

Abstract
Targeted busulfan/cyclophosphamide (TBU/CY) for allogeneic hematopoietic cell transplantation (HCT) carries a high risk of sinusoidal obstruction syndrome (SOS) in patients transplanted for myelofibrosis. We tested the hypothesis that reversing the sequence of administration (from TBU/CY to CY/TBU) will reduce SOS and day +100 non-relapse mortality (NRM). We enrolled 51 patients with myelofibrosis (n=20), acute myeloid leukemia (AML, n=20), or myelodysplastic syndrome (MDS, n=11) in a prospective trial of CY/TBU conditioning for HCT. Cyclophosphamide 60 mg/kg/day IV for two days was followed by daily IV BU for four days, targeted to a concentration at steady state (Css) of 800–900 ng/mL. CY/TBU-conditioned patients had higher exposure to CY (p<0.0001) and lower exposure to 4-hydroxyCY (p<0.0001) compared to TBU/CY-conditioned patients. Clinical outcomes were compared with controls (n=271) conditioned with TBU/CY for the same indications. In patients with myelofibrosis, CY/TBU conditioning was associated with a significantly reduced incidence of SOS (0% vs. 30% after TBU/CY, p=0.006), while SOS incidence was low in both cohorts with AML/MDS. Day +100 mortality was significantly lower in the CY/TBU cohort (2% vs. 13%, p=0.01). CY/TBU conditioning markedly impacted CY pharmacokinetics and was associated with significantly lower incidences of SOS and day +100 mortality, suggesting that CY/TBU is superior to TBU/CY as conditioning for patients with myelofibrosis.
INTRODUCTION
Busulfan followed by cyclophosphamide (BU/CY) is a commonly used high-dose conditioning regimen in allogeneic hematopoietic cell transplantation (HCT). Regimen-related toxicity, graft rejection, and relapse in patients conditioned with BU/CY have been reduced by individualized dosing of BU to a target steady-state concentration (targeted BU/CY, TBU/CY) [1,2]. However, neither BU dose-targeting nor the introduction of intravenous TBU has eliminated hepatic sinusoidal obstruction syndrome (SOS) as a cause of morbidity and mortality [3,4]. BU is not inherently toxic to hepatocytes or to sinusoidal endothelial cells, whereas metabolites of CY, generated within hepatocytes and transported into hepatic sinusoids, are highly toxic to sinusoidal endothelial cells [5-7]. It follows that CY metabolites are the prime cause of regimen-related liver toxicity following the TBU/CY regimen.
There are several possible approaches to minimizing regimen-related toxicity caused by the combination of TBU and CY. One approach is to eliminate CY altogether, for example by using a regimen of fludarabine and BU (FLU/TBU) [8,9]. A second approach is to eliminate variability in CY exposure with pharmacokinetic targeting of CY doses, which is feasible and effective in reducing toxicity [10]. A third, simpler approach is to reverse the order of administration, giving CY first, followed by IV TBU (CY/TBU). The pharmacologic rationale for a CY/ TBU regimen rests on the following observations: 1) BU depletes hepatic glutathione (GSH), and at high concentrations induces oxidative stress in murine hepatocytes in vitro [6]; 2) glutathione is important in both the detoxification of the CY metabolite 4-hydroxycyclophosphamide (HCY) through conversion to glutathionyl-CY and in the elimination of the toxic CY metabolite acrolein [5,11]; 3) restoration of hepatic and sinusoidal endothelial cell GSH levels prevents injury to hepatic sinusoids in several different animal models of toxic liver injury [12]; and 4) studies in patients receiving high-dose conditioning regimens have suggested a reduced risk of hepatotoxicity when BU was given after, rather than before, other conditioning agents [13-15]. Thus, giving BU first appears to potentiate CY toxicity, providing the basis for administering these drugs in reverse order (CY/ TBU) to reduce toxicity.
Here, we report the results of a prospective clinical trial designed to test the hypothesis that reversed-sequence (CY/TBU) conditioning reduces the frequency and severity of hepatotoxicity, compared to the standard sequence of BU followed by CY (TBU/CY). Additionally, we collected pharmacokinetic data to test whether altering the sequence of conditioning agents led to measurable changes in CY metabolism and exposure to CY metabolites. We enrolled two cohorts of patients, one at high risk for toxic sinusoidal liver injury (patients with myelofibrosis) [16] and one at standard risk (patients with myelodysplastic syndrome [MDS] or acute myeloid leukemia [AML]). We compared liver toxicity and outcomes with those in concurrent and historical control patients who received TBU/CY and allogeneic HCT for the same disease indications. The primary outcome was the incidence of moderate/severe SOS after allogeneic HCT.
MATERIALS AND METHODS
Patient selection
Study patients (cases) were enrolled from 1 March 2007 through 30 June 2010 on Fred Hutchinson Cancer Research Center (FHCRC) Protocol 2130. This protocol was approved by the FHCRC Institutional Review Board (IRB) and registered at www.clinicaltrials.gov as NCT00445744. All patients provided written informed consent using forms approved by the IRB. Under the aegis of IRB-approved Protocol 881, a cohort of historical patients (controls) was obtained by retrieving clinical data on consecutive patients with myelofibrosis, AML, or MDS undergoing allogeneic HCT after TBU/CY conditioning between 1 January 2003 and 31 December 2009.
Eligibility criteria
Eligible were patients: a) with primary myelofibrosis (PMF), myelofibrosis secondary to polycythemia vera (PV) or essential thrombocythemia (ET), AML, or MDS; b) aged <61 years if transplanted from unrelated donors, or aged <66 years if transplanted from related donors; c) receiving unmanipulated G-CSF-mobilized peripheral blood mononuclear cell (G-PBMC) or G-CSF-stimulated bone marrow allograft products; d) with a Karnofsky performance status of >70% at the time of HCT; and e) able to provide informed consent. Patients were required to have an HLA-identical related donor or an HLA-matched or 1-HLA-allele-mismatched unrelated donor identified before enrollment.
Exclusion criteria included: a) HIV infection or active viral hepatitis; b) use of medications known to strongly inhibit the cytochrome P450 pathway and which, in the judgment of the attending physician, could not be safely discontinued during conditioning; c) known hypersensitivity to BU or CY; d) hepatic dysfunction as evinced by total serum bilirubin or aspartate aminotransferase >2x the upper limit of normal, or evidence of synthetic dysfunction or cirrhosis; e) renal insufficiency as evinced by creatinine clearance <50% of expected, serum creatinine >2x the upper limit of normal, or dialysis dependence; f) impaired pulmonary function as evidenced by PaO2 <70 mm Hg and DLCO <70% predicted or by PaO2 <80 mm Hg and DLCO <60%, or requirement for continuous supplementary oxygen; and g) impaired cardiac function as evinced by ejection fraction <35% or presence of symptomatic coronary artery disease.
Conditioning regimen
The conditioning regimens for protocol cases and control patients are summarized in Table 1. All patients were conditioned with CY 60 mg/kg/day IV for two consecutive days (total dose, 120 mg/kg) and targeted BU, given for four consecutive days. On the days of CY infusion, patients received MESNA (2-mercaptoethane sulfonate) at milligram doses equal to those of CY as prophylaxis against uroepithelial damage.
Table 1.
Conditioning regimens for case (CY/TBU) and control (TBU/CY) patients.a
| CY/TBU Cases (n=51) | TBU/CY Controls (n=271) | |
|---|---|---|
| Conditioning agents, by transplant day | ||
| -7 | CY IV 60 mg/kgb | BUc |
| -6 | CY IV 60 mg/kg | TBU |
| -5 | BUc | TBU |
| -4 | TBU | TBU |
| -3 | TBU | CY IV 60 mg/kgb |
| -2 | TBU | CY IV 60 mg/kg |
| -1 | Rest | Rest |
| 0 | Allograft infusion | Allograft infusion |
| Busulfan administration route & dosing frequency | ||
| Oral every 6 hours | 0 | 252 (93%) |
| IV every 6 hours | 0 | 15 (6%) |
| IV once daily | 51 (100%) | 4 (1%) |
| Cumulative busulfan dose (mg) | ||
| Oral | Not applicable | 1048 (572–1916) |
| IV | 1098 (580–1510) | 976 (608–1668) |
| Target busulfan Css (ng/ml) | ||
| ≤900d | 0 | 2 (0.7%) |
| 600–900 | 0 | 3 (1.1%) |
| 800–900 | 51 (100%) | 262 (96.7%) |
| >900d | 0 | 4 (1.5%) |
| Busulfan pharmacokinetics | ||
| Dose #1 Css >900 ng/mL | 23 (45%) | 128 (47%) |
| Average daily Csse >900 ng/mL | 1 (2%) | 18 (7%) |
| Average daily Csse (ng/mL) | 856 (811–1191) | 861 (627–968) |
Data presented as median (range) or number (%). Abbreviations: TBU, targeted busulfan; CY, cyclophosphamide; IV, intravenous; Css, busulfan steady-state concentration.
Mesna given concurrently with IV cyclophosphamide to prevent hemorrhagic cystitis.
Phenytoin started one day before busulfan and continued throughout busulfan administration (i.e., day -6 through day -1 for CY/TBU, and day -8 through day -3 for TBU/CY).
Specific target Css detailed in the busulfan dosing and pharmacokinetics section of text.
Cumulative over all 4 days of busulfan administration. For Css, each patient’s busulfan Css over all 4 days was calculated, and then divided by 4 to provide the average daily Css.
Cases (n=51) received CY followed by targeted IV BU (CY/TBU). CY was administered IV at 60 mg/kg/day on days -7 and -6 before HCT. Targeted BU was administered intravenously as Busulfex (Otsuka; Tokyo, Japan) once daily on days -5 through -2, for a total of four daily doses. Prophylactic phenytoin was initiated on day -6 after completion of the second CY dose, and discontinued on day -1; one patient received prophylactic levetiracetam.
Patients in the control cohort (n=271) received targeted BU followed by CY (TBU/CY). In this cohort, BU was administered on days -7 through -4 orally at an initial dose of 1 mg/kg every 6 hours in 252 patients (93%), intravenously at a starting dose of 0.8 mg/kg every six hours in 15 patients (6%), and intravenously at a starting dose of 3.2 mg/kg daily in 4 patients (1%). After the initial weight-based dose of BU, subsequent doses were adjusted to achieve the target plasma steady-state concentrations (Css) described in Table 1. CY was administered at 60 mg/kg/day IV on days -3 and -2. Prophylactic phenytoin was given from day -8 through day -3.
Cyclophosphamide dosing and pharmacokinetics
CY was infused through a central venous catheter. The CY dose was based on adjusted ideal body weight (0.25 × [actual weight – ideal weight] + ideal weight) if actual body weight was greater than ideal body weight [17]. The infusion duration followed FHCRC Standard Practice Guidelines: total CY doses of <5,000 mg were infused over 1 hour, and CY doses ≥ 5,000 mg were infused over 2 hours. CY doses were not adjusted based on pharmacokinetic data.
In cases (CY/TBU) only, blood samples were drawn after each dose of CY from the central venous lines at the end of infusion, and at 2, 4, 8, 16, 20, and 24 hours after the start of the CY infusion. If the CY infusion lasted 1.5 hours or longer, blood samples were instead drawn at the end of infusion and at 3, 5, 8, 16, 20, and 24 hours after the start of the infusion. At each of these time points, blood was aliquoted into two tubes: one containing EDTA for analysis of CY and carboxyethylphosphoramide mustard (CEPM), and the other containing phenylhydrazine HCl to stabilize HCY, as previously described [18]. Samples were refrigerated at the bedside at a target temperature of 4° C unti l transport (within 12 hours) to the Pharmacokinetics Laboratory. Plasma concentrations of CY, HCY, and CEPM were quantified by liquid chromatography and mass spectroscopy methods [10]. Patient exposure to CY and its metabolites was calculated by determining the AUCCY, AUCHCY, and AUCCEPM for the interval 0 to 48 hours using non-compartmental analysis. These AUCs were compared to those previously reported in patients receiving TBU/CY [18]. CY pharmacokinetics were not evaluated in the historical control patients.
Busulfan dosing and pharmacokinetics
In the 51 case patients (CY/TBU), daily IV BU doses were standardized regarding the time of administration, duration of BU infusion, and administration of saline flushes within the IV line for consistent BU pharmacokinetics. In these patients, the first BU dose (day -5) was 4 mg/kg, with body weight calculated as described above [17]. All subsequent BU doses were adjusted to achieve a Css of 800–900 ng/mL.
In the 271 control patients (TBU/CY), the BU administration route and target Css were chosen by the attending physician. The majority of patients received oral BU every six hours (n=252); a minority received IV BU every six hours (n=15) or as a combined single daily dose (n=4). The target Css for most patients (n=262) was 800–900 ng/mL; five patients had target Css ≤900 ng/mL and four patients had target Css >900 ng/mL.
In both cases and controls, blood samples for BU pharmacokinetics (3 mL/sample) were collected in sodium-heparin-containing tubes at the time points previously described [8]. Samples were stored on wet ice or refrigerated until transport to the laboratory, where plasma BU concentrations were analyzed by gas chromatography with mass selective detection as previously described [19]. The dynamic range was from 62 to 4,500 ng/mL and the intraday and interday coefficients of variations were <5% and <8%, respectively.
Individual patient concentration-time data were fit using WinNonlin (version 5.2). The AUC from time 0 to infinity (AUC0 to ∞) was calculated after each dose. Clearance and Css were calculated based on the following equations: clearance = dose divided by AUC, and Css = AUC0 to ∞ multiplied by BU molecular weight (246.3 g/mol) divided by the dosing interval. After calculation of each patient’s clearance, subsequent dose levels were calculated linearly to achieve the target Css, as described previously [17].
Supportive care and prophylaxis
Graft-vs.-host disease (GVHD) prophylaxis consisted of tacrolimus and methotrexate. Tacrolimus was given as a continuous IV infusion beginning on day -1 at an initial dose of 0.03 mg/kg/day, with doses adjusted to achieve trough tacrolimus concentrations at steady-state of 5–15 ng/mL. Tacrolimus was converted from IV infusion to divided oral dosing as soon as tolerated. In the absence of GVHD, tacrolimus was tapered in 20% decrements starting on day +56 after HCT, to be discontinued completely by day +200. In patients with GVHD, tacrolimus was maintained at therapeutic trough concentrations with subsequent tapering and management dictated by the attending transplant physician on the basis of clinical GVHD activity. Methotrexate was given at doses of 10 mg/m2 IV on day +1 (at least 24 hours after donor cell infusion) and on days +3, +6, and +11.
All patients received antifungal, antiviral, and antibacterial prophylaxis per FHCRC standard practice. Hematopoietic growth factors were given only in the event of prolonged neutropenia after day +21. Ursodiol was administered orally to both cases and historical control patients at 12 mg/kg/day, starting two weeks before the initiation of conditioning, per FHCRC standard practice.
Evaluation of outcomes
All case and control patients were evaluated by two investigators (G.B.M. and A.K.) for evidence of SOS after HCT. The diagnosis of SOS was based on the occurrence (by day +20 after HCT) of at least two of the following: hyperbilirubinemia (serum bilirubin > 2.0 mg/dL); hepatomegaly or right upper quadrant pain of liver origin; or weight gain (>2% of dry body weight) due to fluid accumulation [20]. If other possible causes of liver dysfunction were present (e.g., GVHD, sepsis syndrome, drug-induced liver injury), patients were classified as having liver disease of uncertain etiology (LDUE). The severity of SOS was graded as mild (resolving without specific treatment), moderate (requiring diuretics, sodium restriction, or analgesics, but with eventual resolution of abnormalities), or severe (death or non-resolution by day +100).
Overall survival was estimated by the Kaplan-Meier method. The cumulative incidences of non-relapse mortality (NRM) and relapse were estimated by standard methods, treating these outcomes as mutually competing events. Statistical comparisons of survival, NRM, and relapse between groups used Cox regression, restricting the analysis to events within the first 100 days or first two years after HCT, as indicated. The associations of the AUC of CY and its metabolites with these outcomes were evaluated as a test for trend over quartiles using Cox regression. Statistical comparisons of the frequency of SOS were done by the chi-squared test. Comparisons of pharmacokinetic parameters between regimens were carried out using the Wilcoxon rank-sum test. Comparisons of relapse rates were adjusted using the disease-risk criteria described by Kahl et al [21]. Outcomes in patients with AML/MDS were compared to those in patients with myelofibrosis as part of a pre-specified subset analysis.
RESULTS
Patient demographics
Patient and disease characteristics are summarized in Table 2. The median age of cases was 55 (range, 30–65) years. Twenty patients (39%) had myelofibrosis, 11 (22%) had MDS, and 20 (39%) had AML. Two cases had undergone previous allogeneic HCT: one patient with myelofibrosis had rejected an allograft after TBU/CY conditioning 10 years earlier, and a second patient with AML had relapsed after HCT following reduced-intensity conditioning performed three months before study enrollment. The median age in the control cohort of 271 patients was 50 (range, 19-67) years. In this cohort, 33 patients (12%) had myelofibrosis, 143 (53%) had AML, and 95 (35%) had MDS.
Table 2.
Patient characteristics.a
| Characteristic | CY/TBU (n=51) Cases | TBU/CY (n=271) Controls |
|---|---|---|
| Age, in years | 55 (30–65) | 50 (19–67) |
| Diagnosis | ||
| Acute myeloid leukemia | 20 (39%) | 143 (53%) |
| Myelodysplasia | 11 (22%) | 95 (35%) |
| Myelofibrosis | 20 (39%) | 33 (12%) |
| Donor | ||
| Related | 28 (55%) | 96 (35%) |
| Unrelated | 23 (45%) | 175 (65%) |
| HLA-matched | 21 | 98 |
| 1-HLA-allele-mismatched | 2 | 77 |
| Allograft source | ||
| G-PBMC | 51 (100%) | 223 (83%) |
| Bone marrow | 0 (0%) | 48 (17%) |
| CD34+ dose, in cells/kg recipient weight | 13.4 (6.8–28.5) | 9.1 (0.5–45.0) |
| Kahl disease riskb | ||
| Low/moderate | 30 (59%) | 196 (72%) |
| High | 21 (41%) | 75 (28%) |
Data presented as median (range) or number (%).
Abbreviations: CY, cyclophosphamide; TBU, targeted busulfan; HLA, human leukocyte antigen; G-PBMC, granulocyte colony stimulating factor-mobilized peripheral blood mononuclear cells; IV, intravenous.
Kahl disease risk measures risk of relapse after allogeneic hematopoietic cell transplantation [21].
Cyclophosphamide pharmacokinetics
Peak plasma concentrations and AUC of CY and its metabolites HCY and CEPM are summarized in Table 3 and Figure 1. Patients receiving CY/ TBU showed considerable variability in exposure to CY metabolites, including a 3.7-fold variation in AUCCY, a 3.6-fold variation in AUCHCY, and a 4.8-fold variation in AUCCEPM. Pharmacokinetic parameters for patients receiving CY/TBU were compared to those previously obtained in 75 patients receiving TBU/CY conditioning [18]. Given the age-dependent pharmacokinetics of CY [22], these analyses were adjusted for patient age. The median age of the CY/TBU cohort was 55 (range, 30–65) years, while the median age of the historical TBU/CY cohort was 44 (range, 20–66) years [18].
Table 3.
Comparison of pharmacokinetics of CY, HCY and CEPM by conditioning regimen. Peak concentrations (μM) are the highest concentration recorded on that day; AUC (μM•h) is from time 0 to 48 hours. Comparisons are adjusted for age.
| CY/TBU | TBU/CY [18] | P-value | |
|---|---|---|---|
| CY | |||
| Peak [CY], day 1 | 375 ± 60 | 312 ± 171 | <0.0001 |
| Peak [CY], day 2 | 329 ± 66 | 283 ± 124 | <0.0001 |
| AUCCY | 4899 ± 1255 | 2563 ± 1190 | <0.0001 |
|
| |||
| HCY | |||
| Peak [HCY], day 1 | 9 ± 5 | 35 ± 18 | <0.0001 |
| Peak [HCY], day 2 | 20 ± 9 | 36 ± 13 | <0.0001 |
| AUCHCY | 168 ± 48 | 290 ± 98 | <0.0001 |
|
| |||
| CEPM | |||
| Peak [CEPM], day 1 | 12 ± 6 | 27 ± 12 | <0.0001 |
| Peak [CEPM], day 2 | 26 ± 11 | 32 ± 29 | 0.25 |
| AUCCEPM | 475 ± 180 | 522 ± 194 | 0.14 |
Abbreviations: CY, cyclophosphamide; TBU, targeted busulfan; AUC, area under the curve; HCY, 4-hydroxycyclophosphamide; CEPM, carboxyethylphosphoramide mustard.
Figure 1.
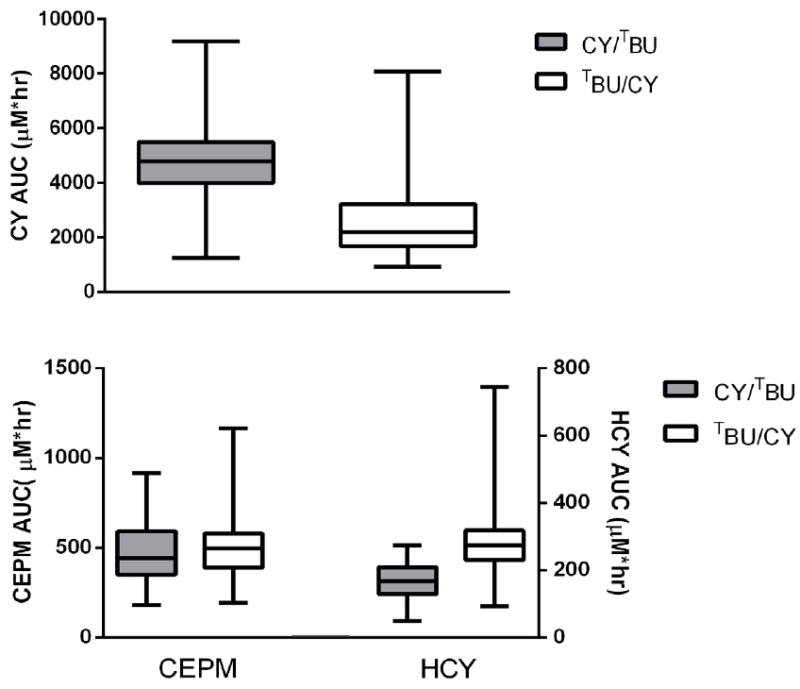
Comparison of CY, HCY, and CEPM exposure by conditioning regimen. AUC0-48hr in patients receiving CY/TBU (gray) and TBU/CY (white). Box designates 25th, 50th, and 75th percentile; whiskers designate 5th and 95th percentiles.
The sequence of CY/TBU administration markedly affected CY pharmacokinetics (Table 3; Figure 1). When CY was given first (CY/TBU), there was a significant increase in AUCCY (4899 vs. 2563 μ(•h, p<0.0001) and a significant decrease in AUCHCY (168 vs. 290 μ(•h, p<0.0001) compared to values with standard TBU/CY. There was also a trend toward reduced AUCCEPM with CY/TBU (475 vs. 522 μU•h, p=0.14). In the CY/TBU cohort, there were no apparent differences in BU Css or in the AUC of CY and its metabolites between patients with myelofibrosis and those with AML/MDS (data not shown). In the CY/TBU cohort, the association of the AUC of CY, HCY, and CEPM with SOS could not be evaluated statistically, since only two cases of SOS occurred. Relapse and NRM were not associated with the AUC of CY and its metabolites. However, higher AUCHCY and AUCCEPM were associated with inferior overall survival (p=0.03 and 0.02, respectively; Table 4).
Table 4.
Relationship between exposure to CY and its metabolites and clinical outcomes among cases conditioned with CY/TBU.a
| Clinical Outcome | Nb | Pharmacokinetic Parameters | ||
|---|---|---|---|---|
| AUCCY | AUCHCY | AUCCEPM | ||
| Non-relapse mortality | 10 | HR=1.15 (p=0.64) | HR=1.67 (p=0.11) | HR=1.40(p=0.25) |
| Relapse | 9 | HR=1.05 (p=0. 84) | HR=1.2 (p=0.53) | HR=1.53 (p=0.10) |
| Overall mortality | 20 | HR=1.28 (p=0.26) | HR=1.74 (p=0.03) | HR=1.67(p=0.02) |
AUC modeled as continuous linear variable, with hazard ratios for AUCCEPM and AUCHCY representing increase in hazard ratio (HR) associated with increase in AUC of 100 μM•h. Hazard ratios for AUCCY represent increase in hazard associated with increase in AUC of 1000 μM•h. Hazard ratios adjusted for age at time of HCT, type of donor, and relapse risk.
Number of events in cohort in 51 cases.
Abbreviations: CY, cyclophosphamide; TBU, targeted busulfan; HR, hazard ratio; AUC, area under the curve; HCY, 4-hydroxycyclophosphamide; CEPM, carboxyethylphosphoramide mustard.
Clinical outcomes in cases
All patients in the CY/TBU cohort initially engrafted (defined by a rise in absolute neutrophil counts to >500 cells/μL for at least three consecutive days) at a median of 17 (range, 11–30) days after HCT. One patient with AML/MDS who received an HLA-allele-mismatched allograft from an unrelated donor suffered late graft failure three months after HCT.
Approximately half of cases (26/51, 51%) did not require parenteral nutrition in the first 20 days after allogeneic HCT. Among patients with myelofibrosis, the median peak serum total bilirubin through day +20 was 2.3 (range, 0.7–30.0) mg/dL. Among patients with AML/MDS, the median peak serum total bilirubin through day +20 was 1.1 (range, 0.5–12.4) mg/dL. The incidence of SOS was 0/20 (0%) in patients with myelofibrosis, and 2/31 (6.5%) in patients with AML/MDS (Table 5). No patient in the CY/TBU cohort developed severe SOS.
Table 5.
Incidences of sinusoidal obstruction syndrome and liver disease of unknown etiology among patients conditioned with TBU/CY vs. CY/TBU.
| CY/TBU (n=51) | TBU/CY (n=271) | |
|---|---|---|
|
| ||
| Myelofibrosis | 20 | 33 |
| No liver disease | 17 (85%) | 19 (58%) |
| LDUE | 3 (15%) | 4 (12%) |
| SOS | 0(0%) | 10 (30%) |
| Mild | 0 | 2 |
| Moderate | 0 | 6 |
| Severe | 0 | 2 |
|
| ||
| AML/MDS | 31 | 238 |
| No liver disease | 26 (84%) | 203 (85%) |
| LDUE | 3 (10%) | 13 (5%) |
| SOS | 2 (6%) | 22 (9%) |
| Mild | 0 | 3 |
| Moderate | 2 | 10 |
| Severe | 0 | 9 |
Abbreviations: CY, cyclophosphamide; TBU, targeted busulfan; LDUE, liver disease of unknown etiology; SOS, sinusoidal obstruction syndrome; AML, acute myeloid leukemia; MDS, myelodysplastic syndrome.
Acute GVHD grades II–IV and grades III–IV occurred in 67% and 8% of cases, respectively, at a median of 28 (range, 8–102) days after HCT. Chronic GVHD developed in 41% of cases at a median of 189 (range, 92–530) days after HCT.
The median follow-up of surviving cases was 19 months, and 32 patients (63%) were alive at last follow-up. Day +100 mortality was 0% in patients with myelofibrosis and 3% in patients with AML/MDS. At two years after HCT, cumulative incidence estimates for overall survival were 68% in patients with myelofibrosis and 56% in patients with AML/MDS; NRM was estimated at 27% and 17% for patients with myelofibrosis and AML/MDS, respectively. The cumulative incidence of relapse was 11% in patients with myelofibrosis and 44% in patients with AML/MDS.
The major causes of death were relapsed malignancy in patients with AML/MDS, and GVHD (with or without concomitant infection) in patients with myelofibrosis. One patient with myelofibrosis died of metastatic prostate cancer, which was diagnosed approximately six months after HCT. One patient with AML/MDS committed suicide at day +102 after HCT.
Comparison of outcomes after CY/TBU vs. TBU/CY
Among patients with myelofibrosis, CY/TBU conditioning was associated with a significantly reduced incidence of SOS as compared to TBU/CY (0% vs. 30%, p=0.006). In patients with AML/MDS, rates of SOS were 6.5% with CY/TBU and 9.2% with TBU/CY (p=0.61). There were no cases of severe SOS in the CY/TBU cohort, compared to 11 in the TBU/CY cohort.
In patients with myelofibrosis, median peak serum total bilirubin levels through day +20 did not differ significantly by conditioning-agent sequence (2.3 mg/dL in the CY/TBU group vs. 2.2 mg/dL in the TBU/CY group, p=0.95). In patients with AML/MDS, the CY/TBU group showed a trend toward lower median peak serum total bilirubin levels through day +20 (1.1 mg/dL vs. 1.4 mg/dL, p=0.07).
Patients conditioned with CY/TBU showed significantly lower day +100 mortality as compared to those conditioned with TBU/CY (2% vs. 12%, p=0.01). For patients with myelofibrosis, the two-year cumulative incidence of relapse was 11% with CY/TBU and 6% with TBU/CY (p=0.62). There were no significant differences in the two-year cumulative incidences of NRM (27% vs. 25%, p=0.91) or overall survival (68% vs. 72%, p=0.78; Figure 2).
Figure 2.
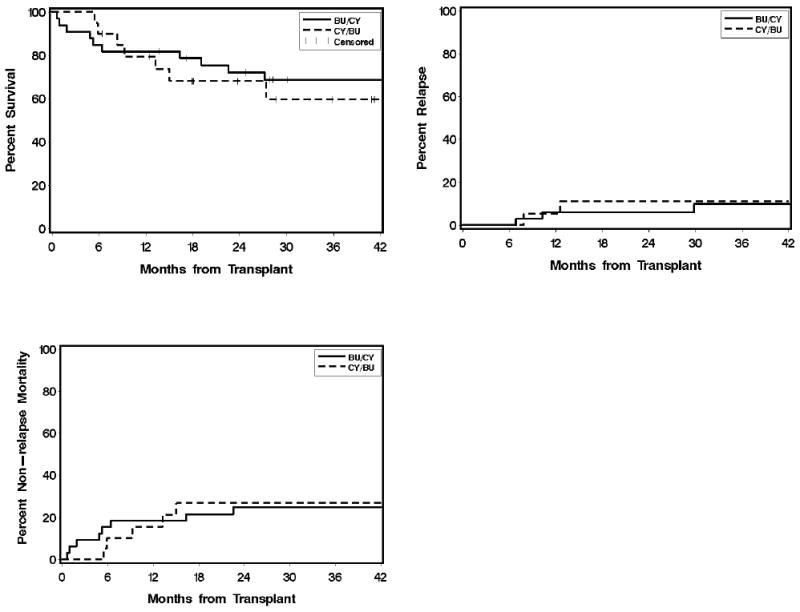
Overall survival, non-relapse mortality, and relapse in patients with myelofibrosis conditioned with CY/TBU (n=20) vs. TBU/CY (n=33). Abbreviations: CY, cyclophosphamide; TBU, targeted busulfan.
For patients with AML/MDS, the two-year cumulative incidences of relapse with CY/TBU vs. TBU/CY were 44% vs. 20% (p=0.008); for NRM, 17% vs. 22% (p=0.84); and for overall survival, 56% vs. 64% (p=0.57; Figure 3). The higher incidence of relapse in AML/MDS patients conditioned with CY/TBU remained statistically significant, albeit attenuated, after adjustment for higher disease risk in this cohort (unadjusted hazard ratio [HR] 2.57, p=0.008; adjusted HR 2.15, p=0.02).
Figure 3.
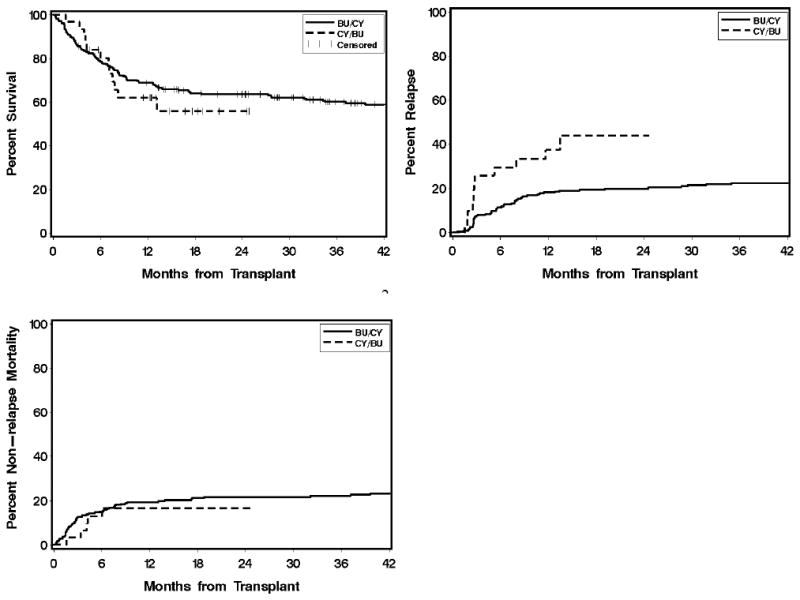
Overall survival, non-relapse mortality, and relapse in patients with AML/MDS conditioned with CY/TBU (n=31) vs. TBU/CY (n=238). Abbreviations: AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; CY, cyclophosphamide; TBU, targeted busulfan.
DISCUSSION
The major findings of this study are: 1) daily IV BU can be safely administered following high-dose CY; 2) the sequence of administration of BU and CY substantially affects CY metabolism; and 3) CY/TBU conditioning is associated with a significantly reduced risk of day +100 mortality, a substantially lower incidence of SOS, and an absence of severe SOS, compared to the standard sequence of TBU/CY. In patients with myelofibrosis, the reduction of SOS incidence from 30% to 0% with a simple reversal of conditioning-agent sequence is both statistically and clinically significant. Busulfan is not inherently hepatotoxic as a single agent in vitro or in human overdoses [6,23]. Our data reinforce the concept that regimen-related liver damage results largely from toxic metabolites of CY, although we recognize reports of hepatotoxicity attributed to BU in combination with fludarabine as well [24,25].
Recent reports support the safety of administered daily IV busulfan with cyclophosphamide [26,27]. However, Williams et al. concluded that daily IV BU at 3.2 mg/kg/day for four days followed by CY 60 mg/kg/day for two days resulted in excessive toxicity: autopsy-confirmed SOS occurred in two of the three patients who received this regimen and had a BU Css >1025 ng/mL [28]. Our cases received the same dose of CY, but in the reverse sequence, followed by daily IV BU at a higher initial dose of 4 mg/kg with subsequent BU doses targeted to a Css of 800–900 ng/mL. This target BU Css range is well below the BU Css ranges of 925–1025 ng/mL previously associated with elevated SOS rates in adults conditioned with BU/CY [2,29,30]. Our data demonstrate acceptable toxicity when CY is administered before targeted daily IV BU at an initial BU dose of 4 mg/kg. Notably, the clearance of daily IV BU did not change during days -5 to -3 in patients receiving CY/TBU [17]. Nevertheless, even in patients receiving CY/TBU, CY metabolism showed substantial interpatient variability when the CY dose, phenytoin dose, and BU Css were held constant between patients (Table 3; Figure 1).
Patients receiving CY/TBU had higher exposure to CY, lower exposure to HCY, and similar exposure to CEPM compared to TBU/CY-conditioned patients. These data agree with our previous report comparing CY/TBI (i.e., CY first) to TBU/CY (i.e., CY after busulfan/phenytoin) [18]. The use of phenytoin as a prophylactic antiepileptic may contribute to this difference. We sought to characterize the clinical significance of the variability in CY pharmacokinetics (Table 4). Reduced HCY exposure may theoretically translate into less immunosuppressive effect, although AUCHCY was not associated with clinical outcomes in patients conditioned with CY/TBI or TBU/CY [7,18]. Regarding toxicity, we previously described AUCCEPM as a reporter for liver toxicity, since it strongly correlates with sinusoidal hepatotoxicity and mortality [7,22]. A pharmacodynamic analysis of SOS with CY metabolites could not be conducted, because only two of the CY/TBU cases developed SOS. There was a statistically significant relationship between overall survival and the AUC of HCY and CEPM (Table 4).
Altering the sequence of conditioning agents is an appealingly simple and inexpensive strategy which uses available, familiar medications. Following preclinical studies [5,31,32], this approach has been translated into clinical trials in humans. Kerbauy et al. described the use of CY/BU conditioning in a cohort of 11 patients and reported lower peak serum aminotransferase levels compared to BU/CY-conditioned historical controls [14]. Of note, peak serum total bilirubin levels were not significantly different. In a larger retrospective cohort of 59 patients conditioned with CY/BU, Cantoni et al. reported lower rates of SOS and transplant-related mortality as compared to a small historical cohort of 16 patients conditioned with BU/CY [15].
Our results extend these earlier retrospective reports in the form of a prospective clinical trial. In addition to prospective enrollment, novel aspects of this study include a focus on patients at high risk of hepatotoxicity (those with myelofibrosis), the availability of a large cohort of concurrent control patients conditioned with TBU/CY, the determination of CY pharmacokinetics, and pharmacokinetic BU targeting to rule out variability in BU exposure as a confounding factor. The target plasma busulfan Css was 800-900 ng/ml for 100% of cases (IV busulfan) and 96.7% of controls (PO and IV busulfan). IV BU has been associated with reduced hepatotoxicity compared to oral BU when dosed by body weight [33]. However, when BU dosing is personalized to a target steady-state concentration, as in our study, outcomes appear to be similar regardless of route of administration [26]. Thus, given the consistent pharmacokinetic targeting of BU in our case and control patients, the route of administration is unlikely to account for the observed differences in outcomes.
We observed a higher risk of relapse in patients with AML/MDS conditioned with CY/TBU, as compared to concurrent AML/MDS patients conditioned with TBU/CY. Some of this risk may relate to confounding variables: patients at high baseline risk of relapse were over-represented in the case cohort, and the relapse rate in the control cohort (20%) was somewhat lower than that generally reported in the literature [34]. Nonetheless, we cannot rule out the possibility that reversing the conditioning-agent sequence may increase the risk of relapse in patients with AML/MDS. Thus, our data do not support the use of this regimen in AML/MDS outside the confines of a well-designed clinical trial.
The major limitation of our study is the use of a concurrent/historical control cohort rather than prospective randomization between CY/TBU and TBU/CY. Our control cohort contained a higher proportion of patients receiving bone marrow (as opposed to G-PBMC) allografts. However, as the most recent available data suggest equivalent outcomes with bone marrow vs. G-PBMC allografts [35], this discrepancy is unlikely to be a significant source of bias in terms of the clinical outcomes of interest. Similarly, our control cohort contained a larger number of patients with HLA-mismatched donors compared to our case cohort. However, after excluding patients with HLA-mismatched donors from both cohorts, CY/TBU conditioning continued to be associated with a significantly lower incidence of SOS in myelofibrosis patients (0% vs. 28%, p=0.01) and lower day +100 mortality (2% vs. 12%, p=0.02), suggesting that our findings were not influenced by this imbalance in donor/recipient HLA matching.
In conclusion, the present data show that reversing the sequence of conditioning agents (from TBU/CY to CY/TBU) before allogeneic HCT was associated with reductions in day +100 mortality and in the incidence of SOS in patients with myelofibrosis. This reduction in hepatotoxicity was likely mediated by reduced exposure to toxic CY metabolites. This change in conditioning sequence, which requires no additional institutional expertise and employs existing medications and technology, can substantially reduce regimen-related toxicity and early mortality and improve outcomes in patients undergoing allogeneic HCT for myelofibrosis.
Acknowledgments
Supported by grants CA09515, CA18029, CA15704, CA162059, and CA76930 from the National Institutes of Health (Bethesda, MD) and by research funding from Otsuka Pharmaceutical Co. (Tokyo, Japan).
We are grateful to the study participants, their caregivers, and the patient care staff for their support of this study. The authors acknowledge Linda Risler for her analytical expertise and Meagan Bemer for her study coordination expertise. The authors also thank Helen Crawford and Bonnie Larson for their assistance with manuscript formatting and citation management.
This work was supported in part by research funding from Otsuka Pharmaceutical Co. (Tokyo, Japan), the makers of Busulfex (IV busulfan), to Andrew Rezvani, Jeannine McCune, and H. Joachim Deeg.
Footnotes
Presented in part at the 51st annual meeting of the American Society of Hematology (New Orleans, LA, 2009).
FINANCIAL DISCLOSURE STATEMENT
The authors have no other relevant financial disclosures to make.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Slattery JT, Clift RA, Buckner CD, et al. Marrow transplantation for chronic myeloid leukemia: the influence of plasma busulfan levels on the outcome of transplantation. Blood. 1997;89:3055–3060. [PubMed] [Google Scholar]
- 2.Slattery JT, Sanders JE, Buckner CD, et al. Graft-rejection and toxicity following bone marrow transplantation in relation to busulfan pharmacokinetics. Bone Marrow Transplant. 1995;16:31–42. [PubMed] [Google Scholar]
- 3.Alyea EP, Kim HT, Ho V, et al. Comparative outcome of nonmyeloablative and myeloablative allogeneic hematopoietic cell transplantation for patients older than 50 years of age. Blood. 2005;105:1810–1814. doi: 10.1182/blood-2004-05-1947. [DOI] [PubMed] [Google Scholar]
- 4.Deeg HJ, Scott BL, Fang M, et al. Five-group cytogenetic risk classification, monosomal karyotype, and outcome after hematopoietic cell transplantation for MDS or acute leukemia evolving from MDS. Blood. 2012;120:1398–1408. doi: 10.1182/blood-2012-04-423046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeLeve LD. Cellular target of cyclophosphamide toxicity in the murine liver: role of glutathione and site of metabolic activation. Hepatology. 1996;24:830–837. doi: 10.1002/hep.510240414. [DOI] [PubMed] [Google Scholar]
- 6.DeLeve LD, Wang X. Role of oxidative stress and glutathione in busulfan toxicity in cultured murine hepatocytes. Pharmacology. 2000;60:143–154. doi: 10.1159/000028359. [DOI] [PubMed] [Google Scholar]
- 7.McDonald GB, Slattery JT, Bouvier ME, et al. Cyclophosphamide metabolism, liver toxicity, and mortality following hematopoietic stem cell transplantation. Blood. 2003;101:2043–2048. doi: 10.1182/blood-2002-06-1860. [DOI] [PubMed] [Google Scholar]
- 8.McCune JS, Woodahl EL, Furlong T, et al. A pilot pharmacologic biomarker study of busulfan and fludarabine in hematopoietic cell transplant recipients. Cancer Chemother Pharmacol. 2012;69:263–272. doi: 10.1007/s00280-011-1736-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pidala J, Kim J, Anasetti C, et al. Targeted i.v. BU and fludarabine (t-i.v BU/Flu) provides effective control of AML in adults with reduced toxicity. Bone Marrow Transplant. 2011;46:641–649. doi: 10.1038/bmt.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCune JS, Batchelder A, Guthrie KA, et al. Personalized dosing of cyclophosphamide in the total body irradiation-cyclophosphamide conditioning regimen: a phase II trial in patients with hematologic malignancy. Clin Pharmacol Ther. 2009;85:615–622. doi: 10.1038/clpt.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geraci JP, Mariano MS, Jackson KL. Radiation hepatology of the rat: microvascular fibrosis and enhancement of liver dysfunction by diet and drugs. Radiat Res. 1992;129:322–332. [PubMed] [Google Scholar]
- 12.Wang X, Kanel GC, DeLeve LD. Support of sinusoidal endothelial cell glutathione prevents hepatic veno-occlusive disease in the rat. Hepatology. 2000;31:428–434. doi: 10.1002/hep.510310224. [DOI] [PubMed] [Google Scholar]
- 13.Meresse V, Hartmann O, Vassal G, et al. Risk factors for hepatic veno-occlusive disease after high-dose busulfan-containing regimens followed by autologous bone marrow transplantation: a study in 136 children. Bone Marrow Transplant. 1992;10:135–141. [PubMed] [Google Scholar]
- 14.Kerbauy FR, Tirapelli B, Akabane H, Oliveira JS. The effect of administration order of BU and CY on toxicity in hematopoietic SCT in humans. Bone Marrow Transplant. 2009;43:883–885. doi: 10.1038/bmt.2008.404. [DOI] [PubMed] [Google Scholar]
- 15.Cantoni N, Gerull S, Heim D, et al. Order of application and liver toxicity in patients given BU and CY containing conditioning regimens for allogeneic hematopoietic SCT. Bone Marrow Transplant. 2011;46:344–349. doi: 10.1038/bmt.2010.137. [DOI] [PubMed] [Google Scholar]
- 16.Wong KM, Atenafu EG, Kim D, et al. Incidence and risk factors for early hepatotoxicity and its impact on survival in patients with myelofibrosis undergoing allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2012 doi: 10.1016/j.bbmt.2012.04.011. [Epub ahead of print 2012 Apr 21] [DOI] [PubMed] [Google Scholar]
- 17.Yeh RF, Pawlikowski MA, Blough DK, et al. Accurate targeting of daily intravenous busulfan with 8-hour blood sampling in hospitalized adult hemtopoietic cell transplant recipients. Biol Blood Marrow Transplant. 2012;18:265–272. doi: 10.1016/j.bbmt.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCune JS, Batchelder A, Deeg HJ, et al. Cyclophosphamide following targeted oral busulfan as conditioning for hematopoietic cell transplantation: pharmacokinetics, liver toxicity, and mortality. Biol Blood Marrow Transplant. 2007;13:853–862. doi: 10.1016/j.bbmt.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 19.Slattery JT, Risler LJ. Therapeutic monitoring of busulfan in hematopoietic stem cell transplantation. Therapeutic Drug Monitoring. 1998;20:543–549. doi: 10.1097/00007691-199810000-00017. [DOI] [PubMed] [Google Scholar]
- 20.McDonald GB, Sharma P, Matthews DE, Shulman HM, Thomas ED. Venocclusive disease of the liver after bone marrow transplantation: diagnosis, incidence, and predisposing factors. Hepatology. 1984;4:116–122. doi: 10.1002/hep.1840040121. [DOI] [PubMed] [Google Scholar]
- 21.Kahl C, Storer BE, Sandmaier BM, et al. Relapse risk among patients with malignant diseases given allogeneic hematopoietic cell transplantation after nonmyeloablative conditioning. Blood. 2007;110:2744–2748. doi: 10.1182/blood-2007-03-078592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiu R, Yao A, Vicini P, et al. Diminishing the risk of nonrelapse mortality in hematopoietic stem cell transplantation: prediction of exposure to the cyclophosphamide metabolite carboxyethylphosphoramide mustard. Clin Pharmacol Ther. 2004;76:270–280. doi: 10.1016/j.clpt.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 23.Jenke A, Freiberg-Richter J, Wilhelm S, et al. Accidental busulfan overdose during conditioning for stem cell transplantation. Bone Marrow Transplant. 2005;35:125–128. doi: 10.1038/sj.bmt.1704697. [DOI] [PubMed] [Google Scholar]
- 24.O’Donnell PH, Artz AS, Undevia SD, et al. Phase I study of dose-escalated busulfan with fludarabine and alemtuzumab as conditioning for allogeneic hematopoietic stem cell transplant: reduced clearance at high doses and occurrence of late sinusoidal obstruction syndrome/veno-occlusive disease. Leukemia & Lymphoma. 2010;51:2240–2249. doi: 10.3109/10428194.2010.520773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perkins JB, Kim J, Anasetti C, et al. Maximally tolerated busulfan systemic exposure in combination with fludarabine as conditioning before allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2012;18:1099–1107. doi: 10.1016/j.bbmt.2011.12.584. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Graiser M, Hutcherson DA, et al. Pharmacokinetic-directed high-dose busulfan combined with cyclophosphamide and etoposide results in predictable drug levels and durable long-term survival in lymphoma patients undergoing autologous stem cell transplantation. Biol Blood Marrow Transplant. 2012;18:1287–1294. doi: 10.1016/j.bbmt.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 27.LeMaistre JA, Bachier C, Smith B, LeMaistre CF, Shaughnessy PJ. Once daily busulfan cyclophosphamide is well tolerated and effective as a preparative regimen for allogeneic hematopoietic stem cell transplant. Journal of Oncology Pharmacy Practice. 2012;18:17–22. doi: 10.1177/1078155210392616. [DOI] [PubMed] [Google Scholar]
- 28.Williams CB, Day SD, Reed MD, et al. Dose modification protocol using intravenous busulfan (Busulfex) and cyclophosphamide followed by autologous or allogeneic peripheral blood stem cell transplantation in patients with hematologic malignancies. Biol Blood Marrow Transplant. 2004;10:614–623. doi: 10.1016/j.bbmt.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 29.Grochow LB. Busulfan disposition: the role of therapeutic monitoring in bone marrow transplantation induction regimens. Semin Oncol. 1993;20:18–25. [PubMed] [Google Scholar]
- 30.Dix SP, Wingard JR, Mullins RE, et al. Association of busulfan area under the curve with veno-occlusive disease following BMT. Bone Marrow Transplant. 1996;17:225–230. [PubMed] [Google Scholar]
- 31.Nilsson C, Forsman J, Hassan Z, et al. Effect of altering administration order of busulphan and cyclophosphamide on the myeloablative and immunosuppressive properties of the conditioning regimen in mice. Exp Hematol. 2005;33:380–387. doi: 10.1016/j.exphem.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 32.Sadeghi B, Jansson M, Hassan Z, et al. The effect of administration order of BU and CY on engraftment and toxicity in HSCT mouse model. Bone Marrow Transplant. 2008;41:895–904. doi: 10.1038/sj.bmt.1705996. [DOI] [PubMed] [Google Scholar]
- 33.Kashyap A, Wingard J, Cagnoni P, et al. Intravenous versus oral busulfan as part of a busulfan/cyclophosphamide preparative regimen for allogeneic hematopoietic stem cell transplantation: decreased incidence of hepatic venoocclusive disease (HVOD), HVOD-related mortality, and overall 100-day mortality. Biol Blood Marrow Transplant. 2002;8:493–500. doi: 10.1053/bbmt.2002.v8.pm12374454. [DOI] [PubMed] [Google Scholar]
- 34.Cornelissen JJ, van Putten WL, Verdonck LF, et al. Results of a HOVON/SAKK donor versus no-donor analysis of myeloablative HLA-identical sibling stem cell transplantation in first remission acute myeloid leukemia in young and middle-aged adults: benefits for whom? Blood. 2007;109:3658–3666. doi: 10.1182/blood-2006-06-025627. [DOI] [PubMed] [Google Scholar]
- 35.Anasetti C, Logan BR, Lee SJ, et al. Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med. 2012;367:1487–1496. doi: 10.1056/NEJMoa1203517. [DOI] [PMC free article] [PubMed] [Google Scholar]