

Abstract
Post-translational modifications such as phosphorylation play a vital role in the regulation of protein function. In our study of the basic Helix-loop-Helix (bHLH) transcription factor HAND1, it was suspected that HAND1 was being phosphorylated during trophoblast giant cell differentiation and that coexpression of a constitutively active kinase with HAND1 resulted in changes in the proteins dimerization profile. In order to accurately document HAND1 phosphorylation and identify the resides being modified, we employed metabolic cell labeling with 32P of tissue culture cells coexpressing a Flag-epitope tagged HAND1 along with a number of active kinases and phosphatase subunits. We generated phosphopeptide maps of the phosphorylated HAND1 using the methods described below and linked these modifications to changes in HAND1 biological function.
Keywords: Phosphorylation, Dimerization, Helix-Loop-Helix Motifs
Introduction
The basic Helix-loop-Helix (bHLH) transcription factors HAND1 and HAND2 are members of the twist family of bHLH proteins (for review (1). HAND factors are expressed in a variety of tissues during murine development including the heart, cardiac neural crest, lateral mesoderm, extraembryonic mesoderm, maternally derived decidua and sympathetic nervous system. bHLH factors activate/repress transcription by forming dimers via their HLH motif and bind DNA through the juxtaposition of the 2 basic domains which recognize a canonical cis-element CANNTG termed an E-box (for review (2)). bHLH proteins can generally be separated into 2 classes. The class A bHLH factors (E-proteins) which are ubiquitously expressed and the tissue-restricted or class B bHLH factors. The established paradigm of function was that a heterodimer composed of a class A and a class B protein was required for transcriptional regulation and in fact many class B factors do not form homodimers or heterodimers with other class B factors efficiently. Recently, we and others recognized that members of the twist family of proteins exhibited a more promiscuous dimerization profile forming homodimers and heterodimers with a wide range of class B factors (3-6). With this in mind, we set out a hypothesis that tissue-specific transcriptional regulation by HAND factors was in fact driven by the specific bHLH dimer complex that could form. This hypothesis begs the question of how is HAND dimerization controlled. Most recently, we determined that the answer to this question is in part addressed by post-translational modification (phosphorylation) of specific residues in the bHLH domain and that these modifications affect the dimerization affinities of HAND proteins that allow for changes in biological function (3). To determine if in fact HAND factors were phosphorylated and to define the specific location of the modified residues, we coexpressed a flag epitope tagged HAND1 (FlagHAND1) with constitutively active kinases and or phosphatases in tissue culture cells (HEK293 and RCHOI), which were subsequently metabolically labeled with 32P-orthophosphate, HAND1 protein was immuno-precipitated using flag antibody, subjected to trypsin digestion, peptides were separated via 2-dimensional phosphopeptide mapping and phosphopeptides were visualized via exposure of the TLC plates to phosphoimager screens. By employing HAND1 deletion and point mutants, this technique allowed for the identification of the specific residues that are phosphorylated.
Materials and Methods
Constructs
PKC-7 is a constitutively active form of PKCα, which was previously described (7). pFC-PKA is a constitutively active PKA (Strategene). pIRES FLAG-HAND1 and HAND1 point mutants are amino FLAG-tag (Sigma) fusions cloned into pIRES NEO (Clonetech). B56α and δ cDNAs were cloned into the expression plasmid pCEP4-l. HAND1 point mutants HAND1 S98A, S109A-T107A &D were generated using the Quick-Change Mutagenesis kit (Strategene) following the manufacturers protocols.
Tissue culture
HEK293 cells were grown in 10% FBS containing DMEM containing antibiotics at 37oC and 5% CO2. A total of 10 μg of the indicated plasmid construct was transfected into cells using a CaPO4 based transfection (8). Briefly DNA was resuspended in 50 μl of H2O. To this 500 μl of 2X HBS was added. While bubbling air constantly with an autopipette, 450 μl of a 2:12 dilution of 2 M CaCl2 was added drop-wise and allowed to sit for 20 minutes at room temperature. Precipitates were then added to cells and allowed to incubate for 4 hours at 37oC and 5% CO2. Media was removed and replaced with 3 ml of 15% glycerol in supplemented DMEM for 1 minute. Media was immediately aspirated, cells were washed in 5 mL of 1X PBS and 10 ml of supplemented DMEM was added and cells were grown for 48 hours.
In vivo labeling-Immunoprecipitations
HEK293 cells were grown and transfected as described above with the indicated constructs. 48 hours post transfection cells were incubated with 1 mCi of 32P orthophosphoric acid (NEN)/ml of phosphate–free DMEM supplemented with dialyzed FBS for 4 hours. Cells were washed in 20 mM HEPES pH 7.4 and 150 mM NaCl followed by lysis in 20 mM NaPO4, 150 mM NaCl, 2 mM MgCl2, 0.1% NP40, 10% glycerol, 3 mg/μl leupeptin, 3 mg/μl pepstatin, 1 mM PMSF, 50 μM NaVO4, 5 mM NaF, 100 nM Okadaic acid, and 5 mM Beta glycerol phosphate. Equal amounts of protein were immunoprecipitated with agarose-conjugated FLAG M2-beads (Sigma) for 2 hours. Samples were washed 3 times in 1X PBS with a tube change on the last wash. Samples were boiled in loading dye, run through a 12% SDS PAGE, dried and exposed to a phosphoimager screen.
2-dimensional phosphopeptides mapping
HEK293 cells were grown, transfected, labeled with 32P, and immunoprecipitated as described above. Dried IP gels were exposed to phosphoimager. Bands were identified and cut by aligning the image with radioactive marker spots. The acrylamide bands corresponding to labeled FLAG-HAND1 were cut away from the dried gel, the filter paper was scraped away from the acrylamide, and rehydrated in 400 μl freshly made 50 mM ammonium bicarbonate. The rehydrated acrylamide was crushed into small pieces and allowed to digest overnight with 30 μg of TPCK-treated trypsin (Worthington) at 37ºC. Digests were spiked the following day. Digested peptides were removed from the crushed acrylamide, washed twice with 50 mM ammonium bicarbonate, and the peptides were concentrated in a Speed Vac (Forma). Digested peptides were washed 4 times with 1 mL ddH2O and then twice in pH 1.9 buffer (2.8% formic acid, 7.8% glacial acetic acid). Samples were then resuspended in 4 μl pH 1.9 buffer, spotted onto cellulose TLC plates and run in the 1st dimension in pH 1.9 buffer on a Hunter Thin Layer Electrophoresis apparatus (HTLE 7000, CBS Scientific, Inc.) for 35 minutes at 1300 V. Plates were dried, rotated 90 degrees, and then run in the 2nd dimension using an isobutyric acid buffer (62.5% isobutyric acid, 1.9% n-Butanol, 4.8% pyridine, 2.9% glacial acetic acid) in a TLC tank. When liquid phase migration was 1 cm from the top of the plates, they were removed, dried, and exposed to a phosphoimager screen for visualization and analysis.
Results and Discussion
We recently demonstrated that HAND1 is phosphorylated during the differentiation of RCHOI trophoblast stem cells and that protein kinase A (PKA), protein kinase C (PKC) and the delta isoform of the B56-regulatory subunit (B56δ) of the protein phosphatase 2A (PP2A) regulate HAND1 phosphorylation status on residues T107 and S109 (3). Key in this study was the ability to determine the exact residues that were being modified in HAND1 so that functional analysis of HAND1 mutants in these residues could be deduced. We employed phosphopeptide analysis to address these questions as it provided a greater sensitivity to changes in HAND1 phosphorylation state than simple immunoprecipitations (IPs) of HAND1 expressed in metabolically labeled cells. We chose to use phosphopeptide mapping over mass spectrophometry as we could perform these experiments ourselves without needing to rely on instrumentation analysis by others. Results show that expression of constitutively active PKC along with HAND1 results in increased phosphorylation of 5 peptides (Fig. 1; 3). Moreover, coexpression of B56α results in no significant change to HAND1 phosphorylation while coexpression of B56δ reduces the phosphorylation on spots 8 and 9 correlating well with B56δ interaction analysis with HAND1 (Fig. 1; 3).
Fig. 1. Identification of HAND1 residues phosphorylated by PKC using phosphopeptide analysis in HEK293 cells.
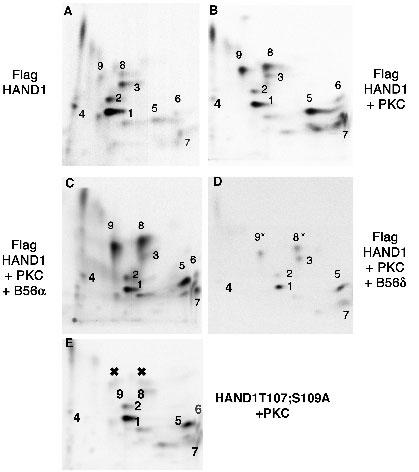
Panels (A) and (B) show the variation of HAND1 phosphorylation when expressed with or without constitutively active PKC. The increased signal intensity of 5-phosphopepties (5-9) indicates increased phosphorylation. Panels (C) and (D) show that coexpression of the non-interacting B56α (C) has no effect on HAND1 phosphorylation by PKC whereas expression of B56δ (D) reduces HAND1 phosphorylation of peptides 8 and 9 (marked *). Panel (E) shows that point mutagenesis of both T107 and S109 to alanine eliminates the phosphorylation of peptides 8 and 9 (marked X) confirming these sites as PKC targets and targets for dephosphorylation by B56δ-containing PP2A.
We next employed serine and threonine mutations based on our understanding of the trypsin restriction map of HAND1. Our designed HAND1 mutants correspond to S→A changes in consensus kinase sites for PKA and PKC. Additionally when designing the mutants, we considered if the mutations were contained within a single trypsin fragment and if other S and or T residues were also present in these peptides (3). In the case of T107 and S109, they were contained in a single fragment, so we decided to mutate these in combination. Phosphopeptide maps of HAND1 and HAND1T107;S109A coexpressed with or without constitutively active PKC show that 2-specific phosphopeptides are absent from the map of HAND1T107;S109A when compared to wildtype HAND1 (Fig. 1). These results correspond with similar previously published experiments in which we coexpressed constitutively active PKA (3). The observation that a double mutation within a single trypsin fragment resulted in the reduction of two phosphopeptides on the map can be interpreted in two ways. One is that the two spots represent mono- and diphosphorylated forms of the protein. The second is that the two peptides are in fact an artifact of incomplete trypsin digestion. Trypsin cuts at basic residues and the HAND1 basic domain contains a high concentration of basic residues. As the HAND1 basic domain is located just amino to T107 and S109 partial trypsin digestion could explain the loss of 2 phosphopeptides in the HAND1T107;S109A mutant shown in Figure 1. This phenomenon was clearly encountered in the mutation of S98 (the only S within its trypsin peptide) where 3-5 partial fragments were reduced in the maps of this HAND1 mutant (3). To try to address this issue directly, we made the single HAND1 mutants T107A and S109A, coexpressed these mutants in cells with constitutive kinase and compared maps to wild type HAND1 (Fig. 2). Results of these experiments show that mutation of T107 to alanine does not significantly alter phosphopeptide pattern of HAND1 when coexpressed with PKA; however, mutation of S109 eliminates phosphorylation of both peptides (Fig. 2). These results suggest that indeed like S98 partial digestion may come into play, but in this case it is also possible that phosphorylation of T107 is required for phosphorylation of S109. To address this question would require mass spectrophometery.
Fig. 2. Single mutations of T107 and S109 suggest that S109 is the main target of PKA and PKC.
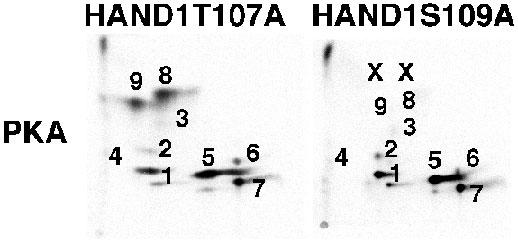
HAND1, HAND1T107A or HAND1S109A were coexpressed with constitutively active PKA in HEK293 cells and subjected to phosphopeptide analysis. Mutation of T107 shows no significant difference in the HAND1 phosphopeptide map specifically peptides 8 and 9 (see Fig. 1). In contrast, mutation of S109 to alanine eliminates phosphopeptides 8 and 9 and recapitulates the data observed in the double mutant. This suggests that peptides 8 and 9 are partial tryptic digests and not mono- and diphosphorylated forms of the protein.
Taken together, the results obtained show that the upregulation of HAND1 phosphorylation that is observed during RCHOI differentiation (3) can be attributed to the modifications of three residues within HAND1. It should be considered that it is possible that other residues within HAND1 may in fact be phosphorylated, but our conditions of mapping are either insufficient to resolve 2 labeled peptides or the peptides run off of the TLC plate in the solvent phase. Employing different solvents for the chromatography used in the 2nd dimension can test this latter possibility. A useful and expedient way to test different solvent conditions is to use in silico mapping simulation programs such as Mobility, which can be found on the Gene Stream web site (http://www.genestream.org/). Based on your trypsin digestion pattern the program will show you which peptides will likely be present on the peptide map using a particular solvent.
Acknowledgments
We thank our coauthors of Molecular Cell 12:1225-1237 Marthe J. Howard, Jennifer R. McDaid, Leanne McIlreavey, Karen M. Dionne, Victoria E. Centonze and Peter Cserjesi. We also thank, Dan Hadzic, Brandon Smiley, YanLi Zhao and Kunal Patel for excellent technical assistance. This work was supported by grants from the National Institutes of Health (R01 HLA61677-04; 2RO1HL61677-05) and March of Dimes Birth Defects Foundation (ABF), and R01 CA80809 (DMV).
Appendix
Protocols
In vivo-labeling
Day -2: The cells, which are to be used for expression should be transfected with the desired DNA by a method that is most efficient for the employed cell line. For HEK293, we employed CaPO4 in 10 cm dishes that are 60% confluent. For RCHOI cells, we used lipofection with lipofectamine 2000 (Invitrogen) in 10 cm dishes following the manufacturer’s protocol.
Day -1: Define an area within the lab for the radioactive work. Using mCi amounts of 32P requires great care. You will need a lead shield, disposable lab coat, booties, eye protection, double gloves, a disposable liquid waste container and lead box to store waste until radiation safety can remove the used isotope from the lab. Although Plexiglas is generally the preferred material for shielding 32P, the thickness of Plexiglas required to protect against 25 to 30 mCi is greater than standard shielding thicknesses. As detailed in the Perkin Elmer technical data accompanying the H3 32PO4, lead is stated for use as shielding for both storage and when working with mCi amounts of isotope. This information is included with each order of 32P and we recommend following all indicated safety procedures outlined in the technical data certificate of analysis information.
Day 0: Aspirate media off transfected cells, wash 2 times with 1X PBS and replace with 3 ml phosphate free media supplemented with dialyzed serum at the percentage appropriate for the cell line employed. At this point, put on all protective gear, inform lab-mates that you are initiating the experiment, and add isotope to a concentration of 1 to 3 mCi/ml. NOTE! If you are doing this in a tissue culture hood turn of the laminar flow blower. The blower facilitates aerosolization of the isotope and can badly contaminate the hood. We add our isotope bench top behind the lead shield, as no microbe will overrun your sample within the incubation period. Place the cell plates in a diaper lined beta shield-plastic box, place the cells in a CO2-water-jacketed incubator, place a small lead shield in front of the box and let the cells grow for 4 hours. NOTE! Although the cells are in a beta shield box within an incubator, significant amounts of radiation may come through the incubator dependent on the sample size and amount of isotope used. Adding the lead shielding as mentioned above and keeping all personnel at a distance from the incubator for this incubation is highly recommended.
After 4 hours take the cells out of the incubator and place them behind the lead shield. Open the box and decant the media into your container. Wash cells 3 times with 10 ml of HEPES Buffer (20 mM HEPES pH7.4, 150 mM NaCl) again decanting the washes in the radioactive waste container. If you wish to use an aspirator set it up such that you have 2-containers in tandem and protect the house vacuum by adding liquid filters to the tubing. Scrape cells down in 800 mL of HEPES buffer, dispense cells in a 1.5 ml microcentrifuge, and spin at 6000 RPM in a microcentrifuge. Decant supernatant into waste container and repeat 2 times.
When cells are washed, resuspend cell pellets in 1 ml of IP buffer (20 mM NaPO4, 150 mM NaCl, 2 mM MgCl2, 0.1% NP40, 10% glycerol, 3 mg/ml leupeptin, 3 mg/ml pepstatin, 1mM PMSF, 50 mM NaVO4, 5 mM NaF, 100nM Okadaic acid, and 5mM Beta glycerol phosphate). Vortex and incubate on ice for 30 minutes, vortex and spin max speed for 20 minutes. Transfer supernatant to a fresh tube.
IP-analysis
Add 50 μl of anti-Flag conjugated agarose beads (FLAG M2- beads, Sigma) to cell lysates incubate for 2 hours at 4 oC rotating constantly.
Spin beads down at 5000 RPM for 5 minutes and aspirate off supernatant.
Wash 3 times with 1 ml of 1X PBS. On the last wash, transfer beads to a fresh tube.
Spin, remove wash buffer, resuspend beads in 80 μL of 1X loading buffer (20% glycerol, 2% SDS, 25 μg/ml bromophenol blue, 125 mM TRIS pH 6.8), boil 5 minutes, and load on an 8-12% SDS-PAGE with 0.75 mm spacers.
Gel purification
Lift gel onto Whatman 3 mm paper and dry for 2 hours at 80oC on a gel dryer. Take a dilution of old 32P, add loading dye, and mark the Whatman 3 mm paper with a pattern to align up the exposure with the gel. (We like to draw lines that correspond the size standards).
Expose for 2 hours on a phosphoimager or 4 hours on film.
Take print out of image (make sure it is actual size) and align the spots so that the gel and image are lined up. With razor blade, cut out the radioactive protein and place it a 1.5 ml eppendorf tube.
Rehydrate gel slice by adding 400 mL of 50 mM NH4HC03 incubate for 5 minutes, take slice out of tube and carefully peel off paper.
Mince slice up into small pieces, place pieces in a fresh siliconized eppendorf, add 400 mL of 50 mM NH4HC03 , and vortex for 1 minute.
Add 30 mL of TPCK-treated trypsin (1 mg/ml, Worthington) incubate overnight at 37 oC.
In the morning vortex, and spike with an additional 30 mL of trypsin. Incubate 4 hours. Note: depending on how basic your protein is you may want to incubate up to 8 hours.
Spin in microcentrifuge for 5 minutes at maximum speed, remove supernatant and put in a fresh siliconized eppendorf tube.
Wash acrylamide pieces twice with 300 mL of 50 mM NH4HC03, adding the washes to the new tube from step 8.
Spin total supernatant at maximum speed for 5 minutes and remove supernatant leaving behind any residual gel/paper using a P1000 equipped with a P-1000 tip and with a tip from a P2 on the end of the P1000 tip. Transfer to a fresh siliconized eppendorf tube.
Spin in a speed vac concentrator until dry.
Wash peptides 4 times with 1 ml of H2O. Speed vac dry after each wash.
Resuspend pellet in 1 ml of pH 1.9 buffer (see material and methods for recipe). Vortex 5 minutes, spin 5 minutes maximum speed, transfer solution to a fresh siliconized eppendorf tube, and speed vac dry.
Wash with 1 ml of pH 1.9 buffer and speed vac dry.
Count samples in scintillation counter via Cerenkov counting.
Spotting TLC and running first dimension
Resuspend the least radioactive pellet in 5 μl of pH 1.9 buffer. Resuspend the remaining samples such that all samples are at the equivalent CPM/μl the lowest sample.
Set up the HTLE 7000 as stated by the manufacturer's instructions. For a more detailed description of apparatus set up see (9).
Fill buffer chambers with 600 ml of pH 1.9 buffer.
Cut 2 plastic sheets at a dimension of 35 cm by 25 cm. Place one on the Teflon sheet, which lies on top of the cooling plate tucking the ends between the cooling plate and buffer tanks.
Cut 2 pieces of Whatman 3 mm paper 20 cm by 28 cm. Fold paper in half lengthwise. Wet paper in pH 1.9 buffer and insert the ends in the slots of the buffer tanks with the folded edge up. Fold the paper over the plastic sheet on top of the cooling plate.
Place the 2nd plastic sheet on top of the cooling plate covering the cooling plate, wicks and a part of the buffer tank.
Add the 2nd Teflon sheet and the neoprene pad. Close and lock the mapper with the safety pins.
Turn on air pressure (10 lb/inch) pushing excess buffer from the wicks.
When done with step 5, disconnect air, open safety latch and remove the neoprene pad, the 2nd Teflon sheet, and 2nd plastic sheet and load the plate.
Spot 5 μl of the sample onto a cellulose TLC plate, 1 μl at a time using a hair dryer to dry the spot completely before adding the next drop. (See Fig. 3 for orientation and where to spot your plate).
Fig. 3. Diagrams showing the layout of the TLC plate and blotter.
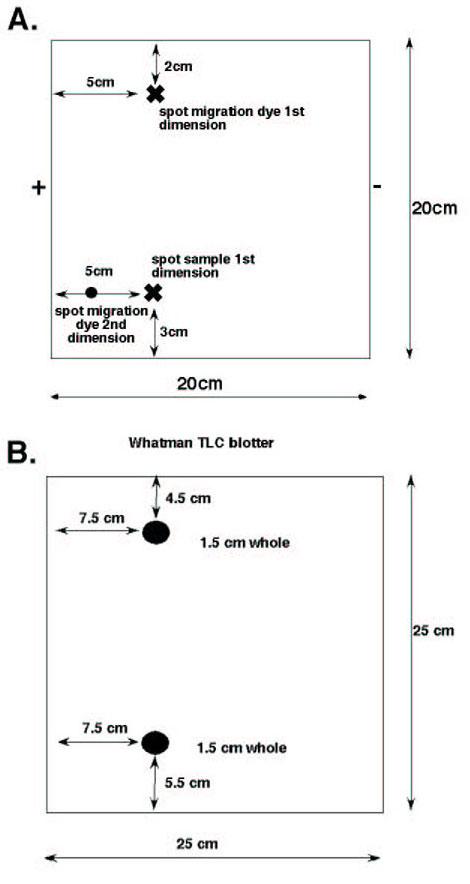
(A) TLC plate layout. X marks the location of where to spot the sample (lower) and where marker dye is applied (upper). Small circle shows location of spotting migration dye for 2nd dimension. + and – indicate the direction of current in relation to the sample for the first dimension. (B) Diagram of Whatman blotter. Dark circles represent 1.5 cm whole that are positioned over the sample and migration dye spots shown in A. Uniform wetting of the TLC plate is important for good peptide migration and consistent results. For exceptional detail on plate and phosphopeptide mapper set up please refer to (9).
Make a blotter for your TLC plate using 2 pieces of Whatman 3 mm paper held together by staples. Make 1.5 mm holes that correspond to the origin spots shown in Figure 3 (you can save and reuse these with proper care).
Wet the blotter in pH 1.9 buffer, wiping off excess liquid. Place blotter over TLC plate lining up holes with spots of origin. Press blotter onto plate around the sample with fingers. Plate should look dull with no shining or puddles of buffer visible on the plate.
Put plate on the phosphopeptide mapper and fold Whatman wick over the edge of the TLC (1 cm on each end) add back the 2nd plastic sheet, the 2nd Teflon sheet and the neoprene pad.
Secure the lid with safety clamps and turn on air and water. Turn on power supply and run 35 minutes at 1300 V. (I would recommend setting the power supply to run for a specific time so all runs are as close to the same as possible). By running electrophoresis of the first dimension for exactly the same amount of time, all maps generated with be essentially identical so changes can be readily observed.
Take plates out and dry. Rotate 90 degrees, and then run in the 2nd dimension using an isobutyric acid buffer (62.5% isobutyric acid, 1.9% n-Butanol, 4.8% pyridine, 2.9% glacial acetic acid in H2O) equilibrated for 24 hours in a TLC tank. When liquid phase migration is 1 cm from the top of the plates (an overnight run), remove plate, dry, and expose to a phosphoimager screen for visualization and analysis.
Equipment
CO2-waterjacketed incubator, tissue culture hood
Basic radiation safety equipment and Geiger counter,
Hunter Thin Layer Electrophoresis apparatus. (HTLE 7000, CBS Scientific, Inc.)
Glass TLC tank with plate stand.
Standard molecular biology equipment microcentrifuge, speed vacuum, concentrator, gel dryer.
References
- Firulli AB. A HANDful of questions: The molecular biology of the HAND-subclass of basic helix-loop-helix transcription factors. Gene. 2003;312C:27–40. doi: 10.1016/S0378-1119(03)00669-3. [DOI] [PubMed] [Google Scholar]
- Massari ME, Murre C. Helix-Loop-Helix Proteins: Regulators of Transcription in Eucaryotic Organisms. Molec Cell Biol. 2000;20(2):429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firulli BA, Howard MJ, McDaid JR, McIlreavey L, Dionne KM, Centonze VE, Cserjesi P, Virshup DM, Firulli BA. PKA, PKC and the Protein Phosphatase 2A Influence HAND factor dimerization: A Mechanisms for Tissue Specific Transcriptional Regulation. Mol Cell. 2003;12:1225–1237. doi: 10.1016/s1097-2765(03)00425-8. [DOI] [PubMed] [Google Scholar]
- Firulli BA, Hadzic DB, McDaid JR, Firulli AB. The basic helix-loop-helix transcription factors dHAND and eHAND exhibit dimerization characteristics that suggest complex regulation of function. J Biol Chem. 2000;275(43):33567–33573. doi: 10.1074/jbc.M005888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott IC, Anson-Cartwright L, Riley P, Reda D, Cross JC. The HAND1 basic helix-loop-helix transcription factor regulates trophobblast differentiation via multiple mechanisms. Molec Cell Biol. 2000;20(2):530–541. doi: 10.1128/mcb.20.2.530-541.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanon I, von Stetina S, Kass J, Baylies MK. Dimerization partners determine the activity of the Twist bHLH protein during Drosophila mesoderm development. Development. 2001;128:3145–3159. doi: 10.1242/dev.128.16.3145. [DOI] [PubMed] [Google Scholar]
- James G, Olson EN. Deletion of the regulatory domain of protein kinase C exposes regions in the hinge and catalytic domains that mediate nuclear targeting. J Cell Biol. 1992;116(4):863–874. doi: 10.1083/jcb.116.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firulli AB, Maibenco DC, Kinniburgh AJ. Triplex forming Ability of a c-myc Promoter element Predicts Promoter Strength. Arch Biochem Biophys. 1994;310(1):236–242. doi: 10.1006/abbi.1994.1162. [DOI] [PubMed] [Google Scholar]
- Van Der Geer P, Luo K, Sefton BM, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis on cellulose thin-layer-plates. 2nd ed. The practical approach series, ed. B.D. Hames. New York, NY: Oxford University Press; 1999, 97-126.