leukemia-p27Kip1 pathway by a somatostatin analogue. Clin. Cancer Res.. 15, 2620–2629. [DOI] [PubMed] [Google Scholar]
- Imamura, T. , 2002. Frequent co-expression of HoxA9 and Meis1 genes in infant acute lymphoblastic leukaemia with MLL rearrangement. Br. J. Haematol.. 119, 119–121. [DOI] [PubMed] [Google Scholar]
- Jamieson, C.H. , Weissman, I.L. , Passegue, E. , 2004. Chronic versus acute myelogenous leukemia: a question of self-renewal. Cancer Cell. 6, 531–533. [DOI] [PubMed] [Google Scholar]
- Jude, C.D. , 2007. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 1, 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva, M.Y. , Jordan, C.T. , 2011. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J. Clin. Oncol.. 29, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov, A.V. , Armstrong, S.A. , 2007. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer. 7, 823–833. [DOI] [PubMed] [Google Scholar]
- Kroon, E. , 1998. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. Embo J.. 17, 3714–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A.R. , 2009. A role for MEIS1 in MLL-fusion gene leukemia. Blood. 113, 1756–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, H.J. , 1999. Frequent co-expression of the HOXA9 and MEIS1 homeobox genes in human myeloid leukemias. Leukemia. 13, 1993–1999. [DOI] [PubMed] [Google Scholar]
- Li, Z. , 2009. Consistent deregulation of gene expression between human and murine MLL rearrangement leukemias. Cancer Res.. 69, 1109–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D.P. , 2013. HOXA9 gene expression in acute myeloid leukemia. Cell Biochem. Biophys.. 10.1007/s12013-013-9586-8 [DOI] [PubMed] [Google Scholar]
- Liu, H. , Cheng, E.H. , Hsieh, J.J. , 2009. MLL fusions: pathways to leukemia. Cancer Biol. Ther.. 8, 1204–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschalek, R. , 2011. Mechanisms of leukemogenesis by MLL fusion proteins. Br. J. Haematol.. 152, 141–154. [DOI] [PubMed] [Google Scholar]
- Matsumoto, A. , 2011. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 9, 262–271. [DOI] [PubMed] [Google Scholar]
- McMahon, K.A. , 2007. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell. 1, 338–345. [DOI] [PubMed] [Google Scholar]
- Milne, T.A. , 2002. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell. 10, 1107–1117. [DOI] [PubMed] [Google Scholar]
- Milne, T.A. , 2005. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. U. S. A.. 102, 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne, T.A. , 2010. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol. Cell. 38, 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Alonso, M.J. , 2005. p21Cip1 and p27Kip1 induce distinct cell cycle effects and differentiation programs in myeloid leukemia cells. J. Biol. Chem.. 280, 18120–18129. [DOI] [PubMed] [Google Scholar]
- Muntean, A.G. , 2010. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell. 17, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntean, A.G. , Hess, J.L. , 2011. The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol.. 7, 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya, T. , Morrison, S.J. , Clarke, M.F. , Weissman, I.L. , 2001. Stem cells, cancer, and cancer stem cells. Nature. 414, 105–111. [DOI] [PubMed] [Google Scholar]
- Saito, Y. , 2010. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci. Transl. Med.. 2, 17ra19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, Y. , 2010. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat. Biotechnol.. 28, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scandura, J.M. , Boccuni, P. , Massague, J. , Nimer, S.D. , 2004. Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc. Natl. Acad. Sci. U. S. A.. 101, 15231–15236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner, S. , 2001. MLL-ENL causes a reversible and myc-dependent block of myelomonocytic cell differentiation. Cancer Res.. 61, 6480–6486. [PubMed] [Google Scholar]
- Seoane, J. , Le, H.V. , Massague, J. , 2002. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 419, 729–734. [DOI] [PubMed] [Google Scholar]
- Slany, R.K. , 2009. The molecular biology of mixed lineage leukemia. Haematologica. 94, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slany, R.K. , Lavau, C. , Cleary, M.L. , 1998. The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol. Cell Biol.. 18, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somervaille, T.C. , Cleary, M.L. , 2006. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 10, 257–268. [DOI] [PubMed] [Google Scholar]
- Thiel, A.T. , 2011. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell. 17, 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemoto, T. , 2005. p57Kip2 is expressed in quiescent mouse bone marrow side population cells. Biochem. Biophys. Res. Commun.. 337, 14–21. [DOI] [PubMed] [Google Scholar]
- van Os, R. , 2007. A limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells. 25, 836–843. [DOI] [PubMed] [Google Scholar]
- Viale, A. , 2009. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature. 457, 51–56. [DOI] [PubMed] [Google Scholar]
- Weissman, I. , 2005. Stem cell research: paths to cancer therapies and regenerative medicine. JAMA. 294, 1359–1366. [DOI] [PubMed] [Google Scholar]
- Weng, A.P. , 2006. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev.. 20, 2096–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, A. , 2008. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Wong, P. , Iwasaki, M. , Somervaille, T.C. , So, C.W. , Cleary, M.L. , 2007. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev.. 21, 2762–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, Z.B. , 2005. The MLL fusion gene, MLL-AF4, regulates cyclin-dependent kinase inhibitor CDKN1B (p27kip1) expression. Proc. Natl. Acad. Sci. U. S. A.. 102, 14028–14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi, H. , 1998. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood. 92, 108–117. [PubMed] [Google Scholar]
- Yamazaki, S. , 2006. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. Embo J.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki, S. , 2009. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood. 113, 1250–1256. [DOI] [PubMed] [Google Scholar]
- Yokoyama, A. , Cleary, M.L. , 2008. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 14, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama, A. , 2005. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 123, 207–218. [DOI] [PubMed] [Google Scholar]
- Yoshihara, H. , 2007. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 1, 685–697. [DOI] [PubMed] [Google Scholar]
- Yu, B.D. , Hess, J.L. , Horning, S.E. , Brown, G.A. , Korsmeyer, S.J. , 1995. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 378, 505–508. [DOI] [PubMed] [Google Scholar]
- Zeisig, B.B. , 2004. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol. Cell Biol.. 24, 617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeleznik-Le, N.J. , Harden, A.M. , Rowley, J.D. , 1994. 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc. Natl. Acad. Sci. U. S. A.. 91, 10610–10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , 2003. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 425, 836–841. [DOI] [PubMed] [Google Scholar]
- Zou, P. , 2011. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 9, 247–261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data