

Abstract
Metabolic coupling, between mitochondria in cancer cells and catabolism in stromal fibroblasts, promotes tumor growth, recurrence, metastasis, and predicts anticancer drug resistance. Catabolic fibroblasts donate the necessary fuels (such as L-lactate, ketones, glutamine, other amino acids, and fatty acids) to anabolic cancer cells, to metabolize via their TCA cycle and oxidative phosphorylation (OXPHOS). This provides a simple mechanism by which metabolic energy and biomass are transferred from the host microenvironment to cancer cells. Recently, we showed that catabolic metabolism and “glycolytic reprogramming” in the tumor microenvironment are orchestrated by oncogene activation and inflammation, which originates in epithelial cancer cells. Oncogenes drive the onset of the cancer-associated fibroblast phenotype in adjacent normal fibroblasts via paracrine oxidative stress. This oncogene-induced transition to malignancy is “mirrored” by a loss of caveolin-1 (Cav-1) and an increase in MCT4 in adjacent stromal fibroblasts, functionally reflecting catabolic metabolism in the tumor microenvironment. Virtually identical findings were obtained using BRCA1-deficient breast and ovarian cancer cells. Thus, oncogene activation (RAS, NFkB, TGF-β) and/or tumor suppressor loss (BRCA1) have similar functional effects on adjacent stromal fibroblasts, initiating “metabolic symbiosis” and the cancer-associated fibroblast phenotype. New therapeutic strategies that metabolically uncouple oxidative cancer cells from their glycolytic stroma or modulate oxidative stress could be used to target this lethal subtype of cancers. Targeting “fibroblast addiction” in primary and metastatic tumor cells may expose a critical Achilles’ heel, leading to disease regression in both sporadic and familial cancers.
Keywords: oncogene, tumor suppressor, RAS, NFkB, TGF-beta, BRCA1, oxidative stress, glycolysis, cancer-associated fibroblast, tumor microenvironment, stromal biomarkers, metabolic symbiosis
Introduction
It is now well-established that (1) oncogene activation, and (2) aberrant growth factor signaling, are sufficient to mediate cell transformation, tumor growth, and metastasis. However, this highly simplified view of cancer does not mechanistically explain the essential role of the tumor stroma or the host cellular microenvironment.
Thus, a more integrated metabolic model of how cancer works will be required for us to invent new effective and non-toxic cancer therapies.1–11 Toward this end, here we will focus on the metabolic-interface between cancer cells and their host microenvironment: cancer-associated fibroblasts or CAFs. We believe that catabolic cancer-associated fibroblasts are a key metabolic “fuel source”, for enabling cancer cell propagation, survival, and systemic dissemination, during metastasis.
Metabolic Symbiosis: Implications for Personalized Medicine and Cancer Therapy
Recent studies directly show that cancer cells extract high-energy nutrients from cancer-associated fibroblasts or other normal adjacent cells, such as adipocytes, via oxidative stress.12-54 This “metabolic symbiosis” would be most essential during early tumorigenesis and later during cancer cell metastasis, when a blood supply is largely absent. We also have termed this type of symbiotic or parasitic relationship “two-compartment tumor metabolism” or “the reverse Warburg effect”.31,33,40,42-45 During metabolic symbiosis, catabolic fibroblasts provide the necessary fuels (such as L-lactate, ketone bodies, glutamine, other amino acids, and free fatty acids) to anabolic cancer cells, to metabolize via their TCA cycle and oxidative phosphorylation (OXPHOS). Energy transfer occurs via a stromal–epithelial “lactate shuttle” to efficiently move nutrients from fibroblasts to tumor cells. To accomplish this energy transfer, fibroblasts excrete L-lactate and ketones using MCT4 transporters.55,56 In contrast, tumor cells take up and re-purpose these fuels using MCT1 transporters.
Here, we discuss the use of 3 new clinical biomarkers of metabolic symbiosis for predicting patient outcome in human breast cancers and other types of cancer. These 3 biomarkers include MCT1, MCT4, and Caveolin-1. Moreover, we highlight recent advances showing that metabolic symbiosis can be modeled in vitro using a co-culture system. By employing this approach, it becomes apparent that oncogenes drive the onset of metabolic symbiosis by re-programming energy metabolism in the tumor microenvironment, via paracrine oxidative stress.
These findings have important implications for achieving the goals of personalized medicine and for designing more effective approaches to anticancer therapy, with a focus on new, highly selective metabolic inhibitors.
Stromal Caveolin-1: A Biosensor of Autophagy in the Tumor Microenvironment
An absence or loss of stromal caveolin-1 (Cav-1) immunostaining is a new biomarker of poor clinical outcome in many different types of cancers, such as breast tumors and DCIS, gastric and prostate carcinomas, as well as in metastatic melanoma lesions.57-68 In breast cancer patients, a loss or stromal Cav-1 is specifically linked to tumor recurrence, metastasis, drug-resistance, and overall poor survival (Fig. 1). More specifically, oxidative stress in cancer-associated fibroblasts leads to the lysosomal/autophagic digestion of Cav-1.30 As such, a reduction or absence of Cav-1 is a sensitive biomarker of oxidative stress, autophagy/mitophagy, and aerobic glycolysis, in the tumor stroma.22,34

Figure 1. Kaplan–Meier analysis of overall survival, using stromal Cav-1 and mct4 to predict clinical outcome. (A) TMA containing a cohort of 185 triple-negative breast cancer patients, with over 20 y of clinical follow-up data, was subjected to immunostaining with antibodies directed against Cav-1 and MCT4. Then, the expression levels of these two protein biomarkers were scored in the tumor stroma. Note that loss of Cav-1 and overexpression of MCT4 are strictly associated with poor clinical outcome. In contrast, patients with high stromal Cav-1 and absent MCT4 show >90% survival at >20 years post-diagnosis. Reproduced, with permission, from reference 70.
Importantly, the prognostic value of stromal Cav-1 in breast cancer patients has now been independently validated in 7 different countries worldwide.57-68
Stromal MCT4: A Biosensor of Oxidative Stress, Glycolysis, and Mitochondrial Dysfunction
Similar to what we have previously observed for Cav-1, MCT4 is new marker of oxidative stress in tumor-associated fibroblasts.55,56 For example, in head and neck cancers, MCT4 is a specific marker of cancer-associated fibroblasts, but it does not label normal fibroblasts.69 Furthermore, in triple-negative breast cancers, stromal MCT4 expression is a strong predictor of poor clinical outcome.70 In this patient cohort, loss of stromal Cav-1 was directly correlated with elevated levels of stromal MCT4 expression and poor survival in triple-negative breast cancer (Fig. 1).70
Functionally, MCT4 mediates the cellular export of L-lactate and ketones from glycolytic cells. Interestingly, MCT4 is highly upregulated under conditions of hypoxia and/or oxidative stress and is a HIF1-α target gene. Thus, MCT4 functions as a new biomarker of oxidative stress, glycolysis, and mitochondrial dysfunction in the tumor microenvironment.
Epithelial MCT1: A Marker of Cell Proliferation, Mitochondrial Power, and Stemness
Previously, we have presented experimental evidence that a “lactate shuttle” can also exist in human cancer tissues.45,55,56,69 Under these conditions, tumor-associated stromal cells express MCT4 and release high-energy mitochondrial fuels (such as L-lactate and ketones) into the tumor stroma. In contrast, tumor cells upregulate MCT1, so they can efficiently import these mito-fuels to use as fuel in the TCA cycle and for OXPHOS in proliferative cancer cells. This form of metabolic-coupling may then allow cancer cells to successfully engraft in pre-clinical animal models, by the metabolic rewiring of their microenvironment.
MCT1 is also a new biomarker of mitochondrial activity and mass, and its expression levels correlate with high proliferation rates in vivo, as we have recently demonstrated.69 More specifically, Ki-67 expression and MCT1 staining are directly linked in both normal mucosa, and head and neck cancer tissues. Also, Ki-67 and MCT1 are both well expressed in the basal stem cell compartment of normal mucosa, suggesting that MCT1 may be a new marker of “stemness” in epithelial cells.69
Exploring the Transition to Malignancy: Modeling Metabolic Symbiosis by Co-Culturing Normal and Oncogene-Transformed Epithelial Cells with Fibroblasts
Recently, we employed a new cell system, to study the potential metabolic consequence of oncogenic stress on the tumor microenvironment.71 For this purpose, we used the HaCaT keratinocyte cell system, which consists of an isogenic set of “normal” and oncogenically transformed epithelial cell lines. In this context, the behavior of normal HaCaT cells was directly compared with transformed HaCaT cells, which harbor the overexpression of activated oncogenes, such as H-Ras (G12V) or NFkB (p65 subunit).
Briefly, normal and transformed HaCaT cells were co-cultured with immortalized human fibroblasts to determine the systemic effects of oncogenes on adjacent stromal cells. Metabolic parameters, such as ROS production/oxidative stress and glucose utilization, were quantitated using sensitive fluorescent probes. As a complementary approach, we also followed the expression of a panel of protein biomarkers, which reflect the onset of metabolic symbiosis (Cav-1, MCT4, and MCT1).71
Surprisingly, epithelial oncogene activation was indeed sufficient to trigger the cancer-associated fibroblasts phenotype, with a loss of stromal Cav-1 and the upregulation of stromal MCT4 expression (Fig. 2). These fibroblasts also showed increased ROS production and elevated glucose uptake, indicative of a shift toward glycolytic metabolism. Thus, we concluded that oncogenes drive the metabolic reprogramming of cancer-associated fibroblasts via oxidative stress71 In accordance with this notion, treatment with N-acetyl-cysteine (NAC), a powerful antioxidant, was sufficient to reverse or prevent the cancer-associated fibroblast phenotype induced by activated oncogenes.

Figure 2. Epithelial oncogenes induce the cancer-associated fibroblast phenotype, in adjacent normal fibroblasts. Cav-1 and MCT4 as stromal biomarkers. (A) HaCaT cells (control [CTRL] vs. Ras-transformed [RAS]) were co-cultured with stromal fibroblasts. Then, the expression of Cav-1 and MCT4 was monitored by immunostaining. Note that Ras-transformed HaCaT cells specifically downregulate Cav-1 and upregulate MCT4 in adjacent fibroblasts, effectively functioning as reporters of the transition to malignancy in cancer cells. E, epithelia. (B) HaCaT cells (CTRL and RAS) were also cultured alone for comparison. Note that MCT4 is upregulated in Ras-transformed cells when they are cultured alone, but then downregulated when they are co-cultured with fibroblasts (as in A). Reproduced and modified, with permission, from reference 71.
Most importantly, these changes in metabolic parameters and protein biomarkers were not induced by normal HaCaT cells. These findings provide the necessary proof-of-concept that stromal Cav-1 and MCT4 can be used as sensitive “biosensors”, to monitor the transition to malignancy, both in vitro and in vivo.71
In addition, under these conditions, MCT1 was selectively upregulated in epithelial cancer cells, during co-culture with fibroblasts (Fig. 3). As such, it appears that oncogenes drive the establishment of a “lactate shuttle” and metabolic symbiosis, to enable the anabolic growth of tumor cells.

Figure 3. Fibroblasts induce the expression of MCT1 in epithelial cancer cells to facilitate metabolic symbiosis. HaCaT–Ras cells were cultured alone (left) or co-cultured with fibroblasts (right) and then subjected to immunostaining with antibodies directed against MCT1. Note that co-culture with fibroblasts induces the expression of MCT1 in Ras-transformed epithelial cells. Thus, co-culture of Ras-transformed cells with fibroblasts induces reciprocal metabolic reprogramming, leading to metabolic symbiosis. Reproduced and modified, with permission, from reference 71.
Virtually identical results were obtained with 2 distinct onco-proteins (RAS and NFkB), directly showing that oxidative stress and inflammation converge on the stromal compartment, leading to glycolytic metabolism in the tumor microenvironment (Fig. 4).

Figure 4. Oncogenes drive oxidative stress and glycolysis in the tumor microenvironment. Summary illustrating that 2 divergent oncogenes (RAS and NFkB) use ROS production and cytokines (inflammation) to induce oxidative stress and glycolysis in adjacent cancer-associated fibroblasts (“the reverse Warburg effect”). Thus, oncogenes act at a distance, to metabolically reprogram the tumor microenvironment.
As a consequence of these findings, we should begin to consider stromal Cav-1 and MCT4 as new metabolic targets for drug development, as they are very specific biomarkers of the catabolic tumor-associated fibroblast phenotype.71 Importantly, many diverse oncogenic stimuli (RAS, NFkB, TGF-β, loss of BRCA1, ethanol exposure, and ROS/hydrogen peroxide) all induce a common metabolic response in the tumor stroma,20,21,56,72,73 which then dramatically changes the expression levels of both Cav-1 and MCT4 (Fig. 5). These studies directly support the “seed and soil” hypothesis, which was initially proposed over 100 years ago now by Stephen Paget.74-77
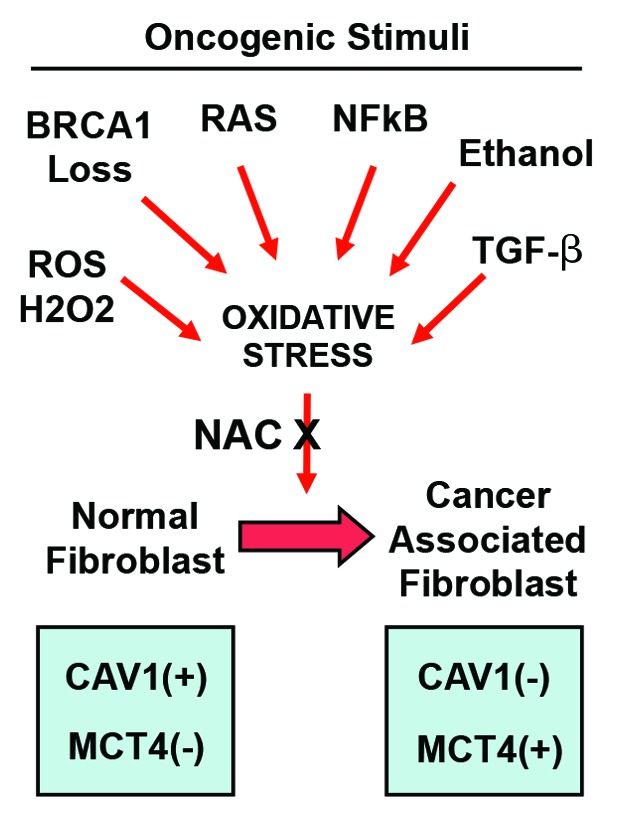
Figure 5. Diverse oncogenic stimuli induce the cancer-associated fibroblast phenotype via oxidative stress. Diagram illustrating that diverse oncogenic stimuli (RAS, NFkB, TGF-β, BRCA1 loss, ethanol exposure, ROS/hydrogen peroxide) all induce oxidative stress in the tumor microenvironment. This, in turn, promotes the catabolic cancer-associated fibroblast phenotype, resulting in a loss of Cav-1 and an increase in MCT4 expression. Treatment with N-acetyl-cysteine (NAC), a powerful antioxidant, is sufficient to reverse or prevent the cancer-associated fibroblast phenotype induced by these divergent oncogenic stressors.
BRCA1 Loss and the Transition to Malignancy: Modeling Metabolic symbiosis in Hereditary Breast and Ovarian Cancers
BRCA1 is a tumor suppressor gene which has several key functions, such as DNA repair and transcriptional regulation, as well as the modulation of cell cycle progression.78 Mutations in the BRCA1 gene dramatically increases a woman’s risk for the development of both breast and ovarian cancers. More specifically, women carrying a BRCA1 mutant allele have a >60% lifetime risk of breast cancer and a >40% lifetime risk of ovarian cancer.79,80 BRCA1 mutations prevent protein production,81 and BRCA1-mutated breast cancers show an absence of nuclear expression, which is associated with a poor prognosis.82,83 Phenotypically, BRCA1-deficient breast cancers are usually high-grade, aggressive and triple-negative, based on gene expression studies and/or immunostaining of paraffin sections.84-86 BRCA1 loss of expression also occurs commonly in sporadic breast cancers, secondary to epigenetic changes, and is often observed in triple-negative or basal-like tumors.80,87-94 As a consequence, loss of BRCA1 protein function may also be a critical driver in both sporadic and familial breast cancers.95,96
Recently, we studied the metabolic effects of BRCA1 loss of function on the tumor microenvironment.20 To recapitulate the tumor stroma, we developed a co-culture model in vitro. Briefly, HCC.1937 breast cancer cells, which harbor mutations that completely inactivate BRCA1 protein expression (BRCA1-null), were co-cultured with hTERT-immortalized stromal fibroblasts.20
Interestingly, HCC.1937 cells drive ROS production in adjacent fibroblasts, and induce the cancer-associated fibroblast phenotype, with the stromal loss of Cav-1 and MCT4 upregulation.20 Remarkably, this CAF phenotype was suppressed either by genetic replacement of the BRCA1 gene in epithelial cancer cells, or by treatment with powerful antioxidants, such as NAC (Fig. 6A).

Figure 6. BRCA1-deficient breast cancer cells induce the cancer-associated fibroblast phenotype. (A) BRCA1-deficient HCC cells were co-cultured with hTERT-immortalized fibroblasts. Then, expression of Cav-1 and MCT4 was monitored by immunostaining. Note that HCC cells drive a loss of stromal Cav-1 and the induction of stromal MCT4. Importantly, this CAF-phenotype was suppressed either by genetic replacement of the BRCA1 gene in the epithelial cancer cells, or by treatment with NAC, a powerful antioxidant. (B) Human breast cancer samples harboring BRCA1 mutations (9 out of 10 examined) also show a loss of stromal Cav-1 and the upregulation of MCT4, as well as strong mitochondrial staining with TOMM20, a marker of mitochondrial mass. For Cav-1 immunostaining, arrowheads point at blood vessels, which do not show a loss of Cav-1, in contrast to adjacent stromal fibroblasts. Images from one representative patient are shown. Reproduced and modified, with permission, from reference 20.
Human breast cancer samples harboring BRCA1 mutations (9 out of 10 examined) also showed a loss of stromal Cav-1 and the upregulation of MCT4, as well as striking mitochondrial staining with TOMM20, a marker of mitochondrial mass (Fig. 6B). Therefore, BRCA1 loss of function drives ROS generation in both mammary epithelial cells and adjacent stromal fibroblasts. This paracrine oxidative stress then promotes the onset of a glycolytic stromal phenotype with metabolic symbiosis, which is reflected by decreased Cav-1 and increased MCT4 expression.20
Quantitatively similar results were also obtained with the BRCA1-deficient ovarian cancer cell line, namely UWB1.289 cells.21 These BRCA1-null ovarian cancer cells produced large amounts of ROS. This, in turn, induced oxidative stress and catabolic metabolism in neighboring stromal fibroblasts (glycolysis, autophagy, and mitophagy).21 These functional changes reflected the onset of the CAF-phenotype and were strictly associated with MCT4 upregulation and Cav-1 loss of expression (Fig. 7A and B), which are markers of “metabolic symbiosis” within the tumor microenvironment. In these UWB fibroblast co-cultures, the CAF phenotype was suppressed by either genetic replacement of the BRCA1 gene in ovarian cancer cells, or by treatment with powerful antioxidants, such as NAC.21

Figure 7. BRCA1-deficient ovarian cancer cells induce the cancer-associated fibroblast phenotype. (A) BRCA1-deficient UWB cells were co-cultured with hTERT-immortalized fibroblasts. Then, the expression of Cav-1 and MCT4 was monitored by immunostaining. Note that UWB cells drive a loss of stromal Cav-1 and the induction of stromal MCT4. (B) Importantly, this CAF-phenotype was suppressed by treatment with NAC, a powerful antioxidant. Genetic replacement of the BRCA1 gene in the epithelial cancer cells also suppressed the CAF-phenotype (not shown). Reproduced and modified, with permission, from reference 21.
ROS production in BRCA1-null ovarian cancer cells also resulted in the paracrine induction of an inflammatory phenotype in adjacent cancer-associated fibroblasts, with the strong activation of an NFkB–luciferase reporter (Fig. 8).21 ROS production and oxidative stress are known activators of NFkB, consistent with the idea that redox signaling promotes an inflammatory phenotype, via the activation of innate immunity.

Figure 8. BRCA1-deficient ovarian cancer cells induce an inflammatory phenotype in adjacent stromal fibroblasts. BRCA1-deficient UWB cells were co-cultured with NIH-3T3 fibroblasts, harboring an NFkB–luciferase reporter, to measure the activation of an inflammatory phenotype. Note that co-culture with BRCA1-deficient UWB cells activates NFkB-mediated gene transcription, in adjacent stromal fibroblasts. Furthermore, this pro-inflammatory phenotype was rescued by recombinant expression of the wild-type BRCA1 gene in UWB cells. The basal state of NFkB activation in NIH-3T3 fibroblasts cultured alone is shown for comparison. Day 0 actually represents 24 h of co-culture. Reproduced and modified, with permission, from reference 21.
Taken together, these functional studies suggest that cancer prevention trials, with antioxidants and/or anti-inflammatories, may be warranted in women with a genetic history of familial BRCA1 mutations.20,21 This novel chemo-prevention approach would be expected to inhibit “metabolic symbiosis” in high-risk BRCA1 patients and reverse the cancer-associated fibroblast phenotype, essentially cutting off the “fuel supply” to cancer cells during tumor initiation.
“Fibroblast Addiction”: A New Therapeutic Target for Cancer Therapy and the Prevention of Drug Resistance
The desmoplastic tissue reaction, or fibroblast-overgrowth, is a generalized response to injury, but can also occur during tumor initiation and metastasis. As such, the desmopastic reaction is a true wound-healing response. However, the exact role of desmoplasia or tissue fibrosis in tumor initiation and progression still remains unknown.
Our recent data suggest that the desmoplastic reaction could actually help allow cancer cells to survive during oncogene activation and to overcome the cellular stress associated with malignant transformation. For example, we and others recently showed that catabolic fibroblasts inhibit oxidative stress in epithelial cancer cells.71,97 During metabolic symbiosis, cysteine (Cys) produced in stromal cells is transferred to cancer cells, where it is converted to glutathione (gamma-Glu-Cys-Gly), a powerful antioxidant.97 This, in turn, functionally protects cancer cells against oncogenic stress,71 which would otherwise induce cell death, via various stress-related mechanism(s), including apoptosis, autophagy, and/or senescence (Fig. 9). Previous studies have also shown that this mechanism may confer drug resistance to anti-estrogens and other chemotherapeutic agents, in epithelial cancer cells.23,24,31,33

Figure 9. Fibroblast addiction: Fibroblasts alleviate oncogenic stress in epithelial cancer cells. Catabolic fibroblasts provide high-energy fuels and precursors for biomass to adjacent cancer cells. As a consequence, fibroblasts protect cancer cells against stress by reducing ROS production, apoptosis, autophagy, and senescence in epithelial cancer cells. Experimental evidence indicates that this “fibroblast addiction” also confers drug resistance to anti-estrogens and other chemo-therapeutic agents by promoting oxidative mitochondrial metabolism in epithelial tumor cells.
These findings suggest that primary cancer cells are “addicted to fibroblasts” for their survival, and that separating them from stromal fibroblasts, may actually catalyze the death of cancer cells. These data could help explain why it is so difficult to generate new “immortal” cancer cell lines from patient samples, as one of the first steps involves the purification of epithelial cancer cells away from stromal fibroblasts.98
Thus, new therapies could be developed to functionally target “fibroblast addiction”. If stromal cells are therapeutically targeted, then an expected result would be acute oncogene-induced stress in epithelial tumor cells, driving the starvation and/or death of cancer cells. This acute stress would likely result in tumor regression.
Conclusions and Future Directions
In summary, we conclude that oncogene activation (RAS, NFkB, TGF-β) and/or tumor suppressor loss (BRCA1) both have very similar functional effects on adjacent stromal fibroblasts, driving the initiation of metabolic symbiosis and the cancer-associated fibroblast phenotype. As such, new therapeutic strategies that metabolically uncouple oxidative cancer cells from their glycolytic stroma, could be used to target this “fibroblast addiction” in both primary and metastatic tumor cells.
Acknowledgments
Dr Ubaldo E. Martinez-Outschoorn was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. Dr Lisanti's and Dr Sotgia’s current affiliation is the University of Manchester (United Kingdom), where they receive funding from the Manchester Cancer Research Centre (MCRC), Breakthrough Breast Cancer (BBC), and The European Research Council (ERC). Drs Michael P Lisanti and Federica Sotgia were also previously supported by the resources of Thomas Jefferson University in Philadelphia, USA. This review is dedicated to the memory of Professor Alton Meister (1922–1995), who taught Dr Lisanti the subject of Biochemistry, as a young MD-PhD student at Cornell University Medical College. Dr Meister made many pioneering contributions to our current understanding of oxidative stress, redox signaling, and glutathione metabolism. For a synopsis of his work, please see: http://www.jbc.org/content/282/38/e30.full?etoc
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25695
References
- 1.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–98. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–73. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 3.Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med. 2013;19:428–46. doi: 10.1016/j.molmed.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–27. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 6.Bissell MJ, Labarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7:17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kenny PA, Lee GY, Bissell MJ. Targeting the tumor microenvironment. Front Biosci. 2007;12:3468–74. doi: 10.2741/2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kenny PA, Nelson CM, Bissell MJ. The Ecology of Tumors: By perturbing the microenvironment, wounds and infection may be key to tumor development. Scientist. 2006;20:30. [PMC free article] [PubMed] [Google Scholar]
- 9.Rønnov-Jessen L, Bissell MJ. Breast cancer by proxy: can the microenvironment be both the cause and consequence? Trends Mol Med. 2009;15:5–13. doi: 10.1016/j.molmed.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Brauer HA, Makowski L, Hoadley KA, Casbas-Hernandez P, Lang LJ, Romàn-Pèrez E, et al. Impact of tumor microenvironment and epithelial phenotypes on metabolism in breast cancer. Clin Cancer Res. 2013;19:571–85. doi: 10.1158/1078-0432.CCR-12-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72:5130–40. doi: 10.1158/0008-5472.CAN-12-1949. [DOI] [PubMed] [Google Scholar]
- 15.Fordyce CA, Patten KT, Fessenden TB, Defilippis R, Hwang ES, Zhao J, et al. Cell-extrinsic consequences of epithelial stress: activation of protumorigenic tissue phenotypes. Breast Cancer Res. 2012;14:R155. doi: 10.1186/bcr3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaudhri VK, Salzler GG, Dick SA, Buckman MS, Sordella R, Karoly ED, et al. Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol Cancer Res. 2013;11:579–92. doi: 10.1158/1541-7786.MCR-12-0437-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther. 2010;10:537–42. doi: 10.4161/cbt.10.6.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lisanti MP, Martinez-Outschoorn UE, Lin Z, Pavlides S, Whitaker-Menezes D, Pestell RG, et al. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: the seed and soil also needs “fertilizer”. Cell Cycle. 2011;10:2440–9. doi: 10.4161/cc.10.15.16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, et al. Accelerated aging in the tumor microenvironment: connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle. 2011;10:2059–63. doi: 10.4161/cc.10.13.16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Outschoorn UE, Balliet R, Lin Z, Whitaker-Menezes D, Birbe RC, Bombonati A, et al. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies. Cell Cycle. 2012;11:4402–13. doi: 10.4161/cc.22776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Outschoorn UE, Balliet RM, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, et al. Hereditary ovarian cancer and two-compartment tumor metabolism: epithelial loss of BRCA1 induces hydrogen peroxide production, driving oxidative stress and NFκB activation in the tumor stroma. Cell Cycle. 2012;11:4152–66. doi: 10.4161/cc.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–76. doi: 10.4161/cc.9.16.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Outschoorn UE, Goldberg A, Lin Z, Ko YH, Flomenberg N, Wang C, et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol Ther. 2011;12:924–38. doi: 10.4161/cbt.12.10.17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, et al. Understanding the metabolic basis of drug resistance: therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle. 2011;10:2521–8. doi: 10.4161/cc.10.15.16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. 2011;10:2504–20. doi: 10.4161/cc.10.15.16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Lisanti MP, Sotgia F. Ketone bodies and two-compartment tumor metabolism: stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle. 2012;11:3956–63. doi: 10.4161/cc.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Ketone body utilization drives tumor growth and metastasis. Cell Cycle. 2012;11:3964–71. doi: 10.4161/cc.22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43:1045–51. doi: 10.1016/j.biocel.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Outschoorn UE, Pavlides S, Sotgia F, Lisanti MP. Mitochondrial biogenesis drives tumor cell proliferation. Am J Pathol. 2011;178:1949–52. doi: 10.1016/j.ajpath.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–33. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 31.Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, et al. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle. 2011;10:4208–16. doi: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness”, driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011;10:1271–86. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012;15:4–5. doi: 10.1016/j.cmet.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515–33. doi: 10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle. 2011;10:1784–93. doi: 10.4161/cc.10.11.15674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle. 2010;9:4297–306. doi: 10.4161/cc.9.21.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485–505. doi: 10.4161/cc.9.17.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle. 2010;9:2201–19. doi: 10.4161/cc.9.11.11848. [DOI] [PubMed] [Google Scholar]
- 39.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY) 2010;2:185–99. doi: 10.18632/aging.100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. 2012;16:1264–84. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 42.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 43.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Mitochondrial oxidative stress drives tumor progression and metastasis: should we use antioxidants as a key component of cancer treatment and prevention? BMC Med. 2011;9:62. doi: 10.1186/1741-7015-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011;13:213. doi: 10.1186/bcr2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, et al. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012;11:1445–54. doi: 10.4161/cc.19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiavarina B, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Tanowitz HB, Pestell RG, et al. Metabolic reprogramming and two-compartment tumor metabolism: opposing role(s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle. 2012;11:3280–9. doi: 10.4161/cc.21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiavarina B, Whitaker-Menezes D, Martinez-Outschoorn UE, Witkiewicz AK, Birbe RC, Howell A, et al. Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth. Cancer Biol Ther. 2011;12:1101–13. doi: 10.4161/cbt.12.12.18703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9:3534–51. doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. 2012;11:3599–610. doi: 10.4161/cc.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012;11:2285–302. doi: 10.4161/cc.20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 2012;11:2272–84. doi: 10.4161/cc.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carito V, Bonuccelli G, Martinez-Outschoorn UE, Whitaker-Menezes D, Caroleo MC, Cione E, et al. Metabolic remodeling of the tumor microenvironment: migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle. 2012;11:3403–14. doi: 10.4161/cc.21701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: evidence for stromal-epithelial metabolic coupling. Cell Cycle. 2010;9:2412–22. doi: 10.4161/cc.9.12.11989. [DOI] [PubMed] [Google Scholar]
- 54.Avena P, Anselmo W, Whitaker-Menezes D, Wang C, Pestell RG, Lamb RS, et al. Compartment-specific activation of PPARγ governs breast cancer tumor growth, via metabolic reprogramming and symbiosis. Cell Cycle. 2013;12:1360–70. doi: 10.4161/cc.24289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10:4047–64. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–83. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF, et al. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther. 2009;8:1071–9. doi: 10.4161/cbt.8.11.8874. [DOI] [PubMed] [Google Scholar]
- 58.Witkiewicz AK, Dasgupta A, Sammons S, Er O, Potoczek MB, Guiles F, et al. Loss of stromal caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol Ther. 2010;10:135–43. doi: 10.4161/cbt.10.2.11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–34. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Witkiewicz AK, Kline J, Queenan M, Brody JR, Tsirigos A, Bilal E, et al. Molecular profiling of a lethal tumor microenvironment, as defined by stromal caveolin-1 status in breast cancers. Cell Cycle. 2011;10:1794–809. doi: 10.4161/cc.10.11.15675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu KN, Queenan M, Brody JR, Potoczek M, Sotgia F, Lisanti MP, et al. Loss of stromal caveolin-1 expression in malignant melanoma metastases predicts poor survival. Cell Cycle. 2011;10:4250–5. doi: 10.4161/cc.10.24.18551. [DOI] [PubMed] [Google Scholar]
- 62.Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle. 2009;8:2420–4. doi: 10.4161/cc.8.15.9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koo JS, Park S, Kim SI, Lee S, Park BW. The impact of caveolin protein expression in tumor stroma on prognosis of breast cancer. Tumour Biol. 2011;32:787–99. doi: 10.1007/s13277-011-0181-6. [DOI] [PubMed] [Google Scholar]
- 64.Qian N, Ueno T, Kawaguchi-Sakita N, Kawashima M, Yoshida N, Mikami Y, et al. Prognostic significance of tumor/stromal caveolin-1 expression in breast cancer patients. Cancer Sci. 2011;102:1590–6. doi: 10.1111/j.1349-7006.2011.01985.x. [DOI] [PubMed] [Google Scholar]
- 65.Simpkins SA, Hanby AM, Holliday DL, Speirs V. Clinical and functional significance of loss of caveolin-1 expression in breast cancer-associated fibroblasts. J Pathol. 2012;227:490–8. doi: 10.1002/path.4034. [DOI] [PubMed] [Google Scholar]
- 66.Sloan EK, Ciocca DR, Pouliot N, Natoli A, Restall C, Henderson MA, et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol. 2009;174:2035–43. doi: 10.2353/ajpath.2009.080924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.El-Gendi SM, Mostafa MF, El-Gendi AM. Stromal caveolin-1 expression in breast carcinoma. Correlation with early tumor recurrence and clinical outcome. Pathol Oncol Res. 2012;18:459–69. doi: 10.1007/s12253-011-9469-5. [DOI] [PubMed] [Google Scholar]
- 68.Ayala G, Morello M, Frolov A, You S, Li R, Rosati F, et al. Loss of caveolin-1 in prostate cancer stroma correlates with reduced relapse-free survival and is functionally relevant to tumour progression. J Pathol. 2013 doi: 10.1002/path.4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Curry JM, Tuluc M, Whitaker-Menezes D, Ames JA, Anantharaman A, Butera A, et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle. 2013;12:1371–84. doi: 10.4161/cc.24092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012;11:1108–17. doi: 10.4161/cc.11.6.19530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martinez-Outschoorn UE, Curry JM, Ko Y-H, Lin Z, Tuluc M, Cognetti D, et al. Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: RAS and NFkB target stromal MCT4. Cell Cycle. 2013;12 doi: 10.4161/cc.25510. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle. 2012;11:3019–35. doi: 10.4161/cc.21384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sanchez-Alvarez R, Martinez-Outschoorn UE, Lin Z, Lamb R, Hulit J, Howell A, et al. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: implications for breast cancer prevention. Cell Cycle. 2013;12:289–301. doi: 10.4161/cc.23109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Langley RR, Fidler IJ. The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer. 2011;128:2527–35. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 76.Fidler IJ, Poste G. The “seed and soil” hypothesis revisited. Lancet Oncol. 2008;9:808. doi: 10.1016/S1470-2045(08)70201-8. [DOI] [PubMed] [Google Scholar]
- 77.Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncol. 2002;3:53–7. doi: 10.1016/S1470-2045(01)00622-2. [DOI] [PubMed] [Google Scholar]
- 78.Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med. 2008;359:2143–53. doi: 10.1056/NEJMra0802968. [DOI] [PubMed] [Google Scholar]
- 79.Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117–30. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–83. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoshikawa K, Honda K, Inamoto T, Shinohara H, Yamauchi A, Suga K, et al. Reduction of BRCA1 protein expression in Japanese sporadic breast carcinomas and its frequent loss in BRCA1-associated cases. Clin Cancer Res. 1999;5:1249–61. [PubMed] [Google Scholar]
- 83.Rakha EA, El-Sheikh SE, Kandil MA, El-Sayed ME, Green AR, Ellis IO. Expression of BRCA1 protein in breast cancer and its prognostic significance. Hum Pathol. 2008;39:857–65. doi: 10.1016/j.humpath.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 84.Foulkes WD, Stefansson IM, Chappuis PO, Bégin LR, Goffin JR, Wong N, et al. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst. 2003;95:1482–5. doi: 10.1093/jnci/djg050. [DOI] [PubMed] [Google Scholar]
- 85.Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene. 2006;25:5846–53. doi: 10.1038/sj.onc.1209876. [DOI] [PubMed] [Google Scholar]
- 86.Stefansson OA, Jonasson JG, Johannsson OT, Olafsdottir K, Steinarsdottir M, Valgeirsdottir S, et al. Genomic profiling of breast tumours in relation to BRCA abnormalities and phenotypes. Breast Cancer Res. 2009;11:R47. doi: 10.1186/bcr2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Turner NC, Reis-Filho JS, Russell AM, Springall RJ, Ryder K, Steele D, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 2007;26:2126–32. doi: 10.1038/sj.onc.1210014. [DOI] [PubMed] [Google Scholar]
- 88.Guendel I, Carpio L, Pedati C, Schwartz A, Teal C, Kashanchi F, et al. Methylation of the tumor suppressor protein, BRCA1, influences its transcriptional cofactor function. PLoS One. 2010;5:e11379. doi: 10.1371/journal.pone.0011379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Magdinier F, Ribieras S, Lenoir GM, Frappart L, Dante R. Down-regulation of BRCA1 in human sporadic breast cancer; analysis of DNA methylation patterns of the putative promoter region. Oncogene. 1998;17:3169–76. doi: 10.1038/sj.onc.1202248. [DOI] [PubMed] [Google Scholar]
- 90.Rio PG, Maurizis JC, Peffault de Latour M, Bignon YJ, Bernard-Gallon DJ. Quantification of BRCA1 protein in sporadic breast carcinoma with or without loss of heterozygosity of the BRCA1 gene. Int J Cancer. 1999;80:823–6. doi: 10.1002/(SICI)1097-0215(19990315)80:6<823::AID-IJC5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 91.Wilson CA, Ramos L, Villaseñor MR, Anders KH, Press MF, Clarke K, et al. Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. Nat Genet. 1999;21:236–40. doi: 10.1038/6029. [DOI] [PubMed] [Google Scholar]
- 92.Catteau A, Harris WH, Xu CF, Solomon E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene. 1999;18:1957–65. doi: 10.1038/sj.onc.1202509. [DOI] [PubMed] [Google Scholar]
- 93.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–9. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- 94.Stefansson OA, Jonasson JG, Olafsdottir K, Hilmarsdottir H, Olafsdottir G, Esteller M, et al. CpG island hypermethylation of BRCA1 and loss of pRb as co-occurring events in basal/triple-negative breast cancer. Epigenetics. 2011;6:638–49. doi: 10.4161/epi.6.5.15667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 96.Metzger-Filho O, Tutt A, de Azambuja E, Saini KS, Viale G, Loi S, et al. Dissecting the heterogeneity of triple-negative breast cancer. J Clin Oncol. 2012;30:1879–87. doi: 10.1200/JCO.2011.38.2010. [DOI] [PubMed] [Google Scholar]
- 97.Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14:276–86. doi: 10.1038/ncb2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180:599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]