

Abstract
During each heartbeat, intercellular electrical coupling via connexin43 (C×43) gap junctions enables synchronous cardiac contraction. In failing hearts, impaired C×43 trafficking reduces gap junction coupling, resulting in arrhythmias. Here we report that internal translation within C×43 (GJA1) mRNA occurs, resulting in truncated isoforms that autoregulate C×43 trafficking. We find at least four truncated C×43 isoforms occur in human heart, with a 20 kDa isoform predominating. In frame AUG codons within GJA1 mRNA are the translation initiation sites and their ablation arrests trafficking of full-length C×43. The 20 kDa isoform is sufficient to rescue this trafficking defect in trans, suggesting it as a trafficking chaperone for C×43. Limiting cap-dependent translation through inhibition of mTOR enhances truncated isoform expression, increasing C×43 gap junction size. The results suggest that internal translation is a mechanism of membrane protein autoregulation, and a potent target for therapies aimed at restoring normal electrical coupling in diseased hearts.
Introduction
Gap junctions, comprised of connexin proteins, provide the intercellular coupling necessary for rapid action potential propagation through the myocardium, triggering synchronized heart contraction (Rohr, 2004; Shaw and Rudy, 1997). Connexin 43 (C×43) is the most commonly expressed of the 21 human connexins, occurs in all organ systems, and is particularly enriched in ventricular cardiomyocytes (Beyer et al., 1987). Six C×43 molecules oligomerize to form transmembrane channels, termed connexons, which couple with apposing connexons on neighboring cells and coalesce into dense gap junction plaques (Unwin and Zampighi, 1980). Given the broad expression pattern of C×43, it is unsurprising that alterations in C×43 gap junction coupling are associated with diverse pathologies including heart disease (Akar et al., 2007; Beardslee et al., 2000; Luke and Saffitz, 1991; Smith et al., 1991), connective tissue disease (Paznekas et al., 2003), and cancer (Solan et al., 2012). In fact, altered C×43 trafficking contributes to the arrhythmias of sudden cardiac death (Kalcheva et al., 2007; Peters et al., 1997; Remo et al., 2011; Shaw and Rudy, 1997; Smyth et al., 2010).
Gap junction turnover, and the C×43 half-life, occur within several hours (Beardslee et al., 1998; Jordan et al., 1999; Shaw et al., 2007). Regulation of C×43 trafficking is therefore a critical and continuous cellular need. Ion channel function and trafficking is usually regulated by related auxiliary protein subunits (Smyth and Shaw, 2010). To date, no such auxiliary molecules have been identified which are specific to connexins, and the mechanisms regulating anterograde transport of C×43 from cell interior to surface remain under investigation (Shaw et al., 2007; Smyth et al., 2010).
Human C×43 is encoded by the GJA1 gene which comprises two exons, the entire coding sequence of the C×43 protein residing within the second exon (Fishman et al., 1991). Only the 5’UTR of GJA1 mRNA has been identified to undergo alternative splicing events (Pfeifer et al., 2004). There are recent data that eukaryotic internal ribosome entry sites (IRES) exist as a form of translational regulation, and the importance of cap-independent translation events is now accepted as a previously underappreciated but powerful mechanism of posttranscriptional protein regulation (Candeias et al., 2006; Ingolia et al., 2011). Interestingly, an IRES element has been reported within the 5’ UTR of GJA1 mRNA (Schiavi et al., 1999), but not within the coding sequence. We are not aware of internal translation sites being identified for C×43 or any mammalian membrane channel.
We previously found that altered C×43 intracellular trafficking contributes to losses in cell-cell coupling in diseased hearts (Smyth et al., 2010; Smyth et al., 2012). In this study, we present the finding that C×43 trafficking is genetically autoregulated by chaperone proteins generated through internal translation events within the coding sequence of C×43 (GJA1) mRNA. In human heart and cell lines, several truncated isoforms of C×43 are detectable which encompass the C-terminus of C×43. Mutagenesis experiments reveal that these isoforms originate from internal translation initiation events, and that loss of their expression arrests full-length C×43 trafficking. We also find that translation of these isoforms is regulated by the PI3K/AKT/mTOR pathway, which promotes cap-dependent translation. Inhibition of mTOR kinase activity increases C×43 gap junction plaque size at cardiomyocyte cell-cell borders. Taken together, these findings introduce a paradigm of membrane protein autoregulation through internal translation and reveal a role for cap-independent translation in maintenance of electrical coupling of the heart.
Results and Discussion
We explored expression of C×43 in the left ventricle of non-diseased human hearts. Western blot using a C×43 C-terminus directed antibody reveals multiple distinct bands (Figure 1A). The dominant band between 40 and 45 kDa is typically reported for the 43 kDa protein, and has been separated into phosphospecific isoforms (Solan and Lampe, 2007). A second dominant band which we term ‘GJA1-20k’ occurs at 18-20 kDa and has been occasionally observed in cell lines (Joshi-Mukherjee et al., 2007). There are as many as five other distinct, but previously unreported, immunoreactive bands detectable below the predicted 43 kDa full length C×43 (we use the term GJA1-43k) protein (Figure 1A, grey arrows). We then investigated, in different human cell lines, whether the C×43 isoforms were also present (Figure S1A). Just as in human heart, the 43 kDa and 20 kDa bands typically predominate and three middle forms are detectable, with variation between each cell line. Of note, we find that the dominant GJA1-20k isoform migrates between 18 and 25 kDa, and can occur as a doublet. We attribute this to altered phosphorylation status of the C×43 C-terminal region (Solan and Lampe, 2007).
Figure 1. Multiple C-terminal isoforms of connexin 43 occur in human heart.
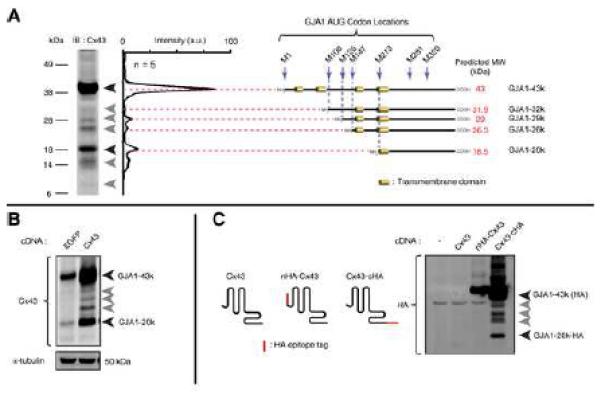
(A) Western blot of non-failing human left ventricle with monoclonal antibody against C×43 C-terminus. Intensity profiles of five separate hearts averaged and plotted in graph with peaks corresponding to major immunodetectable bands (arrows). Schematic: predicted methionine initiated polypeptides and their molecular weights. (B) Western blot of HaCaT cells transfected with cDNA encoding eGFP or C×43. (C) C×43 N- and C-terminus HA tag fusion proteins expressed in 293T cells. Western blot probed with monoclonal anti-HA antibody.
Several mammalian genes have recently been identified to contain internal translation initiation start sites permitting cap-independent translation within their coding regions (Ingolia et al., 2011) and, in zebrafish, C×55.5 can generate distinct protein isoforms (Ul-Hussain et al., 2008a; 2008b). We hypothesized that human GJA1 mRNA occurs as a polycistronic molecule, and that different isoforms of C×43 protein arise from internal translation events. Seven AUG (methionine) codons exist in frame within the coding sequence of C×43. Testing whether each methionine could be a separate start codon, in silico analysis of the GJA1 coding mRNA revealed a striking correlation between predicted protein size, and that of five anti-C×43 bands identified (Figure 1A, schematic).
Transfection of cDNA containing the C×43 coding sequence under transcriptional regulation of a CMV promoter is sufficient to increase expression of four smaller isoforms in addition to full length GJA1-43k (Figure 1B). If the multiple isoforms arise from cleavage of the full length protein, then it would be expected that both C-terminus and N-terminus fragments would exist. However, recognition of truncated isoforms requires antibody against the C-terminus, and not the N-terminus, of C×43 (Figure S1B, left panel, red arrow), indicating the N-terminus of C×43 is contained only in the full length protein, GJA1-43k (Figure S1B, right panel). To confirm C-terminus specificity, we generated N- and C- termini C×43 fusion proteins with the HA epitope tag. Only the C-terminally fused C×43-HA construct generated detectable smaller HA-tagged isoforms dominated by the 20 kDa band as seen in human heart, in addition to full length GJA1-43k-HA (Figure 1C, arrow). We then introduced previously described point mutations, R202E and F199L, which retard trafficking of full length C×43 (Olbina and Eckhart, 2003). Both mutations resulted in loss of gap junction plaque formation and redistribution of C×43 to the cytoplasm (Figure S2A, arrows), yet the presence of truncated isoforms was not affected by the deficit in cytoplasmic trafficking (Figure S2B, arrows) suggesting their origin occurs prior to processing later in the vesicular transport pathway.
To confirm that all C×43 isoforms detected in Figure 1 arise from the same mRNA molecule, three distinct siRNA duplexes were introduced into 293T cells. All three duplexes target regions of the mRNA sequence more 5’ proximal than GJA1-20k, yet all three were capable of ablating expression of the smaller isoforms (Figure 2A). We then transfected in vitro transcribed GJA1 mRNA resistant to siRNA duplex ii in Figure 2A into 293T cells following knockdown of endogenous C×43. Consistent with the possibility of internal translation initiation sites, the exogenously generated GJA1 mRNA was sufficient to encode the major full length (43 kDa) and smaller (20 kDa) forms of C×43 (Figure 2B). If the smaller C×43 isoforms arise from internal translation initiation, then the initial start codon (M1) of the GJA1 gene should not be necessary for their expression. Indeed, ablation of the GJA1 start codon prevents expression of the full length protein, but enhances expression of smaller isoforms (Figure 2C). The in silico studies of Figure 1A predict that in frame internal AUG codons of GJA1 can be initiation sites for the truncated isoforms. We sequentially mutated each internal methionine codon (AUG) to aspartate (GAC). Each mutation resulted in loss of the corresponding predicted C×43 isoform (Figure 2D, arrows). These data are confirmed with fusion of a HA epitope tag to the C-terminus of C×43, where substitution of all six internal methionines with the more innocuous mutation to leucine (C×43-ML-HA) also ablates expression of the smaller isoforms (Figure S2C).
Figure 2. Truncated C×43 isoforms arise from internal translation initiation events.
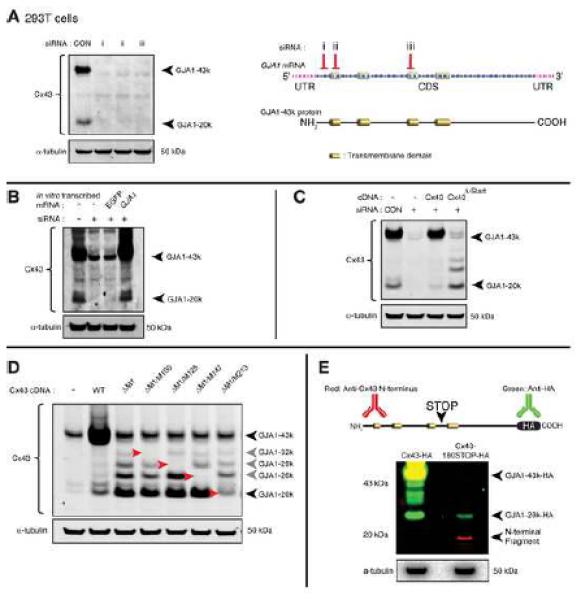
(A) Western blot of endogenous C×43 knockdown in 293T cells using three distinct siRNA duplexes. Schematic illustrating the location of each siRNA targeting sequence within GJA1 mRNA. (B) C×43 Western blot of siRNA-resistant in vitro transcribed mRNA transfection of 293T cells with endogenous C×43 knockdown. (C) C×43 Western blot following siRNA-resistant C×43 cDNA transfection with knockdown of endogenous C×43 in 293T cells. C×43Δ-Start contains AUG to AUA point mutation to ablating initial M1 start codon. (D) C×43 Western blot of transfected C×43 cDNA containing sequential AUG to GAC mutations for internal methionines M100, M125, M147 and M213 in addition to M1. Bands corresponding to internal translation products are highlighted by red arrows. (E) Multiplex Western blot of 293T cells transfected with siRNA-resistant wild-type C×43-HA (left lane) or C×43-180STOP-HA (right lane) which contains a triplet of stop codons at codons 177,178, and 179 (schematic). Blot probed for HA (green) and C×43 N-terminus (red).
Internal translation within the coding sequence of GJA1 mRNA may arise from ribosomes that have read through previous start sites, or by internal ribosomal entry and initiation within the coding sequence proper. We introduced a potent triple-stop (TAGTAATGA) signal by mutation at codons 177, 178, and 179 in the C×43-HA construct (C×43-180STOP-HA). These codons occur proximal to M213 so if GJA1-20k (HA-tagged) is a result of ribosomal read-through its expression would be prevented by the early stop codons. As expected, an 18 kDa N-terminal C×43 fragment corresponding to residues 1-176 is detectable in cells transfected with the C×43-180STOP-HA construct, and full length GJA1-43k-HA protein is absent (Figure 2E, red bands). Probing for HA however, we find that the 177,178,179 stop codons fail to inhibit expression of GJA1-20k (Figure 2E, green bands). These data strongly support ribosomal entry and initiation of translation, as opposed to read-through or cleavage, as the mechanism of GJA1 mRNA internal translation.
C×43 is a molecule with rapid turnover yet no known regulatory subunits. We explored the relationship of internally translated C×43 isoforms with full length GJA1-43k trafficking. Immunofluorescence of 293T cells transfected with wild-type C×43 reveals gap junction plaques at cell-cell borders in 78.7 % of transfected cell pairs (Figure 3A). Introduction of sequential additive AUG to CUG mutations of their corresponding internal start codons (M100L, M125L, M147L, and M213L) reveals that loss of all four internal start codons severely compromises GJA1-43k trafficking. The C×43M4L construct encodes a GJA1-43k protein with all four methionine to leucine mutations, and is largely restricted to cytoplasmic reticular structures consistent with the ER, with only 12 % of cell pairs displaying gap junction plaque formation (Figure 3A, Figure S3A). Given that GJA1-20k arises from M213 in the GJA1 coding sequence and is the most robustly expressed truncated C×43 isoform, we explored whether it was sufficient to rescue transport of GJA1-43kM4L. Indeed, when co-expressed with GJA1-20k-V5 (red), transport of full-length GJA1-43kM4L (green) to gap junctions at the cell surface is rescued, while GJA1-20k-V5 remains cytoplasmic (Figure 3C, Figure 3D). These data indicate that perturbed trafficking of GJA1-43kM4L is not a result of mutation of internal methionines to leucines at the protein level, but rather due to loss of the co-expressed truncated isoform(s).
Figure 3. Truncated C×43 isoforms are necessary for trafficking of full-length C×43 to gap junction plaques.
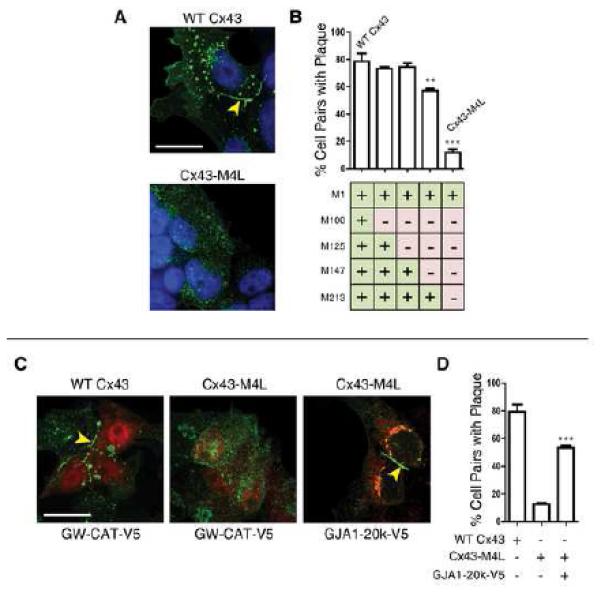
(A) Fixed-cell immunofluorescence of 293T cells expressing siRNA-resistant C×43 (green) with knockdown of endogenous C×43. Nuclei detected with DAPI (blue), arrow: gap junction plaque at cell-cell border. Cell pairs were randomly selected and assessed for presence of gap junction plaque for C×43 constructs containing additive AUG to CUG mutations at internal methionines M100, M125, M147, and M213. C×43M4L encompasses all four AUG to CUG mutations. Percentage of cell pairs with plaque presented in graph in (B). (C) Fixed-cell immunofluorescence of 293T cells co-expressing the control GW-CAT-V5 construct or the GJA1-20k-V5 isoform (red) and siRNA-resistant C×43 (green). Nuclei counterstained with DAPI (blue), arrow: gap junction plaque at cell-cell border. (D) Percentage of cell pairs from (C) assessed for presence of gap junction plaque. Original magnification x 100. Scale bar: 25 µm. Data are presented as mean ±SEM. ***P < 0.001, **P < 0.01.
High resolution immunofluorescence imaging of GJA1-20k-V5 reveals that this isoform is localized primarily to cytoplasmic reticular structures (Figure 3C). This compartment is confirmed as endoplasmic reticulum (ER)/Golgi by immunofluorescence colocalization studies (Figure S3B, Figure S3C), suggesting a role for GJA1-20k early in the C×43 vesicular transport pathway. Indeed, co-immunoprecipitation experiments reveal that full length GJA1-43k complexes with GJA1-20k, and enrichment of GJA1-43k in the ER using Brefeldin A increases this interaction (Figure S3E). To further visualize this interaction and rule out any possible artifact of immunostaining, we co-transfected 293T cells with constructs encoding GJA1-20k-mCherry and GJA1-43kML-eGFP. Comparable data to Figure 3C were obtained, whereby GJA1-20k colocalizes with GJA1-43kML-eGFP in the perinuclear Golgi compartment, but is not incorporated into gap junction plaques at the cell surface with the full length GJA1-43ML-eGFP protein, (Figure S4, arrows). GJA1-20k may therefore act as a chaperone auxiliary protein, regulating trafficking of de novo GJA1-43k molecules through the ER/Golgi.
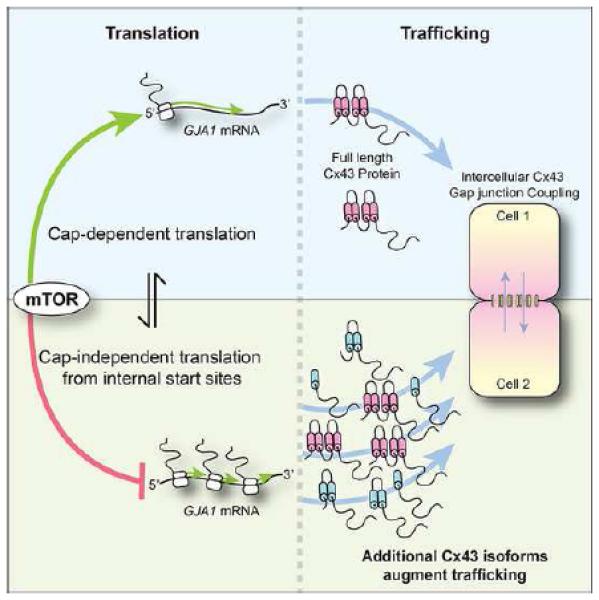
Given the variable expression of truncated C×43 isoforms in human cell lines (Figure S1A), we wondered if their expression is regulated by signaling pathways associated with translation. Using primary neonatal mouse ventricular cardiomyocytes (NMVMs), we found that inhibition of PI3K/AKT signaling using the PI3K inhibitor GDC-0941 (Folkes et al., 2008) increases expression of GJA1-20k (Figure S5). The mammalian target of rapamycin (mTOR) pathway is an established regulator of cap-dependent protein translation integral to PI3K/AKT signaling. Inhibition of mTOR kinase activity using the inhibitor PP242 (Feldman et al., 2009) significantly augments expression of internally translated isoforms in 293T cells transfected with C×43ΔSTART, revealing this pathway as acting independently of the cap and first mRNA start codon and directly inhibits internal or cap-independent translation (Figure 4A). Just as observed with P13K inhibition, PP242 also increases expression of GJA1-20k in NMVMs (Figure 4B). The early formation of additional C×43 isoforms (Figure S2) and their regulation by the PI3K/AKT/mTOR pathway (Figure S4, Figure 4A, Figure 4B) indicate that truncated GJA1 isoforms originate from internal, cap-independent, translation events. Fixed cell immunofluorescence of NMVMs incubated for 16 h with DMSO (left panels) or PP242 (right panels) is presented in Figure 4C. An increase in C×43 (green) gap junction density is apparent in cells treated with PP242, with N-cadherin (red) identifying cell-cell borders. Quantification of C×43 fluorescence intensity profiles of 10-μm vectors bisecting cell–cell borders is presented in Figure 4D. Consistent with data presented in Figure 3, these findings implicate internally translated C×43 isoforms in positive regulation of gap junction formation.
Figure 4. Expression of C×43 truncated isoforms is regulated by the mTOR pathway.

(A) 293T cells transfected with siRNA-resistant C×43 or C×43ΔSTART with knockdown of endogenous C×43 incubated for 8 h in the presence or absence of the mTOR kinase inhibitor PP242. C×43 detected by western blot. (B) Primary neonatal mouse ventricular cardiomycoytes (NMVMs) incubated for 16 h with DMSO or PP242. C×43 detected by Western blot with quantitation of C-terminal fragment relative to full length (43kDa) in graph. (C) Fixed-cell immunofluorescence of cells from (B). Cell borders are identified with N-cadherin (red) for quantification of C×43 (green) gap junction density. Original magnification x 60. Scale bar: 25 μm. (D) Averaged fluorescence intensity profiles of C×43 expression at cell–cell borders. Data are presented as mean ±SEM. **P < 0.01, *P < 0.05.
C×43 has been implicated in cellular processes that are apparently independent of cell-cell communication, including trafficking of other ion channels (Rhett et al., 2012), mitochondrial regulation (Rodriguez-Sinovas et al., 2006), and cell cycle regulation (Olbina and Eckhart, 2003). It is possible that alternatively translated gene products could be responsible for some of these observations. In recent years, regulation of translation has emerged as a potentially major component of cellular protein control, with mTOR pathway recognized as central to promoting cap-dependent translation. The number of identified eukaryotic IRES elements is increasing, with the majority occurring within the 5’ UTR of mRNAs to allow cap-independent translation during mitosis and stress (Komar and Hatzoglou, 2011). Initiation of translation within the mRNA coding sequence has also been reported for proteins such as ECSIT, Myc, and NANOG for example, yielding truncated gene products which can regulate full-length protein function (Ingolia et al., 2011). Many ion channels require subunits to chaperone their journey from the ER to the plasma membrane (Smyth and Shaw, 2010). In this study, we present the finding that GJA1 mRNA is in fact polycistronic, and several N-terminally truncated C×43 isoforms are generated at the translational level. These alternatively translated C×43 ‘subunits’ are necessary for successful transport of the full length protein to the cell surface, and of these, GJA1-20k is sufficient to effect this process. It is also possible that non-C×43 ion channels and other proteins without known chaperones may undergo similar auto-regulation. Cap-independent translation events therefore represent a potential target for protein regulation and therapeutic restoration of normal electrical coupling in diseased hearts.
Experimental Procedures
Human tissue acquisition
With the approval of the UCSF Committee for Human Research, we obtained tissue from hearts from organ donors whose hearts were not transplanted for technical reasons. The California Transplant Donor Network (CTDN) provided the unused donor hearts and obtained informed consent for their use from the next of kin.
Mice
All procedures were reviewed and approved by the University of California Institutional Animal Care and Use Committee.
Molecular Biology
Human GJA1 was obtained from Open Biosystems and cloned into pDONR/221 and pcDNA3.2/V5-Dest using Gateway technology (Life Technologies). Constructs encoding N- and C-terminus HA-epitope tagged C×43, and C-terminal eGFP and mCherry fusion proteins were generated as previously described (Smyth et al., 2012). Mutagenesis was performed using Quickchange Lightning mutagenesis kit according to manufacturer’s instructions (Agilent).
In vitro transcription was undertaken using the T7 mScript Standard mRNA Production System (Cellscript) and pcDNA3.2-huC×43 or pcDNA3.2-eGFP as template.
Stealth siRNA duplexes targeting human GJA1 were obtained from Life Technologies.
NMVMs isolation and culture
P1 neonatal mouse ventricular cardiomyocytes (NMVMs) were isolated and maintained in culture as previously described (Smyth et al., 2012). For inhibitor experiments, cells were insulin starved and FBS dropped to 1% in DMEM/F12 for 8 h prior to addition of GDC-0941 (2.5 μM), PP242 (5 μM), or DMSO. Following 30 min incubation insulin was added to a final concentration of 10 μg/ml and cells were fixed for immunofluorescence or harvested for biochemistry 1 h and 16 h post stimulation.
Western Blotting
Lysates were prepared in RIPA buffer and Western blotting performed as previously described (Smyth et al., 2010; Smyth et al., 2012). Mouse monoclonal anti-C×43 directed against C-(1/3000) and N-terminal (1/2000) regions were obtained from the Fred Hutchinson Cancer Research Center. Mouse monoclonal anti-α-tubulin (1/3000), anti-HA (1/5000), and rabbit polyclonal anti-C×43 (1/3000) were obtained from Sigma. Rabbit polyclonal antibodies against AKT (1/1000) and phosoho-AKTSer473 (1/1000) were obtained from Cell Signaling Technology.
Immunofluorescence
Cells co-transfected with pcDNA3.2-GW-CAT-V5 as a transfection control, or pcDNA3.2-GJA1213-382-V5, together with either wild-type or mutated pcDNA3.2-huC×43-Stop. Eighteen hours post transfection, cells were fixed with 4% PFA and immunostained for C×43 (rabbit polyclonal 1/3000, Sigma) and V5 tag (mouse monoclonal 1/1000, Sigma) with AlexaFluor488 and AlexaFluor555 (Life Technologies) as previously described (Smyth et al., 2010). For mouse monoclonal Golgi apparatus marker 58K (Abcam 1/200) and ER marker PDI (Abcam, 1/200) immunodetection, cells were fixed in ice-cold methanol for 5 min. Monoclonal N-cadherin (BD Biosciences 1/250) was used to detect cell-cell borders in NMVMs.
Slides were imaged using using a Nikon Ti microscope with a ×100/1.49 Apo TIRF objective, Yokogowa CSU-X1 spinning disk confocal unit with 486 and 561 nm laser sources, and Coolsnap HQ2 camera controlled by NIS Elements software.
Quantification of C×43 gap junction plaque formation
Cell pairs with screened in blinded fashion for comparable levels of V5-tag, then assessed for presence of C×43 gap junction plaque. A total of 75 cell pairs were assessed for each condition.
Quantification of C×43 gap junction plaque density
For quantification of C×43 expression at cell-cell borders (Figure 4C) maximum intensity projections of 10 μm confocal z-stacks were generated for N-cadherin (to identify cell-cell borders) and C×43. We then implemented a MATLAB routine which generates 10 μm fluorescence intensity profiles bisecting traced cell-cell borders, automating a quantification technique previously described (Smyth et al., 2012). This routine is freely available upon request.
Statistical analysis
All data are presented as mean ± SEM and the Student’s t test or a one-way ANOVA (with Bonferroni post-test) used accordingly. A P value of less than 0.05 was considered significant.
Supplementary Material
Highlights.
-
1)
Multiple N-terminally truncated isoforms of connexin 43 occur in human heart
-
2)
Internal translation at in frame AUG codons generates smaller connexin 43 isoforms
-
3)
Trafficking of full length connexin 43 is regulated by smaller truncated isoforms
-
4)
The mTOR pathway regulates truncated C×43 isoform translation and gap junction size
Figure 5.
Acknowledgments
This work was supported in part by the NIH grant HL094414 (to R.M.S.) and the American Heart Association Scientist Development Grant SDG3420042 (to J.W.S.). The authors are grateful to Tina S. Fong and Roger Chang (UCSF) for technical assistance in addition to Natalia Jura, Samy Lamouille (UCSF), Ting-Ting Hong, and Shan-Shan Zhang (Cedars-Sinai) for helpful discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akar F, Nass R, Hahn S, Cingolani E, Shah M, Hesketh G, DiSilvestre D, Tunin R, Kass D, Tomaselli G. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. AJP: Heart and Circulatory Physiology. 2007;293:1223–1230. doi: 10.1152/ajpheart.00079.2007. [DOI] [PubMed] [Google Scholar]
- Beardslee M, Laing J, Beyer E, Saffitz J. Rapid turnover of connexin43 in the adult rat heart. Circulation Research. 1998;83:629–635. doi: 10.1161/01.res.83.6.629. [DOI] [PubMed] [Google Scholar]
- Beardslee M, Lerner D, Tadros P, Laing J, Beyer E, Yamada K, Kléber A, Schuessler R, Saffitz J. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circulation Research. 2000;87:656–662. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- Beyer E, Paul D, Goodenough D. Connexin43: a protein from rat heart homologous to a gap junction protein from liver. The Journal of Cell Biology. 1987;105:2621–2629. doi: 10.1083/jcb.105.6.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candeias M, Powell D, Roubalova E, Apcher S, Bourougaa K, Vojtesek B, Bruzzoni-Giovanelli H, and Fåhraeus R. Expression of p53 and p53/47 are controlled by alternative mechanisms of messenger RNA translation initiation. Oncogene. 2006;25:6936–6947. doi: 10.1038/sj.onc.1209996. [DOI] [PubMed] [Google Scholar]
- Feldman M, Apsel B, Uotila A, Loewith R, Knight Z, Ruggero D, Shokat K. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS biology. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman G, Eddy R, Shows T, Rosenthal L, Leinwand L. The human connexin gene family of gap junction proteins: Distinct chromosomal locations but similar structures. Genomics. 1991;10:250–256. doi: 10.1016/0888-7543(91)90507-b. [DOI] [PubMed] [Google Scholar]
- Folkes A, Ahmadi K, Alderton W, Alix S, Baker S, Box G, Chuckowree I, Clarke P, Depledge P, Eccles S, Friedman L, Hayes A, Hancox T, Kugendradas A, Lensun L, Moore P, Olivero A, Pang J, Patel S, Pergl-Wilson G, Raynaud F, Robson A, Saghir N, Salphati L, Sohal S, Ultsch M, Valenti M, Wallweber H, Wan N, Wiesmann C, Workman P, Zhyvoloup A, Zvelebil M, Shuttleworth S. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer . Journal of Medicinal Chemistry. 2008;51:5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- Ingolia N, Lareau L, Weissman J. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K, Solan J, Dominguez M, Sia M, Hand A, Lampe P, Laird D. Trafficking, assembly, and function of a connexin43-Green fluorescent protein chimera in live mammalian cells. Molecular Biology of the Cell. 1999;10:2033–2050. doi: 10.1091/mbc.10.6.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Mukherjee R, Coombs W, Burrer C, de Mora IA, Delmar M, Taffet SM. Evidence for the presence of a free C-terminal fragment of c×43 in cultured cells. Cell Communication & Adhesion. 2007;14:75–84. doi: 10.1080/15419060701402320. [DOI] [PubMed] [Google Scholar]
- Kalcheva N, Qu J, Sandeep N, Garcia L, Zhang J, Wang Z, Lampe P, Suadicani S, Spray D, Fishman G. Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proceedings of the National Academy of Sciences. 2007;104:20512–20516. doi: 10.1073/pnas.0705472105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar A, Hatzoglou M. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle. 2011;10:229–240. doi: 10.4161/cc.10.2.14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke R, and Saffitz J. Remodeling of ventricular conduction pathways in healed canine infarct border zones. Journal of Clinical Investigation. 1991;87:1594–1602. doi: 10.1172/JCI115173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olbina G, Eckhart W. Mutations in the second extracellular region of connexin 43 prevent localization to the plasma membrane, but do not affect its ability to suppress cell growth. Molecular Cancer Research. 2003;1:690–700. [PubMed] [Google Scholar]
- Paznekas W, Boyadjiev S, Shapiro R, Daniels O, Wollnik B, Keegan C, Innis J, Dinulos M, Christian C, Hannibal M, Jabs E. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. The American Journal of Human Genetics. 2003;72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters N, Coromilas J, Severs N, Wit A. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–996. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- Pfeifer I, Anderson C, Werner R, Oltra E. Redefining the structure of the mouse connexin43 gene: selective promoter usage and alternative splicing mechanisms yield transcripts with different translational efficiencies. Nucleic Acids Research. 2004;32:4550–4562. doi: 10.1093/nar/gkh792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remo B, Qu J, Volpicelli F, Giovannone S, Shin D, Lader J, Liu F, Zhang J, Lent D, Morley G, Fishman G. Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circulation Research. 2011;108:1459–1466. doi: 10.1161/CIRCRESAHA.111.244046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhett J, Ongstad E, Jourdan J, Gourdie R. C×43 associates with Na(v)1.5 in the cardiomyocyte perinexus. The Journal of Membrane Biology. 2012;245:411–22. doi: 10.1007/s00232-012-9465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Sinovas A, Boengler K, Cabestrero A, Gres P, Morente M, Ruiz-Meana M, Konietzka I, Miró E, Totzeck A, Heusch G, Schulz R, Garcia-Dorado D. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circulation Research. 2006;99:93–101. doi: 10.1161/01.RES.0000230315.56904.de. [DOI] [PubMed] [Google Scholar]
- Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovascular Research. 2004;62:309–322. doi: 10.1016/j.cardiores.2003.11.035. [DOI] [PubMed] [Google Scholar]
- Schiavi A, Hudder A, Werner R. Connexin43 mRNA contains a functional internal ribosome entry site. FEBS Letters. 1999;464:118–122. doi: 10.1016/s0014-5793(99)01699-3. [DOI] [PubMed] [Google Scholar]
- Shaw R, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circulation Research. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- Shaw R, Fay A, Puthenveedu M, von-Zastrow M, Jan Y, Jan L. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;128:547–560. doi: 10.1016/j.cell.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Green C, Peters N, Rothery S, Severs N. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. The American Journal of Pathology. 1991;139:801–821. [PMC free article] [PubMed] [Google Scholar]
- Smyth J, Hong T, Gao D, Vogan J, Jensen B, Fong T, Simpson P, Stainier D, Chi N, Shaw R. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. The Journal of Clinical Investigation. 2010;120:266–279. doi: 10.1172/JCI39740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth J, Shaw R. Forward trafficking of ion channels: what the clinician needs to know. Heart Rhythm. 2010;7:1135–1140. doi: 10.1016/j.hrthm.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth J, Vogan J, Buch P, Zhang S, Fong T, Hong T, Shaw R. Actin cytoskeleton rest stops regulate anterograde traffic of connexin 43 vesicles to the plasma membrane. Circulation Research. 2012;110:978–989. doi: 10.1161/CIRCRESAHA.111.257964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J, Hingorani S, Lampe P. Changes in connexin43 expression and localization during pancreatic cancer progression. The Journal of Membrane Biology. 2012;245:255–262. doi: 10.1007/s00232-012-9446-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J, Lampe P. Key connexin 43 phosphorylation events regulate the gap junction life cycle. The Journal of Membrane Biology. 2007;217:35–41. doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul-Hussain M, Dermietzel R, Zoidl G. Characterization of the internal IRES element of the zebrafish connexin55.5 reveals functional implication of the polypyrimidine tract binding protein. BMC molecular biology. 2008a;9:92. doi: 10.1186/1471-2199-9-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul-Hussain M, Zoidl G, Klooster J, Kamermans M, Dermietzel R. IRES-mediated translation of the carboxy-terminal domain of the horizontal cell specific connexin C×55.5 in vivo and in vitro. BMC molecular biology. 2008b;9:52. doi: 10.1186/1471-2199-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin P, Zampighi G. Structure of the junction between communicating cells. Nature. 1980;283:545–549. doi: 10.1038/283545a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.