

Abstract
Burkitt lymphoma is characterized by deregulation of c-myc, and therapies targeting c-myc are under investigation as treatments. Histone deacetylase inhibitors are known to abrogate c-myc expression, leading us to examine their effect in a series of Burkitt lymphoma cell lines. While treatment with romidepsin, panobinostat, vorinostat, or belinostat for 48 h resulted in complete cell death in the Ramos and ST486 lines, CA46 and DG75 cells were resistant. In parallel studies, CA46 and DG75 cells were also insensitive to 48 h treatment with the Aurora kinase inhibitors (AKIs) MLN8237 (alisertib), VX-680 (tozasertib), or ZM447439. Bax knockdown is known to lead to HDI resistance, and we found that loss of Bax or both Bak and Bax correlated with resistance to both AKIs and HDIs in the Burkitt cell lines. As proof-of-concept to evaluate the contribution of Bax and Bak to HDI-mediated apoptosis, we found that apoptosis was unaffected in HCT-116 colon carcinoma cells lacking Bak, blunted in cells lacking Bax, and nearly completely abrogated in cells lacking both Bak and Bax compared with wild-type cells. To explore potential clinical variations in Bak and Bax expression, a series of samples from 16 patients diagnosed with Burkitt lymphoma was examined. While the majority of samples were positive for both Bak and Bax, some (3/16) expressed low levels of both proteins. We thus conclude that HDI-mediated and AKI-mediated apoptosis requires mitochondrial engagement, and that baseline Bax and Bak expression may serve as biomarkers for patients with Burkitt lymphoma likely to respond to HDI treatment.
Keywords: Bak, Bax, histone deacetylase inhibitor, vorinostat, romidepsin
Introduction
Histone deacetylase inhibitors (HDIs) have shown promise in the treatment of hematologic malignancies. Vorinostat was the first HDI to receive FDA approval for the treatment of cutaneous T-cell lymphoma,1 and romidepsin was approved for both cutaneous and peripheral T-cell lymphoma.2-5 More recent clinical trials have shown that vorinostat is effective as a single agent in follicular and marginal zone lymphoma,6 mocetinostat has shown promise in the treatment of relapsed Hodgkin lymphoma,7 and transient reductions in leukemic blasts were observed in patients with acute myelogenous leukemia treated with panobinostat.8 In clinical trials with solid tumors, results have been generally disappointing when HDIs are administered as single agents.9
Despite their clinical successes, however, the mechanism of action of HDIs remains elusive. Multiple mechanisms by which HDIs exert their proapoptotic effects have been suggested, such as effects on gene expression, interactions with non-histone proteins, such as Hsp90, or modifications of proteins in the apoptotic pathways.10 Similarly, the mechanisms of resistance to HDIs have not been explored, although some studies have suggested that activation of signaling pathways, increased thioredoxin levels, or increased levels of antiapoptotic proteins such as Bcl-XL or Bcl-2 may play a role.11 We recently demonstrated that upregulation of the mitogen-activated protein kinase pathway led to Bim degradation and resistance to histone deacetylase inhibitors in a model of cutaneous T-cell lymphoma.12 Combination of an HDI with inhibitors of signaling pathways leads to increased apopotosis in several cell line models.13-15
Various reports have shown that HDIs mediate apoptosis through both the intrinsic and extrinsic pathways. Romidepsin has been shown to mediate apoptosis through the tumor necrosis factor (TNF) pathway activation of caspase 8 and downregulation of cellular FLICE-inhibitory protein, but this may be model-specific.16 Vorinostat and entinostat have been shown to increase expression of the death receptors 4 and 5, leading to greater sensitivity to TNF-related apoptosis inducing ligand (TRAIL).17 The intrinsic pathway also plays a major role in HDI-induced apoptosis, as overexpression of the antiapoptic proteins Bcl-2 or Bcl-XL has been shown to attenuate apoptosis.18,19
To further explore the role of apoptosis in HDI-mediated cell death, we sought a model that would readily respond to treatment. Histone deacetylase inhibitors have been shown to be effective in several in vivo models of lymphoma,20-22 and we previously demonstrated romidepsin to be particularly effective in T-cell lymphoma models.23 The transcription factor c-myc has been shown to play a significant role in lymphomagenesis,24,25 leading to increased interest in therapies targeting c-myc expression. Early studies with some HDIs in panels of B-cell lymphoma cell lines demonstrated efficacy in some Burkitt lymphoma cell lines,26,27 possibly due to the fact that HDIs readily downregulate c-myc, which is mutated and deregulated in this disease.28 Thus, we screened four human Burkitt lymphoma cell lines using a short-term treatment with romidepsin that we found more closely represented the clinical results with romidepsin. We found differences in their sensitivity to HDIs and sought to characterize factors contributing to sensitivity. Our findings suggest that baseline expression of apoptotic effector proteins may predict response to HDI treatment.
Results
Short-term romidepsin treatment mediates differential apoptosis in Burkitt lymphoma cell lines
The histone deacetylase inhibitor romidepsin is administered clinically as a 4 h infusion at 14 mg/m2 on days 1, 8, and 15 of a 28 d schedule. In patients, the drug has a half-life of approximately 3 h and a Cmax of 361 ng/ml.2,29 To more closely approximate the dosing schedule of romidepsin, we treated cells for 6 h at a concentration of 1, 3, 10, or 25 ng/ml then incubated cells for an additional 42 h in romidepsin-free medium and examined cellular apoptosis similar to the method of Yu, et al.30 As shown in Figure 1A, Ramos and ST486 cells underwent extensive apoptosis. In CA46 cells, the effect of romidepsin treatment was blunted compared with Ramos and ST486 cells. DG75 cells appeared to be completely resistant to romidepsin-induced apoptosis. To determine whether the observed differences were due to the short-term romidepsin treatment, we treated cells with 25 ng/ml romidepsin continuously for 48 h as shown in Figure 1B. Again, the CA46 and DG75 cell lines demonstrated less apoptosis compared with the Ramos and ST486 lines, which were completely killed by romidepsin treatment. As expression of the ATP-binding cassette transporter ABCB1 has been associated with resistance to romidepsin,31,32 CA46 and DG75 cells were incubated with romidepsin in the presence of the ABCB1 inhibitor valspodar; however, this did not yield an increase in apoptosis (data not shown). We observed an identical pattern of apoptosis when cells were treated with 5 µM vorinostat, 100 nM panobinostat, or 1 µM belinostat, concentrations previously shown to induce apoptosis in cell line models (Fig. 1B). When 10 µM of the pan-caspase inhibitor Q-VD-OPh was added during the 48 h incubation with romidepsin or panobinostat, apoptosis was nearly completely abrogated in the CA46, Ramos, and ST486 cell lines (Fig. 1C), suggesting that the observed apoptosis was caspase-mediated. These results suggested that the apoptosis induced by HDI treatment was caspase-dependent, and that CA46 and DG75 cells might have unique features that render them less sensitive to HDI treatment.

Figure 1. Sensitivity of Burkitt lymphoma cell lines to HDIs.(A)Cells were incubated in the noted concentrations of romidepsin (DP) for 6 h, after which the medium was removed, and cells were allowed to incubate for an additional 42 h in drug-free medium. Cells were then incubated with anti-annexin antibody and propidium iodide and the percentage of annexin positive cells were determined as outlined in the materials and methods. Results are from at least 5 independent experiments.(B)Cells were incubated in the noted concentrations of HDIs continuously for 48 h after which the percentage of annexin-positive cells were determined as in(A)Results from at least four independent experiments are shown. P < 0.006 for all HDI concentrations compared with control.(C)Cells were incubated for 48 h with 10 µM Q-VD-OPh, 25 ng/ml romidepsin, 100 nM panobinostat, or 10 µM Q-VD-OPh concomitantly with romidepsin or panobinostat. The percentage of annexin-positive cells was determined as in (A and B)Results from at least 3 experiments are shown. Differences marked with ****, P < 0.0001. DP, romidepsin; PAN, panobinostat; VOR, vorinostat; BEL, belinostat; QVD, Q-VD-OPh.
Treatment of Burkitt lymphoma cell lines with romidepsin results in increased histone acetylation and decreased expression of c-myc
Treatment with histone deacetylase inhibitors is known to result in increased levels of acetylated histones and decreased levels of c-myc.18,33 To verify that romidepsin treatment had equivalent effects in the cell lines tested, whole-cell lysates were extracted from untreated cell or cells treated with 25 ng/ml romidepsin for 24 h, and immunoblotting was performed for acetylated histone H3 and c-myc as well as for total and cleaved poly (ADP-ribose) polymerase (PARP). This shorter time period was chosen, as extensive cell death at 48 h made protein extraction difficult. As shown in Figure 2A, all cell lines demonstrated increased levels of acetylated histone H3 and decreased c-myc after romidepsin treatment. In addition, we noted almost total PARP cleavage in the Ramos and ST486 cell lines. Less cleaved PARP was observed in CA46 cells, and nearly all PARP was uncleaved in DG75 cells, consistent with the results observed with annexin staining. Immunohistochemistry to detect c-myc was also performed on all the cell lines before and after a 24 h treatment with 25 ng/ml romidepsin. As shown in Figure 2B, all cell lines were positive for c-myc before treatment, and no c-myc was detected after romidepsin treatment. RT-PCR analysis was also performed, and we found that mRNA levels of MYC were decreased in all the cell lines after a 24 h treatment with as little as 3 ng/ml romidepsin (Fig. 2C). Vorinostat and panobinostat treatment also resulted in decreased levels of MYC (data not shown). Only very low levels of ABCB1 were detected in the cell lines, again ruling out expression of ABCB1 as a resistance mechanism. Thus, resistance to HDI-mediated apoptosis in the CA46 and DG75 cells does not appear to be due to the inability of romidepsin or other HDIs to induce histone acetylation or decrease c-myc expression.

Figure 2. Romidepsin causes histone acetylation and a decrease in c-myc levels in Burkitt lymphoma cell lines.(A)Cells were treated with 25 ng/ml romidpesin (DP) for 24 h after which protein was extracted, proteins were separeated by PAGE, and transferred to nitrocellulose membranes. The membranes were subsequently probed for PARP, cleaved PARP, acetylated histone H3, c-myc and GAPDH. Results from one of two independent experiments are shown.(B)Cells were treated as in (A), after which c-myc staining was examined by immunohistohemistry as outlined in the methods section. Magnification, 40×.(C)Cells were treated with the noted concentrations of romidpesin (DP) after which RNA was isolated and PCR analysis was performed for MYC. MYC levels were normalized to rRNA levels. Results from 3 analyses of 2 independent treatments are shown.
Aurora kinase inhibitors are variably effective in Burkitt lymphoma cell lines
Aurora kinase inhibitors have been demonstrated to be particularly effective in Burkitt lymphoma cell lines.34,35 To determine if CA46 and DG75 cells were also resistant to Aurora kinase inhibitors, we treated the Burkitt lymphoma cell lines for 48 h with 250 nM tozasertib or alisertib or 1 µM ZM447439 and examined cellular apoptosis by annexin V and PI staining. As shown in Figure 3, Ramos and ST486 cells readily underwent apoptosis, while CA46 and DG75 cells were nearly completely resistant to Aurora kinase inhibitor-mediated apoptosis. Inhibitors of individual Aurora kinases or pan-Aurora kinase inhibitors seemed to have equivalent affects in the sensitive Burkitt lines.
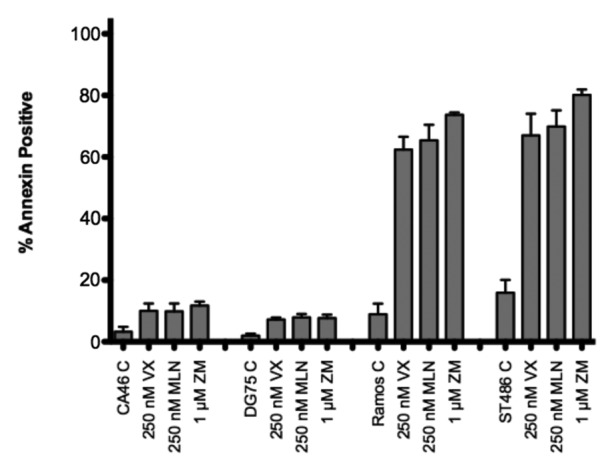
Figure 3. Burkitt lymphoma cell lines are variably sensitive to AKIs. Cell lines were treated with the noted concentrations of AKIs for 48 h, after which cells were stained with annexin V antibody and propidium iodide and the percent annexin-positive cells were determined. Results from at least 3 experiments are shown. P < 0.0001 for all AKI treatments compared with control. VX, VX-680; MLN, MLN8237; ZM, ZM447439.
CA46 cells lack Bax while DG75 cells lack Bak and Bax
Knockdown of the apoptotic protein Bax has been shown to decrease cell sensitivity to HDIs,36 leading us to examine expression of proteins that belong to the intrinsic apoptotic pathway. As seen in Figure 4, Ramos and ST486 cells express both Bax and Bak, while CA46 cells lack Bax, and DG75 cells lack both Bak and Bax. CA46 cells have previously been reported to be Bax-deficient.37 All cell lines were found to express high levels of the proapoptotic protein Bim as well as the antiapoptotic Mcl-1 protein. Notably, levels of Mcl-1 were higher in CA46 and DG75 cells. Bcl-XL was detectable in all cell lines, although levels were higher in Ramos and ST486 cells. Interestingly, Bcl-2 expression, previously shown to confer resistance to HDIs,18 was absent in DG75 cells and low in the other 3 lines. Upon examination of BAX and BAK1 gene expression, we found that all cell lines expressed BAX and BAK1, suggesting that lack of protein expression was due to post-transcriptional effects (data not shown). These results suggested that lack of Bax or lack of both Bak and Bax were associated with resistance of CA46 and DG75 cells, respectively, to HDI-mediated apoptosis. Loss of Bax alone was associated with near complete resistance to Aurora kinse inhibitors in the CA46 line. This was in agreement with a previous report showing that loss of Bak and/or Bax prevents apoptosis mediated by Aurora kinase inhibitors.38 This led us to then examine whether Bax or Bak was required for HDI-mediated apoptosis in a defined model system.
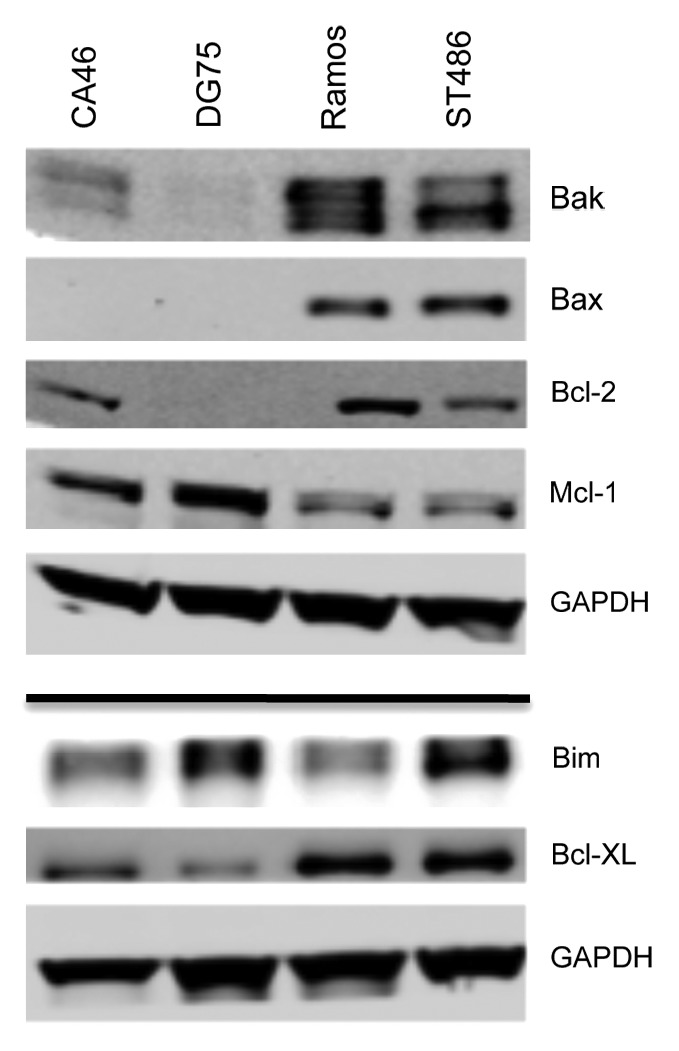
Figure 4. Expression of apoptotic proteins in Burkitt lymophoma cell lines. Whole-cell lysates were extracted from the Burkitt lymphoma lines, after which the proteins were separated by PAGE and transferred to nitrocellulose membranes. The membranes were subsequently probed with antibodies to Bak, Bax, Bcl-2, Bim, Mcl-1, and Bcl-XL. GAPDH served as a loading control. Results from one of two independent experiments are shown.
Cells lacking Bax and Bak are resistant to HDI-mediated apoptosis
To confirm the results obtained with the Burkitt lymphoma lines, we next examined the sensitivity of HCT-116 cells lacking Bak, Bax, or both proteins to HDI treatment. Confirmation of expression of Bak and Bax in the HCT-116 cell lines is shown in Figure 5A. Cells were treated for 48 h with 25 ng/ml romidepsin, 5 µM vorinostat, 100 nM panobinostat, or 1 µM belinostat. HCT-116 cells were sensitive to HDI treatment, with nearly 60% of cells staining positive for annexin V after HDI treatment (Fig. 5B). Loss of Bak expression had little effect on cells staining positive for annexin V, suggesting that Bak was dispensable in the response to HDI treatment. In contrast, cells lacking Bax expression demonstrated approximately a 2-fold reduction in the number of apoptotic cells. Cells lacking both Bak and Bax were nearly 4-fold less sensitive to HDI treatment, with the percentage of annexin positive cells only slightly higher than that found in the untreated cells. The observed results mirrored those in the Burkitt lines. These results suggested that Bax expression was critical to HDI-mediated cell death, and that loss of both Bak and Bak confers near complete resistance to HDI-mediated apoptosis. Apoptosis mediated by HDI treatment thus appears to require mitochondrial engagement.

Figure 5. Effect of HDI treatment on HCT-116 cells lacking Bak, Bax or both.(A)Whole-cell lysates were extracted from parental HCT-116 cells, or cells lacking Bak (BAK−/−), Bax (BAX−/−), or both Bak and Bax (DKO). HUT78 cells also served as a positive control for Bak and Bax. Proteins were separated by PAGE and were transferred to nitrocellulose membranes. The membranes were subsequently probed for Bak or Bax. GAPDH served as a loading control. Results from one of two experiments are shown.(B)HCT-116 cells were treated with the indicated concentrations of HDIs for 48 h, after which cells were incubated with annexin V antibody and propidium iodide. The percent annexin-positive cells were calculated and results from at least 3 independent experiments are shown. P < 0.0001 for all HDI treatments compared with control for HCT-116, BAK−/−, and BAX−/− cells; P < 0.0135 for all HDI treatments compared with control for DKO cells.
Bak and Bax expression in samples from patients with Burkitt lymphoma
We next examined Bak and Bax expression by immunohistochemistry in the Burkitt lymphoma cell lines tested. Results in the cell lines paralleled immunoblot data as CA46 cells were found to express low levels of Bak and no Bax (Fig. 6A and E), DG75 cells were found to express no Bak or Bax (Fig. 6B and F), and Ramos (Fig. 6C and G) and ST486 (Fig. 6D and H) cells expressed high levels of Bak and Bax. To determine if changes in Bak and Bax expression could be observed clinically, we examined a series of 16 samples from patients diagnosed with Burkitt lymphoma as shown in Figure 6; details for the patient samples are provided in Table 1. A range of expression was observed. While most samples expressed readily detectable levels of Bak and Bax (Patient 1, Fig. 6I and L), some samples expressed low levels of both proteins (Paitent 11, Fig. 6J and M), as well as low levels of Bak and high levels of Bax (Patient 6, Fig. 6K and N). Thus, there does seem to be variation in the expression of Bak and Bax in Burkitt lymphoma.

Figure 6. Immunohistochemical detection of Bak and Bax in Burkitt lymphoma cell lines and patient samples. Bak and Bax expression was examind by immunohistochemistry as outlined in the methods section. (A–D) Bak expression in Burkitt cell lines: CA46 (A), DG75 (B), Ramos (C) and ST486 (D). (E–H) Bax expression in Burkitt cell lines: CA46 (E), DG75 (F), Ramos (G) and ST486 (H). Bak (I–K) and Bax (L–N) expression in Burkitt lymphoma samples. More than 75% of tumor cells showed granular cytoplasmic expression for Bak (I) and Bax (L) in case 1 of Burkitt lymphoma; less than 25% of tumor cells were positive for Bak (J) and Bax (M) in case 11; in case 6, the tumor cells showed a discordant expression of Bak and Bax, with low expression (<25%) for Bak (K) and high expression (>75%) for Bax (N); scattered macrophages were also positive for Bak (J and K).
Table 1. Summary of patient data and Bak and Bax expression.
| Patient | Biopsy location | Age | Sex | Bak | Bax |
|---|---|---|---|---|---|
| 1 | LN, supraclavicular | 65 | M | >75% | >75% |
| 2 | Chest/breast mass | 23 | M | >75% | 50–75% w |
| 3 | Small bowel | 61 | M | >75% | 50–75% w |
| 4 | Tonsil | 20 | M | 50–75% | 25–50% w |
| 5 | LN, axilla | 25 | M | 50–75% | 5–25% |
| 6 | LN, cervical | 54 | F | 50–75% | 5–25% |
| 7 | LN, axilla, gall bladder | 46 | M | 25–50% | 5–25% |
| 8 | LN, inguinal | 86 | M | 25–50% | 25–50% |
| 9 | LN, supraclavicular | 65 | M | 5–25% | <5% |
| 10 | LN, cervical | 6 | M | 50–75% | 50–75% |
| 11 | LN, cervical | 25 | M | 5–25% | 5–25% |
| 12 | Cecum, terminal ileum | 10 | M | > 75% | 50–75% |
| 13 | LN, inguinal | 39 | M | 25–50% | 5–25% |
| 14 | Nasal cavity | 6 | M | 5–25% | 5–25% |
| 15 | Adnexal mass | 28 | F | >75% | 50–75% w |
| 16 | LN, cervical | 16 | M | 25–50% | 25–50% |
Samples were stained and expression expressed in quartiles as outlined in the “Materials and Methods” section. LN, lymph node.
Discussion
The lack of success of HDIs in solid tumors has led to increased interest in the potential mechanisms of intrinsic resistance to these compounds. In determining the sensitivity of a series of Burkitt lymphoma cell lines to romidepsin treatment, we found that 2 of the 4 lines examined exhibited decreased apoptosis not only to romidpesin, but to a series of HDIs currently in clinical trials as well as to a series of AKIs. The CA46 cell line was found to lack Bax, and the DG75 line lacked both Bak and Bax, thus explaining the observed resistance. This was confirmed in HCT-116 cells lacking Bax or both, which exhibited a similar apoptosis profile, as was observed with the Burkitt lines, underscoring the importance of Bax in mediating apoptosis induced by HDI treatment as well as implicating the intrinsic pathway as the predominant apoptotic pathway. Our results suggest that the mitochondria are involved in HDI-mediated apoptosis.
Loss of Bax and Bak has resulted in resistance to apoptosis from a number of stimuli. Mouse embryonic fibroblasts deficient in Bak and Bax were resistant to apoptosis mediated by treatment with staurosporine, etoposide, tunicamycin, thapsigargin, brefeldin A, or UV irradiation compared with wild-type cells.39 Similarly, cytochrome c release was nearly completely prevented in HCT-116 cells deficient in Bak and Bax following treatment with camptothecin, indomethacin, 5-fluorouracil, cisplatin, or ABT-737.40 HCT-116 cells deficient in Bax alone had decreased cytochrome c release compared with wild-type cells when treated with camptothecin, indomethacin, 5-fluorouracil, or ABT-737,40 which corroborates our findings that HDI-mediated apoptosis is blunted in lymphoma cells lacking Bax, and that loss of both Bak and Bax results in resistance to HDI-mediated apoptosis.
Interestingly, mutations in the coding sequence of BAX are found in cell lines and tumor samples with high microsatellite instability (MSI). Rampino et al. reported frameshift mutations in both BAX alleles in cell lines and tumor samples with MSI; such cell lines were found to lack expression of Bax protein.41 Colorectal carcinomas as well as gastric cancers and endometrial cancers with high MSI were frequently found to have frameshift mutations in the BAX gene.42-44 Mutations in BAX have also been reported in hematopoietic samples with MSI,45 but this appears to occur less frequently when compared with colorectal carcinoma samples. While resistance to apoptosis has been noted in cell lines with MSI that display BAX frameshift mutations,45 MSI itself has not been found to be a negative prognostic factor. Paradoxically, MSI has been associated with a better prognosis in gastric cancer46 and colorectal cancer,47 although response to 5-fluorouracil-based regimens appears to be worse in tumors with MSI.47 However, these studies did not look at BAX mutation specifically. Ionov and colleagues examined BAX mutations in patients with gastric or colon cancer, and found that patients with tumors harboring BAX mutations had significantly shorter survival than those without mutations, suggesting that Bax loss might impair apoptotic activity in tumors.48
Studies have linked lower Bax expression to worse clinical outcome in several cancer types. Reductions in BAX gene expression in breast cancer were found to be associated with poor response to combination chemotherapy with cyclophosphamide, fluorouracil, and epirubicin, and shorter survival.49 Low expression of Bax was also found to correlate with poor survival of patients with metastatic or recurrent gastric cancer who received FOLFOX chemotherapy.50 Kang et al. reported that low expression of Bax was the most significant predictor of poor survival in patients with esophageal cancer treated with fluorouracil and cisplatin with ratiotherapy.51 Median survival was lower for Bax negative patients with transitional cell carcinoma of the bladder.52 On the other hand, high Bax expression was also found to be a good prognostic factor in acute myeloid leukemia.53
Burkitt lymphoma is a very aggressive non-Hodgkin lymphoma that has a unique distribution in the world. Endemic Burkitt is commonly found in Africa and is linked to early EBV infection and likely endemic malaria infection, while sporadic Burkitt lymphoma is found elsewhere.54 It has been a remarkably chemosensitive disease that, treated with aggressive chemotherapy, has a high rate of cure. Over 85% of patients have a complete remission after an aggressive chemotherapy regimen, such as the Magrath or modified Magrath regimen.55,56 Recent data with EPOCH chemotherapy suggest an even higher rate of complete remission. However, relapse does occur in a subset. Also, older patients who require dose reduction or those with suboptimal therapy due to resource availability suffer poorer outcomes.55 In this context, it is of interest that romidepsin and other HDIs may be particularly effective. These agents effect a dramatic reduction in c-myc expression at the RNA and protein level, and they cause cell death after a short exposure. While some may argue that there is no role for a new therapeutic, it should be noted that romidepsin may serve as a targeted therapy for the reduction of c-myc in this disease and provide activity in a single agent that could then be tolerated in older patients or utilized in a more resource-poor environment. These agents deserve a second look in this disease. It is furthermore of interest that Bak and Bax expression appear to be so important in resistance to romidepsin, in that Bak and Bax loss could serve as a biomarker of patients not expected to have a good response to single agent romidepsin and identify those patients for more aggressive therapy. This study raises the question of whether it is time to look at a targeted therapy for Burkitt lymphoma.
In summary, we show that apoptosis mediated by HDI treatment occurs via the intrinsic apoptotic pathway. Burkitt lymphoma is a disease with inherent sensitivity to HDIs and may offer a less toxic treatment alternative. Bax and concurrent Bak and Bax loss may result in decreased efficacy of HDIs and could be used as markers to screen patients whose disease would respond to therapy with HDIs.
Materials and Methods
Chemicals
Romidepsin was obtained from the Developmental Therapeutics Department, National Cancer Institute (NCI). MLN8237 (alisertib), VX-680 (tozasertib). ZM447439, vorinostat, belinostat and panobinostat were from Chemietek. Q-VD-OPh was purchased from R&D Systems.
Cell lines
Epstein-Barr virus-negative CA46, DG75, Ramos and ST486 Burkitt lymphoma cell lines were obtained from the American Type Culture Collection. HCT-116 cells were obtained from the NCI Anticancer Drug Screen. HCT-116 cells lacking Bax were a kind gift of Dr Bert Vogelstein.57 Cells lacking Bak or both Bak and Bax were obtained from the laboratory of Dr RJ Youle.40 All cell lines were maintained in RPMI-1640 (Mediatech, Inc) with 10% fetal calf serum and antibiotics.
Flow cytometry
The percentage of apoptotic cells was determined using fluorescein isothiocyanate-conjugated annexin V antibody (BioLegend) and propidium iodide. Cells were plated in 6-well dishes and treated with the desired concentrations of HDIs or AKIs for the noted times. Cells were then harvested and stained with annexin V antibody and propidium iodide in 1× binding buffer (Becton Dickinson). Two-color flow cytometry analysis was used to generate dot-plots, from which percent annexin-positive cells was determined using FloJo v 6.4.7 (Tree Star, Inc).
Immunoblot analysis
Treated cells were pelleted and washed once with cold phosphate-buffered saline. Cell pellets were subsequently resuspended in cold lysis buffer (40 mM Hepes pH 7.5, 120 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 0.5 mM EDTA, 1% Triton X-100) containing protease inhibitor (Sigma-Aldrich) and phosphatase inhibitors (50 mM NaF and PhosphoSTOP Phosphatase Inhibitor Cocktail Tablets, Roche), briefly sonicated and placed on ice for 15 min. Unlysed cells and nuclei were removed by centrifugation at 14000 RPM for 15 min at 4°C. Cell lysates (50 µg of protein) were loaded onto precast 4–12% (w/v) NuPAGE Novex Bis-Tris Mini gels (Invitrogen), subjected to electrophoresis, and electrotransferred onto nitrocellulose membranes (Millipore). Membranes were probed with antibodies against the following proteins: Bax, Bak, Bim, Bcl-XL, MCL-1, poly (ADP ribose) polymerase (PARP), and cleaved PARP (all from Cell Signaling Technology); acetylated histone H3 (Millipore) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (American Research Products). Proteins were visualized with the Odyssey System (LI-COR) using a 1:4000 dilution of the IRDye 800CW goat anti-mouse secondary antibody or IRDye 700CW goat anti-rabbit secondary antibody (LI-COR).
RT-PCR analysis
Total RNA was isolated using Trizol® reagent according to the manufacturer’s instructions (Invitrogen). Reverse transcription was performed on 1 µg total RNA. cDNA amplification was done using primers specific for ABCB1, MYC, BAX, and BAK1. Amplification of rRNA (rRNA) served as an internal control. Quantitative real-time PCR was done using TaqMan Master mix (Roche) in a LightCycler 480 Instrument (Roche). PCR amplification was done at 95 °C for 10 min followed by 45 cycles of 95 °C for 10 s and 60 °C for 10 s. Fluorescence signal was acquired at the end of the elongation step of every PCR cycle (72 °C for 1 s). PCR results were normalized by rRNA expression, and the fold changes were determined dividing expression values of the genes in the resistant cells by expression values of the genes in the parental cells. Primers used were rRNA forward 5′- TTACCCTACTGATGATGTGTTGTTG -3′, reverse 5′- CCTGCGGTTCCTCTCGTA -3′; ABCB1 forward 5′- CGGATGATCTTCTTCCAAGATTTC -3′, reverse 5′- TCCCCTTCAAGATCCATCC -3′; MYC forward 5′- CACCAGCAGCGACTCTGA -3′ reverse 5′- GATCCAGACTCTGACCTTTTGC -3′; BAX forward 5′- CCATCATGGGCTGGACAT -3′, reverse 5′- CACTCCCGCCACAAAGAT -3′; BAK1 forward 5′- CCTGCCCTCTGCTTCTGA -3, reverse 5′- CTGCTGATGGCGGTAAAAA -3′.
Patient samples
Anonymized samples of histologically documented Burkitt lymphoma were obtained from consultation cases sent to the National Cancer Institute Laboratory of Pathology. The Institutional Review Board of the National Cancer Institute approved this study.
Immunohistochemistry
Immunohistochemical studies were conducted on 5 µm formalin-fixed, paraffin-embedded cell line or patient sample sections using Bax and Bak or c-myc, rabbit monoclonal antibodies (Epitomics). For Bak and Bax, after deparaffinization and rehydration, the slides were retrieved using low pH Dako Target retrieval solution (Dako) in a pressure cooker, in a microwave (Hot start 6 min), followed by 15 min blocking with 5% Tris goat serum and incubation with primary antibodies at a concentration of 1:500, for 1 h. The I-View DAB detection kit (Ventana Medical Systems) was used. As positive control, human tonsil sections were run and showed appropriate staining patterns. Cells were considered positive if they showed a cytoplasmic granular staining for Bax and/or Bak. Reactivity for Bax and Bak in human Burkitt lymphoma cases was scored in quartiles (5–25%; 25–50%, 50–75%, > 75%). Samples were considered negative if less than 5% of cells were positive. For c-myc, an automated system was used for incubation and detection (Benchmark XR, Ventana Medical Systems) in combination with the XT UltraView DAB v3 with amplifier kit (Ventana). Slides were incubated for 2 h at 37 °C with primary antibody at a 1:50 dilution. Images were taken using an Olympus Bx50 microscope, at 40× magnification (objective Olympus plan 40×/0.65), with an Olympus DP71 camera (adaptors: U-TV1x-2 and Olympus U-CMAD3). The images were captured using Olympus DP controller software and imported through Adobe Photoshop CS4, Version 11.0.
Statistical analysis
The significance of differences in apoptosis induced by various drug treatments was determined by a two-tailed Student t test. Differences with a P value of < 0.05 were considered significant.
Acknowledgments
We thank Dr Bert Vogelstein for the use of the Bax-deficient HCT-116 cells and Dr RJ Youle for the HCT-116 cells that were deficient in Bak or both Bak and Bax. The authors also thank Drs Wyndham Wilson and Kieron Dunleavy for helpful discussion.
This work was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
This work was supported in part by a CRADA between the NCI and Celgene Corporation.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government.
Glossary
Abbreviations:
- HDI
histone deacetylase inhibitor
- AKI
Aurora kinase inhibitor
- MSI
microsatellite instability
Disclosure of Potential Conflicts of Interest
This work was supported in part by a CRADA between the NCI and Celgene Corporation.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25914
References
- 1.Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–15. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 2.Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, Zain J, Prince HM, Leonard JP, Geskin LJ, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410–7. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piekarz RL, Frye R, Prince HM, Kirschbaum MH, Zain J, Allen SL, Jaffe ES, Ling A, Turner M, Peer CJ, et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117:5827–34. doi: 10.1182/blood-2010-10-312603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero D, Borchmann P, Morschhauser F, Wilhelm M, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30:631–6. doi: 10.1200/JCO.2011.37.4223. [DOI] [PubMed] [Google Scholar]
- 5.Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485–91. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 6.Kirschbaum M, Frankel P, Popplewell L, Zain J, Delioukina M, Pullarkat V, Matsuoka D, Pulone B, Rotter AJ, Espinoza-Delgado I, et al. Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29:1198–203. doi: 10.1200/JCO.2010.32.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Younes A, Oki Y, Bociek RG, Kuruvilla J, Fanale M, Neelapu S, Copeland A, Buglio D, Galal A, Besterman J, et al. Mocetinostat for relapsed classical Hodgkin’s lymphoma: an open-label, single-arm, phase 2 trial. Lancet Oncol. 2011;12:1222–8. doi: 10.1016/S1470-2045(11)70265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G, Sharma S, et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res. 2006;12:4628–35. doi: 10.1158/1078-0432.CCR-06-0511. [DOI] [PubMed] [Google Scholar]
- 9.Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. 2012;4:505–24. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schrump DS. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: mechanisms and potential clinical implications. Clin Cancer Res. 2009;15:3947–57. doi: 10.1158/1078-0432.CCR-08-2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robey RW, Chakraborty AR, Basseville A, Luchenko V, Bahr J, Zhan Z, Bates SE. Histone deacetylase inhibitors: emerging mechanisms of resistance. Mol Pharm. 2011;8:2021–31. doi: 10.1021/mp200329f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakraborty AR, Robey RW, Luchenko VL, Zhan Z, Piekarz RL, Gillet JP, Kossenkov AV, Wilkerson J, Showe LC, Gottesman MM, et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: rationale to combine romidepsin with an MEK inhibitor. Blood. 2013;121:4115–25. doi: 10.1182/blood-2012-08-449140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Venkannagari S, Fiskus W, Peth K, Atadja P, Hidalgo M, Maitra A, Bhalla KN. Superior efficacy of co-treatment with dual PI3K/mTOR inhibitor NVP-BEZ235 and pan-histone deacetylase inhibitor against human pancreatic cancer. Oncotarget. 2012;3:1416–27. doi: 10.18632/oncotarget.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shao Y, Aplin AE. BH3-only protein silencing contributes to acquired resistance to PLX4720 in human melanoma. Cell Death Differ. 2012;19:2029–39. doi: 10.1038/cdd.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stauber RH, Knauer SK, Habtemichael N, Bier C, Unruhe B, Weisheit S, Spange S, Nonnenmacher F, Fetz V, Ginter T, et al. A combination of a ribonucleotide reductase inhibitor and histone deacetylase inhibitors downregulates EGFR and triggers BIM-dependent apoptosis in head and neck cancer. Oncotarget. 2012;3:31–43. doi: 10.18632/oncotarget.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aron JL, Parthun MR, Marcucci G, Kitada S, Mone AP, Davis ME, Shen T, Murphy T, Wickham J, Kanakry C, et al. Depsipeptide (FR901228) induces histone acetylation and inhibition of histone deacetylase in chronic lymphocytic leukemia cells concurrent with activation of caspase 8-mediated apoptosis and down-regulation of c-FLIP protein. Blood. 2003;102:652–8. doi: 10.1182/blood-2002-12-3794. [DOI] [PubMed] [Google Scholar]
- 17.Frew AJ, Lindemann RK, Martin BP, Clarke CJ, Sharkey J, Anthony DA, Banks KM, Haynes NM, Gangatirkar P, Stanley K, et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc Natl Acad Sci U S A. 2008;105:11317–22. doi: 10.1073/pnas.0801868105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vrana JA, Decker RH, Johnson CR, Wang Z, Jarvis WD, Richon VM, Ehinger M, Fisher PB, Grant S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene. 1999;18:7016–25. doi: 10.1038/sj.onc.1203176. [DOI] [PubMed] [Google Scholar]
- 19.Newbold A, Lindemann RK, Cluse LA, Whitecross KF, Dear AE, Johnstone RW. Characterisation of the novel apoptotic and therapeutic activities of the histone deacetylase inhibitor romidepsin. Mol Cancer Ther. 2008;7:1066–79. doi: 10.1158/1535-7163.MCT-07-2256. [DOI] [PubMed] [Google Scholar]
- 20.Sasakawa Y, Naoe Y, Inoue T, Sasakawa T, Matsuo M, Manda T, Mutoh S. Effects of FK228, a novel histone deacetylase inhibitor, on human lymphoma U-937 cells in vitro and in vivo. Biochem Pharmacol. 2002;64:1079–90. doi: 10.1016/S0006-2952(02)01261-3. [DOI] [PubMed] [Google Scholar]
- 21.Hartlapp I, Pallasch C, Weibert G, Kemkers A, Hummel M, Re D. Depsipeptide induces cell death in Hodgkin lymphoma-derived cell lines. Leuk Res. 2009;33:929–36. doi: 10.1016/j.leukres.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 22.Lindemann RK, Newbold A, Whitecross KF, Cluse LA, Frew AJ, Ellis L, Williams S, Wiegmans AP, Dear AE, Scott CL, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci U S A. 2007;104:8071–6. doi: 10.1073/pnas.0702294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piekarz RL, Robey RW, Zhan Z, Kayastha G, Sayah A, Abdeldaim AH, Torrico S, Bates SE. T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance. Blood. 2004;103:4636–43. doi: 10.1182/blood-2003-09-3068. [DOI] [PubMed] [Google Scholar]
- 24.Sander S, Rajewsky K. Burkitt lymphomagenesis linked to MYC plus PI3K in germinal center B cells. Oncotarget. 2012;3:1066–7. doi: 10.18632/oncotarget.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calado DP, Sasaki Y, Godinho SA, Pellerin A, Köchert K, Sleckman BP, de Alborán IM, Janz M, Rodig S, Rajewsky K. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol. 2012;13:1092–100. doi: 10.1038/ni.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucas DM, Alinari L, West DA, Davis ME, Edwards RB, Johnson AJ, Blum KA, Hofmeister CC, Freitas MA, Parthun MR, et al. The novel deacetylase inhibitor AR-42 demonstrates pre-clinical activity in B-cell malignancies in vitro and in vivo. PLoS One. 2010;5:e10941. doi: 10.1371/journal.pone.0010941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sculley TB, Buck M, Gabrielli B, Parsons PG, Krauer KG. A histone deacetylase inhibitor, azelaic bishydroxamic acid, shows cytotoxicity on Epstein-Barr virus transformed B-cell lines: a potential therapy for posttransplant lymphoproliferative disease. Transplantation. 2002;73:271–9. doi: 10.1097/00007890-200201270-00021. [DOI] [PubMed] [Google Scholar]
- 28.Wang R, Brunner T, Zhang L, Shi Y. Fungal metabolite FR901228 inhibits c-Myc and Fas ligand expression. Oncogene. 1998;17:1503–8. doi: 10.1038/sj.onc.1202059. [DOI] [PubMed] [Google Scholar]
- 29.Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, Brooks R, Piekarz RL, Tucker E, Figg WD, et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res. 2002;8:718–28. [PubMed] [Google Scholar]
- 30.Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen DM, Chen GA, Schrump DS. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst. 2002;94:504–13. doi: 10.1093/jnci/94.7.504. [DOI] [PubMed] [Google Scholar]
- 31.Xiao JJ, Huang Y, Dai Z, Sadée W, Chen J, Liu S, Marcucci G, Byrd J, Covey JM, Wright J, et al. Chemoresistance to depsipeptide FK228 [(E)-(1S,4S,10S,21R)-7-[(Z)-ethylidene]-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8,7,6]-tricos-16-ene-3,6,9,22-pentanone] is mediated by reversible MDR1 induction in human cancer cell lines. J Pharmacol Exp Ther. 2005;314:467–75. doi: 10.1124/jpet.105.083956. [DOI] [PubMed] [Google Scholar]
- 32.Robey RW, Zhan Z, Piekarz RL, Kayastha GL, Fojo T, Bates SE. Increased MDR1 expression in normal and malignant peripheral blood mononuclear cells obtained from patients receiving depsipeptide (FR901228, FK228, NSC630176) Clin Cancer Res. 2006;12:1547–55. doi: 10.1158/1078-0432.CCR-05-1423. [DOI] [PubMed] [Google Scholar]
- 33.Ueda H, Nakajima H, Hori Y, Goto T, Okuhara M. Action of FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum no. 968, on Ha-ras transformed NIH3T3 cells. Biosci Biotechnol Biochem. 1994;58:1579–83. doi: 10.1271/bbb.58.1579. [DOI] [PubMed] [Google Scholar]
- 34.Mori N, Ishikawa C, Senba M, Kimura M, Okano Y. Effects of AZD1152, a selective Aurora B kinase inhibitor, on Burkitt’s and Hodgkin’s lymphomas. Biochem Pharmacol. 2011;81:1106–15. doi: 10.1016/j.bcp.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 35.Ikezoe T, Takeuchi T, Yang J, Adachi Y, Nishioka C, Furihata M, Koeffler HP, Yokoyama A. Analysis of Aurora B kinase in non-Hodgkin lymphoma. Lab Invest. 2009;89:1364–73. doi: 10.1038/labinvest.2009.106. [DOI] [PubMed] [Google Scholar]
- 36.Shao W, Growney JD, Feng Y, O’Connor G, Pu M, Zhu W, Yao YM, Kwon P, Fawell S, Atadja P. Activity of deacetylase inhibitor panobinostat (LBH589) in cutaneous T-cell lymphoma models: Defining molecular mechanisms of resistance. Int J Cancer. 2010;127:2199–208. doi: 10.1002/ijc.25218. [DOI] [PubMed] [Google Scholar]
- 37.Gutiérrez MI, Cherney B, Hussain A, Mostowski H, Tosato G, Magrath I, Bhatia K. Bax is frequently compromised in Burkitt’s lymphomas with irreversible resistance to Fas-induced apoptosis. Cancer Res. 1999;59:696–703. [PubMed] [Google Scholar]
- 38.Li M, Jung A, Ganswindt U, Marini P, Friedl A, Daniel PT, Lauber K, Jendrossek V, Belka C. Aurora kinase inhibitor ZM447439 induces apoptosis via mitochondrial pathways. Biochem Pharmacol. 2010;79:122–9. doi: 10.1016/j.bcp.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 39.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C, Youle RJ. Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1's inhibitory effect on Bak. Oncogene. 2012;31:3177–89. doi: 10.1038/onc.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–9. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 42.Ouyang H, Furukawa T, Abe T, Kato Y, Horii A. The BAX gene, the promoter of apoptosis, is mutated in genetically unstable cancers of the colorectum, stomach, and endometrium. Clin Cancer Res. 1998;4:1071–4. [PubMed] [Google Scholar]
- 43.Percesepe A, Kristo P, Aaltonen LA, Ponz de Leon M, de la Chapelle A, Peltomäki P. Mismatch repair genes and mononucleotide tracts as mutation targets in colorectal tumors with different degrees of microsatellite instability. Oncogene. 1998;17:157–63. doi: 10.1038/sj.onc.1201944. [DOI] [PubMed] [Google Scholar]
- 44.Vassileva V, Millar A, Briollais L, Chapman W, Bapat B. Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability. Cancer Res. 2002;62:4095–9. [PubMed] [Google Scholar]
- 45.Brimmell M, Mendiola R, Mangion J, Packham G. BAX frameshift mutations in cell lines derived from human haemopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene. 1998;16:1803–12. doi: 10.1038/sj.onc.1201704. [DOI] [PubMed] [Google Scholar]
- 46.Corso G, Pedrazzani C, Marrelli D, Pascale V, Pinto E, Roviello F. Correlation of microsatellite instability at multiple loci with long-term survival in advanced gastric carcinoma. Arch Surg. 2009;144:722–7. doi: 10.1001/archsurg.2009.42. [DOI] [PubMed] [Google Scholar]
- 47.Pino MS, Chung DC. Microsatellite instability in the management of colorectal cancer. Expert Rev Gastroenterol Hepatol. 2011;5:385–99. doi: 10.1586/egh.11.25. [DOI] [PubMed] [Google Scholar]
- 48.Ionov Y, Yamamoto H, Krajewski S, Reed JC, Perucho M. Mutational inactivation of the proapoptotic gene BAX confers selective advantage during tumor clonal evolution. Proc Natl Acad Sci U S A. 2000;97:10872–7. doi: 10.1073/pnas.190210897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krajewski S, Blomqvist C, Franssila K, Krajewska M, Wasenius VM, Niskanen E, Nordling S, Reed JC. Reduced expression of proapoptotic gene BAX is associated with poor response rates to combination chemotherapy and shorter survival in women with metastatic breast adenocarcinoma. Cancer Res. 1995;55:4471–8. [PubMed] [Google Scholar]
- 50.Jeong SH, Han JH, Kim JH, Ahn MS, Hwang YH, Lee HW, Kang SY, Park JS, Choi JH, Lee KJ, et al. Bax predicts outcome in gastric cancer patients treated with 5-fluorouracil, leucovorin, and oxaliplatin palliative chemotherapy. Dig Dis Sci. 2011;56:131–8. doi: 10.1007/s10620-010-1280-8. [DOI] [PubMed] [Google Scholar]
- 51.Kang SY, Han JH, Lee KJ, Choi JH, Park JI, Kim HI, Lee HW, Jang JH, Park JS, Kim HC, et al. Low expression of Bax predicts poor prognosis in patients with locally advanced esophageal cancer treated with definitive chemoradiotherapy. Clin Cancer Res. 2007;13:4146–53. doi: 10.1158/1078-0432.CCR-06-3063. [DOI] [PubMed] [Google Scholar]
- 52.Hussain SA, Ganesan R, Hiller L, Murray PG, el-Magraby MM, Young L, James ND. Proapoptotic genes BAX and CD40L are predictors of survival in transitional cell carcinoma of the bladder. Br J Cancer. 2003;88:586–92. doi: 10.1038/sj.bjc.6600765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ong YL, McMullin MF, Bailie KE, Lappin TR, Jones FG, Irvine AE. High bax expression is a good prognostic indicator in acute myeloid leukaemia. Br J Haematol. 2000;111:182–9. doi: 10.1046/j.1365-2141.2000.02315.x. [DOI] [PubMed] [Google Scholar]
- 54.Magrath I. Epidemiology: clues to the pathogenesis of Burkitt lymphoma. Br J Haematol. 2012;156:744–56. doi: 10.1111/j.1365-2141.2011.09013.x. [DOI] [PubMed] [Google Scholar]
- 55.Magrath I. Towards curative therapy in burkitt lymphoma: the role of early african studies in demonstrating the value of combination therapy and CNS prophylaxis. Adv Hematol. 2012;2012:130680. doi: 10.1155/2012/130680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lacasce A, Howard O, Lib S, Fisher D, Weng A, Neuberg D, Shipp M. Modified magrath regimens for adults with Burkitt and Burkitt-like lymphomas: preserved efficacy with decreased toxicity. Leuk Lymphoma. 2004;45:761–7. doi: 10.1080/1042819031000141301. [DOI] [PubMed] [Google Scholar]
- 57.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–92. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]