

Abstract
There is growing evidence that many host proteins involved in innate and intrinsic immunity are regulated by SUMOylation, and that SUMO contributes to the regulatory process that governs the initiation of the type I interferon (IFN) response. The tumor suppressor p53 is a modulator of the IFN response that plays a role in virus-induced apoptosis and in IFN-induced senescence. Here we demonstrate that IFN treatment increases the levels of SUMOylated p53 and induces cellular senescence through a process that is partially dependent upon SUMOylation of p53. Similarly, we show that vesicular stomatitis virus (VSV) infection induces p53 SUMOylation, and that this modification favors the control of VSV replication. Thus, our study provides evidence that IFN signaling induces p53 SUMOylation, which results in the activation of a cellular senescence program and contributes to the antiviral functions of interferon.
Keywords: antiviral activity, interferon, p53, senescence, SUMO
Introduction
Interferons (IFNs) are a critical initial defense barrier that dampens pathogen growth.1 In addition, IFNs affect many other cellular functions, such as cell growth, and manifest anti-oncogenic activity.2-4 Tumor suppressor p53 is a direct transcriptional target of type I IFNs (IFN-α/β) contributing to the antitumor activity and antiviral actions mediated by the interferon pathway.4,5 Type I IFNs stimulate p53 expression,4 acetylation at lysine residue 320, and phosphorylation at serine 15,5 resulting in growth arrest and senescence.5 Cell infection with vesicular stomatitis virus (VSV) also upregulates p53 and induces both its acetylation and phosphorylation modifications, which prime virus-infected cells for enhanced apoptosis.4,6
The activity of p53 can be modulated by covalent interaction with small ubiquitin-related modifiers (SUMO), although the result of this interaction may depend upon the promoter context, stimuli, or cell type studied, as previously suggested.7 In this sense, there are reports showing enhanced,8-11 no effect,7 or decreased p53 transcription activity12,13 after SUMOylation.
Here we identify IFN treatment and infection with VSV as novel inducers of p53 SUMOylation. We examined the contribution of p53 SUMOylation to the cytostatic activity mediated by IFN and evaluated the role of this modification on the antiviral activity exerted by p53. Loss of p53 SUMOylation significantly reduced its ability to induce apoptosis in response to infection with VSV, which resulted in enhanced viral replication. In addition, a p53 SUMOylation mutant also showed reduced ability to induce cell senescence in response to IFN treatment. These findings provide new insights into the mechanisms that mediate the antiproliferative and antiviral activities of type I IFNs.
Results
IFN treatment induces p53 SUMOylation
p53 SUMOylation is induced upon hydrogen peroxide treatment and other genotoxic stresses,7,9,11 and type I IFN has been shown to induce the DNA damage-signaling pathway.5 Therefore, we decided to evaluate whether IFN treatment induced posttranslational modification of p53 by SUMO. To this end, HEK-293 cells were transfected with pcDNA, Ubc9 and His6-SUMO1 or Ubc9 and His6-SUMO2, and then treated with 500 or 1000 U/ml of IFN-α for 16 h. Whole-cell extracts and Histidine purified proteins were then analyzed by western blot. As shown in Figure 1A, incubation of cells with 500 or 1000 U/ml of IFN induced a clear increase in the levels of p53-SUMO1 and p53-SUMO2 proteins. Similar results were observed when HEK-293 cells transfected as described above were analyzed at an earlier time point after treatment with 500 U/ml of IFN-α. A clear increase in the levels of p53-SUMO1 and p53-SUMO2 proteins was detected at 8 h after IFN treatment. However, the level of p53-SUMO2 protein was even higher at 16 h after IFN treatment (Fig. 1B). All together, these results indicate that IFN induces the conjugation of p53 to SUMO1 and SUMO2.
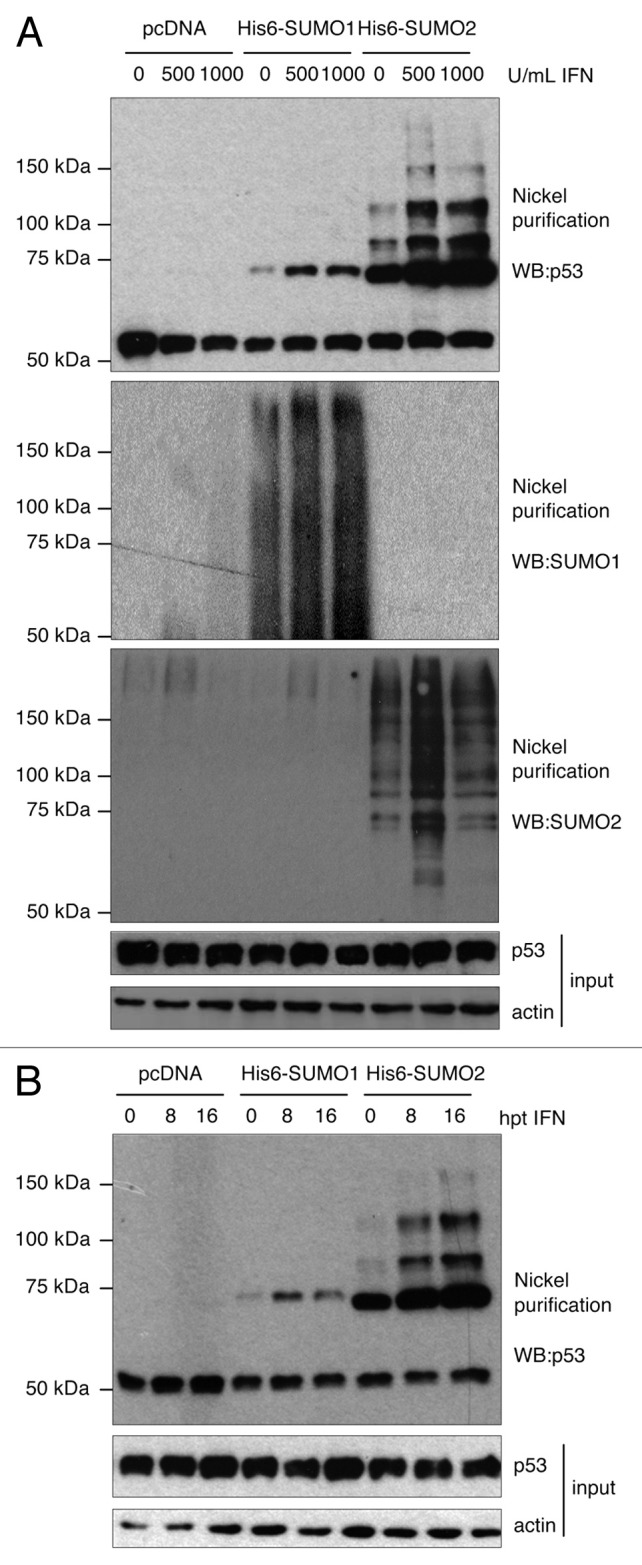
Figure 1. IFN-α treatment of HEK-293 cells induces p53 SUMOylation. (A) Stimulation of p53 SUMOylation after treatment for 16 h with different concentrations of IFN-α. (B) Stimulation of p53 SUMOylation after treatment with 500 U/ml of IFN-α for different periods of time.
VSV infection induces the conjugation of SUMO1 and SUMO2 to the lysine residue 386 in p53
The expression of IFN-α and -β is upregulated in response to viral infection.14 For this reason, we decided to evaluate whether infection with VSV can also induce the SUMOylation of p53. To this end, HEK-293 cells were transfected with pcDNA, Ubc9 and His6-SUMO1 or Ubc9 and His6-SUMO2, and 36 h after transfection cells were infected with VSV at an MOI of 1 PFU/cell. At 4 h after infection, whole-protein extracts and Histidine purified proteins were analyzed by western blot using an anti-p53 antibody. As shown in Figure 2A, infection with VSV induced an increase in the levels of p53-SUMO1 and p53-SUMO2 proteins. To corroborate these results and to analyze whether the induction of p53 SUMOylation occurs early after infection, we performed a time-course analysis of p53 SUMOylation after VSV infection. As shown in Figure 2B, a slight increase in the intensity of the p53-SUMO1 band was already detected as early as 2 hpi. However, the level of this band was even higher at 4 hpi, when we detected the maximum amount of SUMO1- and SUMO2-modified p53 proteins. To further corroborate these data, we decided to analyze the co-localization of both p53 and SUMO in cells infected with VSV. It has been previously reported that p53 and SUMO1 co-localize in the nucleoli of MCF7 and in PML nuclear bodies of HEK-293 cells.7 Accordingly, we decided to analyze the localization of p53 and SUMO in both cell lines. Cells were transfected with GFP-p53 and 36 h after transfection cells were infected with VSV at an MOI of 1 PFU/ml. At 4 h after infection cells were immunostained with anti-SUMO1 or anti-SUMO2 antibody, and the localization of both SUMO1 or SUMO2 and GFP-p53 was determined by confocal microscopy analysis. Mock-infected cells showed GFP-p53 homogenously distributed throughout the nucleus excluding nucleoli, while SUMO appeared as discrete nuclear dots (Fig. 2C). We observed a clear translocation of both SUMO2 and GFP-p53 to the nucleoli in around 50% of the MCF7-infected cells, resulting in co-localization of both proteins (Fig. 2C, left panel). In HEK-293 cells SUMO1 and GFP-p53 also co-localized in the nucleus of infected cells, even though SUMO1 was not translocated to the nucleolus.

Figure 2. VSV infection induces the conjugation of SUMO1 and SUMO2 to the lysine residue 386 in p53. (A) p53 SUMOylation after 4 h of VSV infection. (B) Time-course experiment showing p53 SUMOylation during VSV infection. (C) Co-localization of GFP-p53 and SUMO2 or SUMO1 in VSV infected MCF7 cells (left panel) and HEK-293 cells (right panel), respectively. (D) VSV infection induces the SUMOylation of p53-WT, but not of p53-K386R mutant.
Lysine 386 is the main SUMO acceptor in p53.8,11 For this reason, we decided to analyze whether the induction of p53-SUMO conjugation by VSV infection occurred in this particular lysine residue. For this, p53-null mouse embryo fibroblasts (MEFs) were co-transfected with wild-type (p53-WT) or the SUMOylation mutant (p53-K386R) p53, in combination with pcDNA, Ubc9 and His6-SUMO1 or Ubc9 and His6-SUMO2, and 36 h after transfection cells were infected with VSV as described above. Whole-cell extracts and Histidine purified proteins were then analyzed by western blot using anti-p53 antibody. We detected an increase in the levels of p53-SUMO1 and p53-SUMO2 proteins in those cells transfected with p53-WT after infection with VSV, as expected (Fig. 2D). However, no SUMO-p53 bands were detected in the cells transfected with p53-K386R, independently of VSV infection. These results demonstrate that VSV infection induces an increase in the conjugation of both SUMO1 and SUMO2 to the lysine residue 386 in p53. It is noteworthy that in all cases we observed significantly higher levels of p53 protein conjugated to SUMO2 than to SUMO1, likely due to the limited amount of SUMO 1 vs. SUMO2 protein available for conjugation.
SUMO contributes to the antiviral effect mediated by p53
Activation of p53 in response to virus infection induces transcriptional activation of some of its classical target genes, such as Puma,4 and of some IFN-inducible genes, such as ISG54.15 To elucidate the influence of p53 SUMOylation on p53-mediated transcriptional transactivation induced in response to virus infection, we generated derivatives of the p53-null human lung carcinoma cell line H1299 expressing p53-WT or the p53-K386R mutant. Cells were infected with a recombinant VSV-expressing GFP (rVSV-GFP), and at 3 h after infection, cells expressing GFP were isolated by fluorescence-activated cell sorting (FACS) and subjected to RNA extraction to measure the transactivation of p53-response genes by real-time quantitative PCR analysis (qRT-PCR). As shown in Figure 3A, VSV infection induced the transactivation of the pro-apoptotic Puma gene and the IFN-inducible gene ISG54 in cells expressing wild-type p53, as expected. However, this transactivation was clearly reduced in those cells transfected with empty vector or the SUMOylation-defective p53-K386R mutant (Fig. 3A). These results suggest that SUMOylation positively regulates p53-dependent transcriptional activity in response to virus infection. Tumor suppressor p53 contributes to virus-induced apoptosis, and alterations in p53 expression or activity may have profound effects on virus replication.4,6,15,16 To evaluate the consequences of blocking p53 SUMOylation on apoptosis induction in response to virus infection, H1299 cells transfected with pcDNA, p53-WT, or p53-K386R were infected with rVSV-GFP at an MOI of 0.1 PFU/ml, and 12 h after infection, we determined the number of apoptotic GFP-positive cells using Annexin-V staining as a readout. As shown in Figure 3B, and as expected, more than 20% of the infected cells expressing p53-WT entered apoptosis in comparison with pcDNA-transfected cells, in which less than 5% of the virus-infected cells underwent apoptosis. The percentage of apoptotic p53-K386R-expressing cells in response to VSV infection was significantly lower than the percentage observed in p53-WT cells and higher than the one detected in cells transfected with pcDNA (Fig. 3B). These results indicate that SUMOylation is required, at least to some extent, for p53-dependent apoptosis in response to virus infection. Finally, to examine the role of p53 SUMOylation on p53-mediated antiviral activity, cells transfected with pcDNA, p53-WT, or p53-K386R were infected with VSV, and at 48 h after infection we determined the virus titers in cell supernatants. As expected, expression of wild-type p53 induced a statistically significant decrease in viral titer in comparison with pcDNA-transfected cells (Fig. 3C). Importantly, the viral titer obtained in cells expressing the p53-K386R mutant was significantly higher than the one detected in p53-WT expressing cells and lower than the one recovered from pcDNA transfected cells (Fig. 3C). Altogether, these results indicate that SUMOylation is an event required for the full transcriptional activation of p53-dependent genes and apoptosis induction in response to virus infection and the subsequent control of virus replication.

Figure 3. SUMOylation is required for the full antiviral activity mediated by p53. (A) SUMOylation is required for p53-dependent transcriptional activity in response to VSV infection. The levels of mRNA expression are shown as induction relative to uninfected cells transfected with pcDNA. Presented are the mean values and SD of 3 replicates. The right panel represents the expression levels of p53 in the cells. (B) SUMOylation is required for p53-dependent apoptosis in response to virus infection. Bar graphs show the percentage of apoptotic cells (+/−SD) of triplicate samples. (C) SUMOylation is required for the control of virus replication mediated by p53. Data represents means ± SD for data obtained in 3 independent experiments. *P < 0.05, ** P < 0.005, ***P < 0.0005, Student t test.
SUMOylation of p53 contributes to the senescence program induced by IFN
Tumor suppressor p53 is a major mediator of premature cell senescence in response to IFN-β treatment5,17 or after overexpression of SUMO2/3.9 In addition, it has been proposed that p53 SUMOylation contributes to the induction of senescence observed after PIASγ overexpression.18 We then speculated that the increase in p53 SUMOylation induced by IFN might play a role in the induction of senescence triggered by this cytokine. To evaluate this possibility, we analyzed the clonogenic capacity of the H1299 derivative cells expressing pcDNA, p53-WT, or p53-K386R in the absence or presence of IFN-α. The number of colonies formed by cells treated with IFN-α was significantly lower than the number obtained in untreated control cells (P < 0.05), independently of the expression or not of p53-WT or p53-K386R (Fig. 4A). However, the p53-WT expressing cells were significantly (P < 0.05) more sensitive to IFN-α compared with pcDNA- or p53-K386R-expressing cells (Fig. 4A). We then analyzed the senescence-associated β-galactosidase (SA-β-Gal) activity. Consistent with previous studies, we observed that treatment of p53-WT expressing cells with IFN induced the appearance of SA-β-Gal-positive cells (Fig. 4B). In contrast, the number of cells transfected with pcDNA that were positive for SA-β-Gal staining was lower and did not increase significantly after IFN treatment (Fig. 4B). In addition, the percentage of cells expressing the SUMOylation mutant p53-K386R that were positive for SA-β-Gal staining after IFN treatment was significantly lower than in p53-WT-expressing cells and did not differ significantly to untreated cells (Fig. 4B). Of note, the percentage of SA-β-Gal positive cells detected in untreated p53-WT or p53-K386R cells was higher than in cells transfected with pcDNA. In addition, western blot analysis revealed that IFN treatment induced the upregulation of PML, independently of the transfection with pcDNA, p53-WT, or p53-K386R (Fig. 4C). However, we only detected the upregulation of p21 in the IFN-treated p53-WT-expressing cells and not in the pcDNA- or p53-K386R-transfected cells. Finally, as it has been well established that E2F-dependent transcription is repressed in cells undergoing senescence,19-22 qRT-PCR to monitor the expression of the E2F target gene MCM6 was performed. We only detected downregulation of MCM6 in response to interferon treatment in the p53-WT-expressing cells (0.5-fold relative to untreated p53-WT-expressing cells) and not in the pcDNA- or p53-K386R-transfected cells. Altogether, these results indicate that SUMOylation of p53 contributes to the senescence program induced by IFN, although it may be not required for other senescence-inducing stimuli.

Figure 4. IFN treatment induces cellular senescence in a p53 SUMOylation-dependent manner. (A) Increased sensitivity of p53-WT cells to IFN-α. The clonogenic capacity of H1299 cells treated with IFN-α was assessed. The colony number relative to untreated cells (average +/− SD of triplicates) is shown. *P < 0.05, Student t test when comparing the reduction in the clonogenicity of pcDNA or p53-K386R- and p53-WT-expressing cells. (B) SUMOylation of p53 contributes to the senescence program induced by IFN. Representative pictures of SA-β-Gal staining (upper-left panel) and quantification of data, showing the percentage of SA-β-Gal-positive cells (upper-right panel). Data shown average +/−SD from 2 independent experiments. Fold change in the fraction of p53-WT- or p53-K386R-expressing cells that were positive for SA-β-Gal staining after IFN-α treatment relative to the change observed in the control cells (lower-right panel) (average +/−SD from 4 independent experiments) *P < 0.05, ***P < 0.0005, Student t test. Western blot analysis of protein extracts from cells treated or not with IFN-α (lower-left panel). Numbers under the bands, densitometric analysis showing fold change in p21 levels compared with untreated cells.
Discussion
Type I IFN induces the transcription of multiple genes leading to immunomodulatory as well as growth-inhibitory effects.23,24 Tumor suppressor p53 is a direct transcriptional target of type I IFNs, contributing to their antitumor and antiviral actions.4,5 In this study we demonstrate that both treatment with type I IFN and infection with VSV, a negative sense single-stranded RNA virus that is extremely sensitive to the antiviral actions of the IFNs, induce p53 SUMOylation. These observations, together with the results showing that pronounced SUMOylation of p53 was detected very early after infection, suggest that SUMO modification might be involved in the positive regulation of the antiviral immunity exerted by p53. A role for SUMO modification in p53 antiviral activity is also consistent with the observation that a p53 SUMOylation mutant fails to control VSV infection as efficiently as the p53-WT protein. There are several examples demonstrating that VSV infection can alter the SUMOylation status of host cell proteins,25,26 and that this modification can contribute to the control of virus replication,25,27 suggesting that SUMOylation is a positive regulator of innate immunity. Thus, the IFN-induced SUMOylation of p53 is part of the complex host innate antiviral response.
In addition, we showed that IFN-induced senescence in H1299 cells stably expressing p53 was dependent to a great extent upon SUMOylation of p53. Similarly, other authors have also proposed a contribution of p53 SUMOylation to the induction of senescence after PIASg overexpression.18 These findings raise the possibility of modulating p53 SUMOylation as a potential mechanism to regulate cell senescence.
p53 SUMOylation is induced in response to hydrogen peroxide treatment9 and other genotoxic stresses.7,11 Both, IFN treatment and VSV infection can activate a DNA damage response in the cell.4,5 Thus, it is conceivable that DNA damage works as a mediator for the induction of p53 SUMOylation, although the exact molecular mechanism is not known. At the molecular level, SUMOylation of p53 can be induced by MDM2, ARF, and L11,28,29 PIAS1,13,30,31 PIASxb,13 PIASg,12,18 E1B-55k,32 RAX/PACT,33 PKC activation,34 or by direct targeting of p53 to the nucleolus.28 Due to the high number of molecular targets of IFN, several pathways may account for the IFN-dependent effect on p53 SUMOylation. Additional analyses are required to elucidate the exact mechanisms that mediate this effect.
In summary, our results open up the possibility that both the antiviral and antiproliferative effects that depend on the cross-talk between the p53 and IFN pathways may be promoted by strategies directed toward the upregulation of SUMOylated p53, which may lead to novel anticancer and antiviral therapies. Consequently, viral interference with SUMOylation of p53 could diminish defense mechanisms and/or confer a more favorable host cell environment for viral propagation. Identification of viral factors with this capability will shed light on this.
Materials and Methods
Cell lines and transfections
MEFs derived from null p53 mice or WT mice, and the cell lines MCF-7, HEK-293, BSC40, and H1299 were maintained in DMEM supplemented with 10% heat inactivated-fetal calf serum (Gibco), 5 mmol/L L-glutamine (Invitrogen), and 1% penicillin-streptomycin solution (Sigma, 10 000 U/mL penicillin and 10 mg/mL streptomycin). Transfection of HEK-293 and MCF-7 was done using X-treme (Roche), and H1299 cells and p53 null MEFs were transfected with lipofectamine 2000, following the manufacturer’s instructions. Infections were performed using VSV of Indiana strain or recombinant VSV expressing GFP (rVSV-GFP) kindly provided by Dr Adolfo Garcia-Sastre (Mount Sinai School of Medicine), and virus yields were measured by plaque assays in BSC-40 cells. To obtain the p53-WT- or p53-K386R-expressing cell lines, H1299 cells were transfected with pcDNA, pcDNA-p53-WT or pcDNA-p53-K386R and selected for 2 weeks on 1 mg/ml G418 (Invitrogen). G418-resistant clones were either pooled or isolated as individual clones. Both, pooled and at least one individual clone for each of the transfected constructs were used in each experiment.
Plasmids and reagents
pcDNA-p53 pcDNA-p53-K386R, pcDNA-His6-SUMO1, pcDNA-His6-SUMO2, and pcDNA-SV5-Ubc9 have been previously described.11,35,36 Interferon α was purchased from GenScript.
Western blot analysis and antibodies
For western blot analysis, cells were washed in PBS, scraped in SDS-gel loading buffer and boiled for 5 min. Proteins of total extracts were separated by SDS-PAGE and transferred to nitrocellulose membrane. The following antibodies were used: anti-SUMO1 (FL-101) (sc-9060, Santa Cruz), anti-SUMO2 (51-9100, Zymed Laboratories), anti-actin (69100, MP Biomedicals), anti-p53 (DO-1) (sc-126, Santa Cruz), anti-PML (H238) (sc-5621, Santa Cruz), anti-p21 (F-5) (sc-6264, Santa Cruz), anti-VSV M protein (EB0011, KeraFAST), and Alexa 594-conjugated anti-rabbit antibody (A-11037, Molecular Probes). Quantification of band intensities was performed by using ImageJ software and normalized by the tubulin densitometry values.
Fluorescence-activated cell sorting
Cells were infected with VSV-GFP at subjected to one-way cell sorting of GFP+ cells using a FACS-Aria instrument (BD).
Immunofluorescence staining
Immunofluorescence staining and confocal analysis were performed as previously described.37 Analysis of the samples was performed on a Leica TCS SP5 confocal laser microscope using simultaneous scans to avoid shift between the optical channels. Images were exported using Adobe Photoshop CS2 version 9.0.2.
Purification of His-tagged conjugates
Purification of His-tagged conjugates using Ni2+-NTA-agarose beads was performed as described.38
Quantitative reverse transcription-PCR (qRT-PCR) analysis
For qRT-PCR analysis, RNA was isolated from cells using TRIzol (Invitrogen). Quantitative RT-PCR was performed using 100 ng of sample RNA and SYBR green (Roche) in an ABI PRISM 7900HT instrument following manufacturer’s instructions. Specific MCM6 primers were previously reported.22
Apoptosis determination
Cells were infected with rVSV-GFP at a multiplicity of infection (MOI) of 0.1 PFU/ml. At 12 h after infection, GFP-positive cells were harvested and stained with Annexin-V-APC and propidium iodide. Cells were then analyzed in a FACs canto II instrument.
Cell proliferation, clonogenic assay, and senescence determination
For clonogenic assay, cells were seeded at 1000 cells per 60 cm tissue culture dishes in triplicate and incubated with or without IFN-α (1000 U/mL) for 7 d. Cells were fixed with methanol, and colonies were stained with 0.5% crystal violet solution. For SA-β-Gal activity, cells were plated in 12 multi-well, 0.5 × 104 cells per well, and treated with 1000 U/mL of IFN-α for 3 d, or left untreated, and then stained with the senescence β-galactosidase kit (Cell Signaling) following the manufacturer’s instructions. Percentage positive staining was determined by dividing the number of β-Gal-positive cells into the total number within 10 random fields from duplicate dishes.
Statistical analysis
For statistical analysis between control and different groups the Student t test was applied. The significance level chosen for the statistical analysis was P < 0.05
Acknowledgments
Funding at the laboratory of CR is provided by BFU-2011-27064. MC is a Miguel Servet investigator funded by ISCIII (CP11/00273). LMV is supported by Juan de la Cierva Programme. CFC-H is supported by La Caixa fellowship. AS is supported by a FPU fellowship. JMV is supported by a pre-doctoral fellowship from Xunta de Galicia. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Glossary
Abbreviations:
- IFN
interferon
- VSV
vesicular stomatitis virus
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25868
References
- 1.Levy DE, García-Sastre A. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 2001;12:143–56. doi: 10.1016/S1359-6101(00)00027-7. [DOI] [PubMed] [Google Scholar]
- 2.Gresser I, Belardelli F. Endogenous type I interferons as a defense against tumors. Cytokine Growth Factor Rev. 2002;13:111–8. doi: 10.1016/S1359-6101(01)00035-1. [DOI] [PubMed] [Google Scholar]
- 3.Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their actions. Annu Rev Biochem. 1987;56:727–77. doi: 10.1146/annurev.bi.56.070187.003455. [DOI] [PubMed] [Google Scholar]
- 4.Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K, et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424:516–23. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- 5.Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol Biol Cell. 2006;17:1583–92. doi: 10.1091/mbc.E05-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muñoz-Fontela C, González D, Marcos-Villar L, Campagna M, Gallego P, González-Santamaría J, Herranz D, Gu W, Serrano M, Aaronson SA, et al. Acetylation is indispensable for p53 antiviral activity. Cell Cycle. 2011;10:3701–5. doi: 10.4161/cc.10.21.17899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwek SS, Derry J, Tyner AL, Shen Z, Gudkov AV. Functional analysis and intracellular localization of p53 modified by SUMO-1. Oncogene. 2001;20:2587–99. doi: 10.1038/sj.onc.1204362. [DOI] [PubMed] [Google Scholar]
- 8.Gostissa M, Hengstermann A, Fogal V, Sandy P, Schwarz SE, Scheffner M, Del Sal G. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 1999;18:6462–71. doi: 10.1093/emboj/18.22.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li T, Santockyte R, Shen RF, Tekle E, Wang G, Yang DC, Chock PB. Expression of SUMO-2/3 induced senescence through p53- and pRB-mediated pathways. J Biol Chem. 2006;281:36221–7. doi: 10.1074/jbc.M608236200. [DOI] [PubMed] [Google Scholar]
- 10.Muller S, Berger M, Lehembre F, Seeler JS, Haupt Y, Dejean A. c-Jun and p53 activity is modulated by SUMO-1 modification. J Biol Chem. 2000;275:13321–9. doi: 10.1074/jbc.275.18.13321. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez MS, Desterro JM, Lain S, Midgley CA, Lane DP, Hay RT. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999;18:6455–61. doi: 10.1093/emboj/18.22.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson V, Davis GE, Maxwell SA. A putative protein inhibitor of activated STAT (PIASy) interacts with p53 and inhibits p53-mediated transactivation but not apoptosis. Apoptosis. 2001;6:221–34. doi: 10.1023/A:1011392811628. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt D, Müller S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci U S A. 2002;99:2872–7. doi: 10.1073/pnas.052559499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sen GC. Viruses and interferons. Annu Rev Microbiol. 2001;55:255–81. doi: 10.1146/annurev.micro.55.1.255. [DOI] [PubMed] [Google Scholar]
- 15.Muñoz-Fontela C, Macip S, Martínez-Sobrido L, Brown L, Ashour J, García-Sastre A, Lee SW, Aaronson SA. Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med. 2008;205:1929–38. doi: 10.1084/jem.20080383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munoz-Fontela C, Garcia MA, Garcia-Cao I, Collado M, Arroyo J, Esteban M, Serrano M, Rivas C. Resistance to viral infection of super p53 mice. Oncogene. 2005;24:3059–62. doi: 10.1038/sj.onc.1208477. [DOI] [PubMed] [Google Scholar]
- 17.Hebner C, Beglin M, Laimins LA. Human papillomavirus E6 proteins mediate resistance to interferon-induced growth arrest through inhibition of p53 acetylation. J Virol. 2007;81:12740–7. doi: 10.1128/JVI.00987-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bischof O, Schwamborn K, Martin N, Werner A, Sustmann C, Grosschedl R, Dejean A. The E3 SUMO ligase PIASy is a regulator of cellular senescence and apoptosis. Mol Cell. 2006;22:783–94. doi: 10.1016/j.molcel.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 19.Dimri GP, Hara E, Campisi J. Regulation of two E2F-related genes in presenescent and senescent human fibroblasts. J Biol Chem. 1994;269:16180–6. [PubMed] [Google Scholar]
- 20.Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/S0092-8674(03)00401-X. [DOI] [PubMed] [Google Scholar]
- 21.Rowland BD, Denissov SG, Douma S, Stunnenberg HG, Bernards R, Peeper DS. E2F transcriptional repressor complexes are critical downstream targets of p19(ARF)/p53-induced proliferative arrest. Cancer Cell. 2002;2:55–65. doi: 10.1016/S1535-6108(02)00085-5. [DOI] [PubMed] [Google Scholar]
- 22.Vernier M, Bourdeau V, Gaumont-Leclerc MF, Moiseeva O, Bégin V, Saad F, Mes-Masson AM, Ferbeyre G. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 2011;25:41–50. doi: 10.1101/gad.1975111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brierley MM, Fish EN. Review: IFN-alpha/beta receptor interactions to biologic outcomes: understanding the circuitry. J Interferon Cytokine Res. 2002;22:835–45. doi: 10.1089/107999002760274845. [DOI] [PubMed] [Google Scholar]
- 24.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–64. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 25.González-Santamaría J, Campagna M, Ortega-Molina A, Marcos-Villar L, de la Cruz-Herrera CF, González D, Gallego P, Lopitz-Otsoa F, Esteban M, Rodríguez MS, et al. Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis. 2012;3:e393. doi: 10.1038/cddis.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kubota T, Matsuoka M, Chang TH, Tailor P, Sasaki T, Tashiro M, Kato A, Ozato K. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J Biol Chem. 2008;283:25660–70. doi: 10.1074/jbc.M804479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ran Y, Liu TT, Zhou Q, Li S, Mao AP, Li Y, Liu LJ, Cheng JK, Shu HB. SENP2 negatively regulates cellular antiviral response by deSUMOylating IRF3 and conditioning it for ubiquitination and degradation. J Mol Cell Biol. 2011;3:283–92. doi: 10.1093/jmcb/mjr020. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, Chen J. MDM2-ARF complex regulates p53 sumoylation. Oncogene. 2003;22:5348–57. doi: 10.1038/sj.onc.1206851. [DOI] [PubMed] [Google Scholar]
- 29.Stindt MH, Carter S, Vigneron AM, Ryan KM, Vousden KH. MDM2 promotes SUMO-2/3 modification of p53 to modulate transcriptional activity. Cell Cycle. 2011;10:3176–88. doi: 10.4161/cc.10.18.17436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kahyo T, Nishida T, Yasuda H. Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol Cell. 2001;8:713–8. doi: 10.1016/S1097-2765(01)00349-5. [DOI] [PubMed] [Google Scholar]
- 31.Miyauchi Y, Yogosawa S, Honda R, Nishida T, Yasuda H. Sumoylation of Mdm2 by protein inhibitor of activated STAT (PIAS) and RanBP2 enzymes. J Biol Chem. 2002;277:50131–6. doi: 10.1074/jbc.M208319200. [DOI] [PubMed] [Google Scholar]
- 32.Pennella MA, Liu Y, Woo JL, Kim CA, Berk AJ. Adenovirus E1B 55-kilodalton protein is a p53-SUMO1 E3 ligase that represses p53 and stimulates its nuclear export through interactions with promyelocytic leukemia nuclear bodies. J Virol. 2010;84:12210–25. doi: 10.1128/JVI.01442-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennett RL, Pan Y, Christian J, Hui T, May WS., Jr. The RAX/PACT-PKR stress response pathway promotes p53 sumoylation and activation, leading to G₁ arrest. Cell Cycle. 2012;11:407–17. doi: 10.4161/cc.11.2.18999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heo KS, Fujiwara K, Abe J. Disturbed-flow-mediated vascular reactive oxygen species induce endothelial dysfunction. Circ J. 2011;75:2722–30. doi: 10.1253/circj.CJ-11-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell. 1998;2:233–9. doi: 10.1016/S1097-2765(00)80133-1. [DOI] [PubMed] [Google Scholar]
- 36.Vertegaal AC, Andersen JS, Ogg SC, Hay RT, Mann M, Lamond AI. Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteins revealed by quantitative proteomics. Mol Cell Proteomics. 2006;5:2298–310. doi: 10.1074/mcp.M600212-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Campagna M, Herranz D, Garcia MA, Marcos-Villar L, González-Santamaría J, Gallego P, Gutierrez S, Collado M, Serrano M, Esteban M, et al. SIRT1 stabilizes PML promoting its sumoylation. Cell Death Differ. 2011;18:72–9. doi: 10.1038/cdd.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marcos-Villar L, Lopitz-Otsoa F, Gallego P, Muñoz-Fontela C, González-Santamaría J, Campagna M, Shou-Jiang G, Rodriguez MS, Rivas C. Kaposi’s sarcoma-associated herpesvirus protein LANA2 disrupts PML oncogenic domains and inhibits PML-mediated transcriptional repression of the survivin gene. J Virol. 2009;83:8849–58. doi: 10.1128/JVI.00339-09. [DOI] [PMC free article] [PubMed] [Google Scholar]