

A-type nuclear lamins, all encoded by the LMNA gene through alternative splicing, are targets for mutation in a wide range of rare diseases, including a dominant mutation that enhances production of lamin A splice variant progerin, causing Hutchinson-Gilford progeria syndrome (HGPS).1 Often ignored, however, is the complex and poorly understood connection between A-type lamins and cancer.2 Expression of the 2 main A-type lamins (Lamins A and C) vary in inconsistent patterns in a wide range of cancers.
More recently, enhanced progerin expression has been associated with prostate cancer development, an intriguing finding, since this splice variant of lamin A occurs at low frequency in normal cells and has been associated with aging.1 A new study by Jung et al. reports a role for progerin in modulating p53 activity in renal cell carcinomas (RCCs), and, moreover, that progerin expression is controlled by the von Hippel-Lindau tumor suppressor (VHL).3 This is particularly interesting, since RCCs, like many cancers, are highly age-associated, and increasing influence of progerin with age could, at least in part, explain this phenomenon.
Motivation for the study started with the puzzling observation that VHL inactivation is an early event in RCC tumor progression, inconsistent with its known role in promoting degradation of HIF-1α and thereby blocking the pro-angiogenic response to hypoxia.4 Putting together this observation with other clues, for instance that p53 declines functionally in RCCs generally without mutation, and that nuclear irregularities similar to those in HGPS (associated with progerin expression) are a common feature of RCCs, Jung et al. tested the hypothesis that VHL may regulate these pathways.3
Following these threads with extensive experimental studies, the authors found that RCCs lacking VHL have high progerin protein content in the absence of changes in LMNA mRNA levels, leading them to test and generate support for a model whereby the E3 ubiquitin ligase activity of VHL is required for progerin turnover.3 Excess progerin, in turn, binds to and sequesters p14/ARF, permitting p53 degradation through MDM2. Interestingly, this activity occurs independently of HIF-2α, another VHL regulated factor that can inhibit p53 function.5,6 Together, these findings connect VHL to the p53 pathway and ascribe to it additional tumor suppressor activities.
Cellular production of normal lamin A involves the farnesylation of a cysteine residue in the C terminus of the protein.1 However, unlike other farnesylated proteins, such as Ras, the lamin A farnesylation event is transient, with proteolytic cleavage prior to the cysteine removing the modification in the mature protein. Progerin, however, lacks this cleavage site and remains permanently farnesylated, a feature that contributes to progeroid pathologies. Farnesyl transferase inhibitors, developed to inhibit Ras farnesylation, have been tested widely in cancer with limited success. The study by Jung et al. suggests that progerin may be another meaningful target of farnesyl transferase inhibitors in a subset of cancers.3
HGPS cells are reported to have both elevated DNA damage and an abnormal DNA damage response.1 However, the connection between progerin and p53 has not been made previously. This may be related to the nature of the cell being interrogated. For instance, primary fibroblasts engineered to express progerin have proliferation defects that can be suppressed through inhibition of p53 function by expression of HPV E6.7 Moreover, the progeroid phenotypes of a mouse HGPS model can be partially rescued by deletion of p53.8 The implication is that p53 remains at least partially active in these settings, even though progerin is expressed. One possibility is that the ability of progerin to interact with p14/ARF and suppress p53 depends on the immortalization status of the cells. Primary cells expressing progerin would maintain p53 inducibility, but not a subset of tumor cells, including RCCs. Consistently, Jung et al. identified a leukemia cell line where progerin expression was elevated, leading to inhibition of p53.3 However, they were also able to detect enhanced p53 expression in HGPS fibroblasts when progerin expression was reduced by siRNA, indicating that this regulatory pathway is at least partially intact. More studies will be needed to understand the complex interplay between progerin and p53 pathway components.
Aging is the biggest risk factor for most forms of cancer, yet the molecular reasons underlying this phenomenon have yet to be ascertained. What changes in the aging organism enable cancer progression? While several possibilities, including dysregulated gene expression programs and enhanced inflammation, are likely factors, the study by Jung et al. raises a new possibility that progerin can interfere directly with normal events that mediate repair of DNA lesions.3 Further studies will be important to explore this intriguing possibility. (Fig. 1)
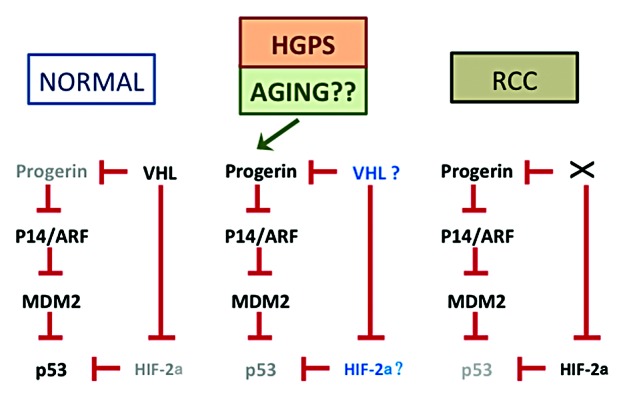
Figure 1. Model for interactions between progerin and p53 pathway. This diagram, reproduced and enhanced from Jung et al.,3 demonstrates the proposed interactions between progerin and p53 in different settings. In normal cells (left), low progerin activity permits a normal p53 response pathway. In HGPS cells (middle), high progerin expression leads to reduced p53 levels, although evidence exists that p53 can still be active in the presence of progerin in this context. Aging may also lead to enhanced progerin levels or activity, with similar consequences for p53. The fates of VHL and HIF-2a require further exploration. In renal cell carcinomas (right), loss of VHL leads to enhanced progerin and HIF-2a levels, both of which inhibit p53 through apparently independent pathways.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26158
References
- 1.Schreiber KH, et al. Cell. 2013;152:1365–75. doi: 10.1016/j.cell.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chow KH, et al. Nat Rev Cancer. 2012;12:196–209. doi: 10.1038/nrc3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jung YS, et al. Cell Cycle. 2013;12 doi: 10.4161/cc.25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majmundar AJ, et al. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts AM, et al. Cancer Res. 2009;69:9056–64. doi: 10.1158/0008-5472.CAN-09-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertout JA, et al. Proc Natl Acad Sci U S A. 2009;106:14391–6. doi: 10.1073/pnas.0907357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kudlow BA, et al. Mol Biol Cell. 2008;19:5238–48. doi: 10.1091/mbc.E08-05-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varela I, et al. Nature. 2005;437:564–8. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]