

Abstract
Tardive dyskinesia (TD) is a serious, often disabling, movement disorder that is caused by medications that block dopamine receptors (i.e., neuroleptics, anti-emetics). There is currently no standard treatment approach for physicians confronted with such patients. This may be the result of notions that TD is disappearing because of the switch to second-generation antipsychotic agents and that it is largely reversible. In this article we demonstrate that second-generation antipsychotics do, indeed, cause TD and, in fact, the frequency is likely higher than expected because of growing off-label uses and a tripling of prescriptions written in the last 10 years. In addition, studies demonstrate that TD actually remits in only a minority of patients when these drugs are withdrawn. Furthermore, neuroleptic agents are often utilized to treat TD, despite prolonged exposure being a risk factor for irreversibility. The outcome of these trends is a growing population afflicted with TD. We review non-neuroleptic agents that have shown positive results in small, early-phase, blinded trials, including tetrabenazine, amantadine, levetiracetam, piracetam, clonazepam, propranolol, vitamin B6, and Ginkgo biloba. Other options, such as botulinum toxin and deep brain stimulation, will also be discussed, and a suggested treatment algorithm is provided. While these agents are reasonable treatment options at this time there is a need, with a concerted effort between neurology and psychiatry, for full-scale drug development, including multicenter, randomized, blinded trials to confirm the effectiveness of the agents that were positive in phase 2 trials and the development of newer ones.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-013-0222-5) contains supplementary material, which is available to authorized users.
Keywords: Tardive dyskinesia, Prevalence, Reversibility, Tetrabenazine, Amantadine, Levetiracetam
Introduction
Tardive dyskinesia (TD) is a serious, often disabling, movement disorder caused by all medications that block dopamine receptors, including clozapine [1–4]. TD not only occurs in the psychiatrically ill, but also in nonpsychiatric patients exposed to antipsychotics or other dopamine receptor blocking agents such as metoclopramide. As a result, it has been the subject of intense medico-legal scrutiny [5]. While there have been numerous attempts to review varied agents in the treatment of TD [1, 6], until very recently there were no evidence-based guidelines for physicians confronted with this disorder. In 2013, the American Academy of Neurology published the first evidence-based guideline on the treatment of tardive syndromes, a much-needed addition to the literature on this subject [7].
The term “tardive” dyskinesia was coined in 1964 by Faurbye et al. [8] based upon the observation that some patients develop dyskinetic movements as a late complication of neuroleptic exposure. Diagnostic and Statistical Manual of Mental Disorders V criteria suggest 3 months of exposure is needed except in the elderly, where 1 month suffices. However, it is well known that this disorder may appear relatively early in the course of treatment, even after just a few doses [9]. Classical TD involves oral–buccal–lingual masticatory movements [10] characterized by repetitive jaw, face and lingual movements (commonly protrusion), and may involve puffing of the cheeks, lip-smacking, puckering, and pursing [10]. Severe cases can be associated with difficulty speaking, chewing and swallowing, and tongue mutilation. TD movements can also result in limb chorea, trunk movements as rhythmic rocking or thrusting motions (referred to as pelvic dyskinesia when pelvic muscles are involved), and respiratory dyskinesia with altered rhythmical patterns leading to hyper- and hypoventilation that can be life threatening [11]. Other subtypes, occurring alone or combined, include tardive dystonia, akathisia, tics, tremor, and myoclonus [6, 12]. The pathophysiology of TD remains elusive, though multiple hypotheses have been proposed (summarized in Table 1). Other than neuroleptic exposure, risk factors for developing TD include older age [13, 14], African–American ethnicity [15, 16], the presence of diabetes mellitus [17, 18], and the occurrence of acute dystonic reactions or drug-indiced parkinsonism [19, 20]. Female sex, prior brain damage, cocaine abuse, alcohol consumption, and a diagnosis of bipolar disorder are less consistently correlated with TD [2]. Treatment-related risk factors include longer exposure and high doses of the offending agents [19, 21].
Table 1.
| Proposed mechanism | Support |
|---|---|
| D2 receptor up-regulation with subsequent hypersensitivity | Rodent models demonstrate universal, reversible D2 receptor up-regulation after exposure to dopamine receptor blockers (DRBs); findings not supported by human studies using imaging and immunohistochemistry |
| GABA insufficiency | GAD and GABA levels are diminished in the substantia nigra and external pallidum in animal models and in the spinal fluid of humans with TD; levels correlate with vacuous chewing movements (VCMs); findings not consistently replicated; human studies using GABA agonists have been disappointing |
| Increased opioids (encephalin and dynorphin) | Increased mRNA levels reported following exposure to DRBs; administration of opioid receptor antagonists into basal ganglia of rodents attenuates VCMs |
| Glutamate and excitotoxicity | Spinal fluid of humans with TD shows higher levels of N-acetylaspartate, N-acetylaspartylglutamate, and aspartate than controls; levels correlate with TD severity |
| Oxidative stress and cell injury | Neuroleptics (FGA > SGA) may have a direct toxic effect by inhibiting complex I of the electron transport chain; increased reactive oxygen species seen in rats exposed to haloperidol; reduced erythrocyte copper, zinc-superoxide dismutase activity in humans with TD |
| Genetic susceptibility | Family studies demonstrate concordance for TD among first-degree relatives; multiple associations of candidate gene polymorphisms have been identified |
GABA = gamma aminobutyric acid; GAD = glutamic acid decarboxylase; TD = tardive dyskinesia; mRNA = messenger RNA; FGA = first-generation antipsychotics; SGA = second-generation (atypical) antipsychotics.
Initially, physicians were reluctant to accept TD as a drug-related disorder based on the fact that movements similar to TD occur in untreated psychotic patients and elderly individuals and the odd clinical course of worsening with drug withdrawal [2, 22, 23]. It took nearly 2 decades before TD was universally accepted as an iatrogenic disorder and before the first therapeutic trials were reported [24, 25]. We believe that the development of meaningful therapies has been further delayed by the notions that second-generation agents have a lower propensity to induce TD potentially leading to a decrease in its perceived importance and a belief that it is on the verge of disappearing; that TD is generally reversible in a majority of patients; and the idea that use of neuroleptics is an appropriate treatment strategy for TD, even in nonpsychiatric patients. In this article we will address each of these flawed views to demonstrate that TD is actually a growing problem and follow up with a discussion on potentially effective therapeutic options.
Is TD Disappearing?
It has been generally believed that the frequency of TD is decreasing based on the pharmacology of the second-generation (atypical) antipsychotics (SGA). Certainly, an examination of the number of publications on the subject (which are shrinking) would support this perception [26]. Available incidence studies relate primarily to psychiatric agents and psychotic populations, while the incidence and prevalence of TD in nonpsychiatric patients is unknown, even for drugs used as commonly as the gastrointestinal medication metoclopramide, where the estimates for frequency range from <1 % to nearly 30 % [10]. The figures for such cases are generally not included in frequency estimates, but they are important considering more than 7 million prescriptions are written for this drug annually [27].
In a review of 56 studies conducted between 1959 and 1979, Kane et al. [28] reported an average point prevalence for TD development in neuroleptic exposed patients of 20 % (range 0.5–65.0 %). Subtracting the estimated frequency of spontaneous dyskinesia (5 %) they concluded that the true prevalence was probably around 15 % [28]. Another review of 76 studies conducted through 1989 indicated that the overall TD prevalence was closer to 24 % (range 3.3–62.0 %) [29]. A prospective examination in 908 psychiatric patients with a median 7-month exposure to neuroleptics showed an annual incidence estimate for TD of 5 % [30], and the impression that, at least for the first 4–5 years of exposure, the cumulative incidence of TD increases linearly with duration of exposure. In 1993, Glazer et al. [31] presented long-term risk estimates for TD in a prospective cohort study of 362 psychiatric outpatients who did not have TD at baseline and were maintained on neuroleptics. They also estimated the risk of TD to be 5 % per year and long-term risks of 25 % after 5 years, 49 % after 10 years, and 68 % after 25 years. These figures have been confirmed by others [19, 32]. In the elderly, the incidence rates are substantially higher: more than 25 % per year in 1 study [19] and 12 % in a prospective study of 266 outpatients over the age of 45 years [14].
Clozapine, the first SGA, became available in the USA in 1989 after the Food and Drug Administration approved its use for treatment-resistant schizophrenia [33]. Throughout the 1990s and early 2000s, 10 additional SGAs became available and largely replaced the first-generation agents for the treatment of psychosis. The SGAs have also been used extensively (through off-label prescribing) as mood stabilizers, adjunctive agents in the treatment of refractory depression, anti-anxiety agents, treatment for agitation in dementia and disruptive behavior in young patients, and in other conditions [34, 35]. Based upon preclinical data, the SGAs were expected to carry a much smaller risk of extrapyramidal side effects (including TD) than the first-generation agents, thereby giving rise to the hope that TD would, ultimately, disappear.
However, prevalence rates in studies utilizing SGAs were, in fact, similar to those seen with older agents [2]. A recent comparative study provided prevalence rates of 13 % among patients on SGAs and 32 % among patients on first-generation agents (p = 0.0001), indicating that TD is less common but continues to be a problem [36]. However, in another study of a long-term stay hospital where patients were assessed from 2003 to 2007 the frequency of TD was 28 % [37]. Incidence rates have been similar with first- and second-generation agents. In a recent review of 12 trials published between 2004 and 2008, the annual incidence of TD was found to be 3.9 % for SGAs and 5.5 % for first-generation drugs [36]. Several other studies have found no change in the incidence at all [38–40].
Some of the apparent inconsistencies between studies may result from the use of different drugs and doses. Clozapine is the prototypical atypical agent with a lower frequency of acute drug-induced movement disorders [2]. It is clear that clozapine can cause TD [3, 4], but while it is suspected that the frequency is the lowest among all SGAs [41], the actual incidence remains unknown as this drug is not approved as first-line treatment, which means all patients exposed to clozapine have already been exposed to other neuroleptic agents. This is not the case for other SGAs that clearly have a higher predilection for all drug-induced movement disorders than clozapine. There are several problems, with many of the incidence studies involving SGAs showing dramatic decreases and others showing similar rates including the use of relatively short-term study durations and hence drug exposures, comparisons with high-dose haloperidol treatment as the “gold standard”, and the fact that most patients were previously exposed to, or continue to be treated with, first-generation agents [2, 42].
While the use of SGAs may be associated with slightly lower incidence and prevalence rates, TD as an entity is clearly not disappearing. In fact, given the increasing off-label use of many of the offending agents because of perceived safety, resulting in more than 54 million antipsychotic prescriptions written in 2011, and >3-fold increase over 10 years [34] (see also the Agency of Healthcare Research and Quality; Statistical Brief 275), the overall number of patients affected with TD is on the rise. Such figures suggest, conservatively, nearly 5 million people exposed and 700,000 developing TD (not including those exposed to anti-emetics).
Is TD Reversible?
Studies of the natural history of TD have reported widely variable remission rates (1–62 % [43–46]) (Table 2) depending on patient age, psychiatric diagnosis, course of the psychiatric disorder, and duration of therapy [21, 46]. The studies examining reversibility have been conducted mostly in schizophrenic patients who continued to receive neuroleptics, while few studies have examined remission of TD after complete discontinuation of the offending agents [46, 47]. For instance, in the study reporting 62 % resolution [45], and in other studies [48], patients were still treated with neuroleptic agents, and TD remission was associated with worsening parkinsonism. It is possible that the neuroleptic treatment partially masked ongoing TD [1]. The most notable attempt to examine the course of TD in cases where neuroleptics were removed [46] was a prospective study of 49 psychiatric patients in whom 36 could actually stop the drugs. In this study only 2 % had complete resolution and 20 % showed improvement. In our retrospective cohort study of 108 TD patients in whom the inciting medications were completely withdrawn [49] (mean age 59 years, 69 % women, 94 % nonschizophrenic diagnosis), the mean duration of antipsychotic exposure was 4.8 years. Of these patients, 42 % were exposed to anti-emetic drugs (87 % metoclopramide), 36 % SGAs, and 7 % first-generation neuroleptics. TD completely resolved in only 13 % of these patients 3 years after discontinuing the inciting drug, with only 2 % of the total cohort resolving without the addition of therapeutic agents. The rest were treated with variable agents.
Table 2.
Summary of studies examining reversibility in tardive dyskinesia
| Author, year [ref.] | Patients (n) | Diagnosis | Duration of follow-up | Prospective or retrospective | Continued dopamine antagonists? | % Remission |
|---|---|---|---|---|---|---|
| Bergen et al., 1989 [43] | 101 | All psychiatric 90 % schizophrenia |
4 y | Prospective | Yes in 90 % | 1 % |
| Casey, 1985 [44] | 27 | All psychiatric | 5 y | Prospective | 63 % continued 37 % discontinued |
30 % |
| Fernandez et al., 2001 [45] | 53 | All schizophrenia | 14 y | Prospective | Yes | 62 % |
| Modestin et al., 2008 [48] | 83 | All psychiatric 69 % schizophrenia or schizoaffective |
8 y | Prospective | Yes in 90 % | 47 % decrease in prevalence; Of 8 patients off neuroleptics 1 remitted |
| Glazer et al., 1990 [46] | 36 | All psychiatric | 40 wks | Prospective | No | 2 % remission 20 % improvement |
| Zutshi et al., 2012 [49] | 108 | 44 % psychiatric 56 % nonpsychiatric |
2–3 y | Retrospective | No | 2 % remission with no symptomatic treatment 13 % remission overall |
Should Neuroleptics be Used as a Therapeutic Agent for TD?
Increased age and duration of exposure of neuroleptics are associated with irreversibility of TD [50–53]. It would then be prudent, especially in the elderly, to limit exposure to these drugs. Intuitively, the best first step in treating TD, once it has occurred, is to remove the dopamine-blocking agents (if this is possible) [54]. However, there is insufficient evidence that this leads to improvement [7]. In addition, such withdrawal of neuroleptics may worsen TD, at least initially [55–58] contributing to the belief (and, in turn, the practice) that neuroleptics can be used as symptomatic treatment for TD. The suppressive effects of first-generation neuroleptics on TD can be substantial (>60 % improvement), but several studies have suggested that this is only a temporary effect [25, 59], triggering a spiraling of the dose upwards over time. The SGAs quetiapine, clozapine, risperidone, and olanzapine [60–64] have also been used to treat TD with similarly unknown efficacy and safety in the long term [21]. Other long-term studies do not support the ability of SGAs to suppress TD [46]. From these data, it can be concluded that neuroleptics should not be used as therapeutic agents to treat TD. In psychiatric patients in whom neuroleptics cannot be withdrawn, switching from first-generation antipsychotics to SGAs or to lower-potency drugs may be helpful, though there is insufficient evidence to support the use of one SGA over the others [7]. Intuitively, however, the SGAs quetiapine and clozapine, which have low occupancy and fast dissociation from dopamine D2 receptors, would appear to be the best candidates.
Potential Directions in Therapy
Several articles have attempted to review all agents examined for the treatment of TD [1, 21, 65]. Some of these drugs demonstrated efficacy (for classical TD) in small, blinded, randomized trials, and these agents represent reasonable initial treatment targets not only for patients currently, but for the development of larger controlled trials that are needed to better define treatment approaches. They include tetrabenazine, amantadine, levetiracetam, piracetam, clonazepam, propranolol, vitamin B6, and Ginkgo biloba (Table 3). They are the primary subject of this section. A suggested treatment algorithm is featured in Fig. 1. Other tardive syndromes (aside from classical TD) may require different treatment approaches, though this requires further research.
Table 3.
Summary of the results of blinded trials in drugs that represent appropriate targets as non-neuroleptic treatment for tardive dyskinesia
| Drug | Authors, year [ref.] | Number of patients (n) and type | Design | Primary outcome | Results |
|---|---|---|---|---|---|
| Tetrabenazine | Kazamatsuri et al., 1972 [24, 25] | 24 psychotic hospitalized | 12-week double-blind, placebo- controlled crossover | Frequency of movements in 3 mins | 60 % reduction |
| Tetrabenazine | Ondo et al., 1999 [67] | 19 total 12 psychotic 7 gastrointestinal patients |
20-week open treatment | Blinded video AIMS score change | 54 % improvement |
| Amantadine | Angus et al., 1997 [81] | 16 psychotic hospitalized | 17-week double-blind, placebo- controlled crossover | AIMS change | 15 % improvement |
| Amantadine | Pappa et al., 2010 [82] | 22 schizophrenia outpatients | 5-week double-blind, placebo- controlled crossover | AIMS change Responders ≥ 20 % improvement |
22 % improvement 45 % responders |
| Levetiracetam | Woods et al., 2008 [83] | 50 schizophrenia outpatients |
12-week double-blind, placebo-controlled, parallel group | AIMS change | 25 % improvement |
| Piracetam | Libov et al., 2007 [90] | 40 schizophrenia and schizoaffective patients | 9-week double-blind placebo-controlled crossover | ESRS change | Significant reduction |
| Clonazepam | Thaker et al., 1990 [91] | 19 psychiatric outpatients | 12-week double-blind, placebo-controlled crossover | Blinded video Maryland Psychiatric Research Center Movement Disorder Scale change | 37 % improvement |
| Propranolol | Schrodt et al., 1982 [95] | 4 psychiatric outpatients |
20-week double-blind, crossover, alternating periods of active drug or placebo | Blinded video AIMS score change | Long-term improvement in 2 patients |
| Vitamin B6 | Lerner et al., 2007 [99] | 50 schizophrenic or schizoaffective inpatients | 26-week double-blind placebo-controlled crossover | ESRS change | Significant reduction |
| Ginkgo biloba | Zhang et al., 2011 [100] | 157 schizophrenia outpatients |
13-week double-blind, placebo-controlled, parallel group | AIMS change Responders ≥ 30 % improvement |
31 % improvement 51 % responders |
AIMS = Abnormal Involuntary Movement Scale; ESRS = Extrapyramidal Symptom Rating Scale
Fig. 1.
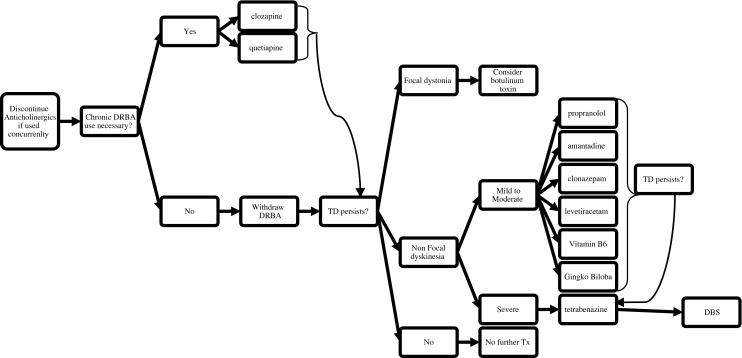
Treatment algorithm for tardive dyskinesia (TD) (adapted with permission from Fig. 21.1 in [10])
For many movement disorder specialists, the dopamine-depleting agent vesicular monoamine transporter 2 inhibitor (VMAT2 inhibitor) tetrabenazine is the current treatment of choice for moderate-to-severe forms of TD [6], especially since its 2008 Food and Drug Administration approval for use in Huntington’s disease [66]. However, to date, only 2 small, blinded trials in TD have been reported. One study, from 1972, examined 24 hospitalized psychotic patients [24]. There was a 4-week placebo run-in period during which antipsychotics were withdrawn, followed by a 14-week tetrabenazine treatment period (up to 150 mg/day) and a second placebo period lasting 2 weeks. TD disappeared in 8 patients, was markedly reduced in 6, and slightly decreased or unchanged in 6. Comparison with placebo showed a reduction in the frequency of TD movements by 60 %. The second study, from 1999, involved 19 TD patients who were treated for approximately 20 weeks in an open-label study with up to 150 mg of tetrabenazine [67]. Mean scores on the blinded videotaped Abnormal Involuntary Movement Scale (AIMS) assessment improved by 54 % (p < 0.001). Eleven patients rated themselves as markedly improved, 6 as moderately improved, and 2 as mildly improved. The results are supported by larger open and retrospective experiences [68, 69]. Usefulness of this drug is sometimes limited, however, by the potential for serious adverse effects, most prominently drowsiness, parkinsonism, akathisia, and depression with suicidal ideation, particularly in the elderly [68], and further work is needed before it can be recommended for routine use in TD [70]. This is level C evidence [7]. Similar results have been reported in small, blinded trials with reserpine—another dopamine-depleting agent—in the early 1980s [71, 72]. However, this drug is rarely utilized because it causes both peripheral and central adverse effects, and the depression can be protracted [73]. It is suggested that the evidence is insufficient [7].
Amantadine as a treatment for TD came about because of early open-label reports of effectiveness in TD [74–76] and levodopa-induced dyskinesia [77–79]. The mechanism of action is unclear, but has been postulated to relate to N-methyl-D-aspartate receptor-blocking properties of this drug [80]. Two small, blinded trials have been reported. One, from 1997, demonstrated improvement in 16 chronically ill psychiatric patients who were hospitalized long term [81]. After a 2-week baseline period, a 7-week placebo-controlled, randomized period was started followed by a 1-week washout and a 7-week crossover period. The primary outcome variable was the median AIMS score during the 3 weeks when they received 300 mg/day (vs placebo). The study demonstrated a 15 % reduction in the overall AIMS score (p = 0.05) and 10 of the 16 patients improved by ≥20%. A second trial, reported in 2010 [82], involved 22 outpatients with schizophrenia and TD who received amantadine (400 mg/day) or matching placebo in a double-blind, randomized trial for 2 weeks followed by a 4-day washout and a 2-week crossover phase. The primary outcome was the change in AIMS score at the end of each treatment phase. Results demonstrated a statistically significantly reduced average total AIMS score (from 13.5 before treatment to 10.5 after treatment) and reduced severity AIMS (from 2.04 before treatment to 1.54 after treatment). The overall change was 22 %, with 10 patients improving by >20 %. Little or no placebo effect was seen in these 2 trials. There was no worsening of psychotic symptoms. This is also level B evidence [7].
Levetiracetam is an anti-epileptic that has also been shown to improve TD [83]. The mechanism of action is unclear, but may relate to N-type calcium channel blockade [84] or to its binding to synaptic vesicle protein 2A, which may play a role in stability of synaptic vesicles containing neurotransmitters such as dopamine. The drug has been shown to improve TD in open studies [85–87] and levodopa-induced dyskinesia in Parkinson’s disease [88]. This study was a double-blind, placebo-controlled, randomized, 12-week, parallel group study. Severity of TD was measured with AIMS. Fifty patients were enrolled—25 randomized to each group. At 12 weeks the active group (mean dose 1900 mg/day) improved 44 % in the AIMS score, while the placebo group improved 19 % (p = 0.02). Three adverse effects occurred in the active group and were seen in 2 patients each, including ataxia, irritability, and decreased appetite [83].
Piracetam, a chemically similar drug to levetiracetam used predominantly for myoclonus in Europe and Asia, was initially shown to improve TD symptoms more than 30 years ago [89]. More recently, it has been shown to be effective in a randomized, double-blind, placebo-controlled crossover study [90]. In this study, 40 patients with schizophrenia or schizoaffective disorder and TD were given an oral formulation while being maintained on their usual antipsychotic regimen. After being randomized to 4 weeks of placebo or piracetam (4800 mg/day), a 1-week washout occurred prior to entering the 4-week crossover phase. The primary outcome measure was change in score on the Extrapyramidal Symptom Rating Scale (ESRS). The mean decrease in the TD subscale was 3 points in the piracetam group in contrast to deterioration of condition in the placebo group by −0.2 points (p = 0.003).
The use of clonazepam as a treatment for TD was explored in the 1970s and 1980s with a blinded trial ultimately published in 1990 [91]. Interest was based on its indirect gamma aminobutyric acidA agonist effect [91]. This controlled trial was a randomized, double-blind, placebo-controlled crossover study that included 19 psychiatric outpatients. Patients went through a 1-week placebo run-in, followed by 4 weeks of randomized therapy, a 2-week washout, a 4-week crossover therapy phase followed by a 1-week placebo washout. Results demonstrated a 37 % reduction in Maryland Psychiatric Research Center Movement Disorder Scale dyskinesia score with reversal on placebo. Mild-to-moderate sedation was reported in 6 patients and ataxia in 3. The antidyskinetic effect was seen at 2–3.5 mg per day. In 5 patients followed long term, the effectiveness waned after 5–8 months. This is level B evidence [7].
Propranolol was examined as a treatment of TD many years ago, but its use was recently revisited [92]. The reports, involving 31 patients, have been mostly open-label single cases or case series with a single double-blind, crossover study design [93–95]. Many of the open-label experiences reported rapid often dramatic response (including complete reversal) to low doses of the drug. The 1 controlled study was a double-blind, intensive case design and utilized 10 times the dose of the open trials (up to 800 mg/day) for unclear reasons. The patients were examined weekly up to 20 weeks with the AIMS scale and videotaped. Patients were treated with alternating periods of active drug or placebo. The study showed variable response in the blinded portion of the study, but significant long-term improvement in an open-label follow up [95]. Interest in propranolol waned when it was found that it increased neuroleptic plasma levels, possibly leading to suppression of symptoms [96, 97]. However, it is noteworthy that many of the responsive patients were, in fact, not treated with neuroleptics at the time [92].
Vitamin B6 is an antioxidant that showed promise for treating TD in early case series and a small, blinded trial [98]. A more recent, larger, randomized, double-blind, placebo-controlled trial also supports its efficacy [99]. In this 26-week study, 50 inpatients with schizophrenia or schizoaffective disorder and TD were randomly assigned to 1200 mg of vitamin B6 or placebo. After 12 weeks of treatment and a 2-week washout, they entered the 12-week crossover phase. The primary outcome was change in ESRS. The mean decrease in ESRS clinical global impression scores from baseline to endpoint was 2.4 points in patients treated with vitamin B6 and 0.2 points in patients treated with placebo (p < 0.0001). The mean decrease in the parkinsonism subscale score was 18.5 points and 1.4 points, respectively (p < 0.00001), and the mean decrease in the dyskinesia subscale score was 5.2 points and −0.8 points, respectively (p < 0.0001). Evidence for this treatment is considered insufficient (level U) [7].
Finally, extract of Ginkgo biloba (EGb-761), a potent antioxidant, was recently examined as a treatment of TD in 157 Chinese inpatients with schizophrenia [100]. This was a single site (Veterans Affairs Hospital), double-blind, placebo-controlled, parallel group trial of 240 mg/day of EGb-761. All patients were male and under age 60 years with TD present for at least 1 year. Patients went through a 1-week single-blind placebo run-in where any patients with a 25 % placebo effect were removed from randomization. The rest were treated for 12 weeks with placebo or EGb at a 1:1 ratio. The primary outcome measures were AIMS total score and the proportion of patients with >30 % improvement (responders). One hundred and fifty-two patients (97 %) completed the trial. AIMS scores decreased significantly in patients with TD receiving EGb-761 treatment relative to the placebo group (2.13 ± 1.75 vs −0.10 ± 1.69; p < 0.0001). In the active group 51 % responded, while only 5 % in the placebo group responded (p < 0.001). No worsening of psychopathology or cognition was seen. There were no significant adverse effects. This is considered level B evidence [7].
Many other medications have been examined in open trials. For these, the data are inadequate, inconclusive, and/or negative and not worthy of further investigation. These agents are listed in Table 4. For completeness, it is worth mentioning that a few small, open-label trials support the use of branched-chain amino acids (valine, isoleucine, and leucine) in treating TD [101–103]; however, these positive results should be interpreted with caution given the lack of blinded studies. Query of clinicaltrials.gov revealed only 3 additional TD treatments currently under investigation. They are pyridoxal 5’-phosphate, D-serine, and NBI-98854 (a novel VMAT2 inhibitor).
Table 4.
Summary of therapeutic agents studied in tardive dyskinesia with inadequate, inconclusive, and/or negative results [1, 10, 21]
| Cholinergic agents | Deanol,* lecithin,* and meclofenoxate* |
| Cholinesterase inhibitors | Galantamine and donepezil |
| Catecholaminergic agents | Celiprolol* and tiapride |
| Gamma aminobutyric acid agonists | Baclofen,* valproate,* and progabide |
| N-methyl-D-aspartate inhibitors | Memantine and talampanel |
| Dopamine agonists | Bromocriptine* |
| Alpha antagonists | Clonidine* |
| Antioxidants | Vitamin E,* eicosapentaenoic acid,* and melatonin* |
| Calcium channel blockers | Nifedipine and diltiazem |
| Miscellaneous agents | Buspirone, lithium, and gamma-linoleic acid, zolpidem |
*Negative trial results
Alternatives to medical therapy for TD include botulinum toxin injections and deep brain stimulation (DBS). There are multiple reports of improvement after botulinum toxin injections, particularly in cases where severe lingual movements were targeted with genioglossus injections [104–106]. One small, single-blind study showed a significant benefit in patients who remained on stable doses of neuroleptics [107]. Multiple case reports and series have shown improvement in TD, and, in particular, tardive dystonia, after DBS of the globus pallidus interna [108–110]. Rare case reports of successful thalamic and subthalamic DBS also exist [111]. The reported benefits after DBS vary widely, but most cases have experienced an improvement of at least 50 % in TD symptoms. Onset of benefit can be immediate or delayed by up to 6 months, and some published cases suggest that the benefit can be sustained for many months or years. A wide range of DBS parameters have been utilized, and no consensus recommendations exist. These overall positive results with botulinum toxin and DBS should be interpreted with caution given the lack of large studies utilizing double blinding, control groups, or standardized therapy parameters. The level of evidence is considered insufficient (level U).
Conclusion
TD, initially described nearly 60 years ago, is a serious complication of dopamine receptor-blocking drug therapy in both psychiatric and nonpsychiatric patients. It appears to be irreversible in most patients and increasing exposure is likely leading to increasing prevalence beyond the definition of rare disease. While discontinuation of the offending agents is an important part of the approach to treating TD, this is often not possible in disease states such as schizophrenia and gastroparesis. There are no approved medical therapeutic options for existing TD and limited guidelines. There is clearly a great need for the development of effective therapies. Several potential non-neuroleptic candidates for therapy have been tested in small, blinded trials and demonstrated some level of efficacy, as described above. Certainly, these represent reasonable therapeutic options today and we recently suggested an algorithm utilizing these drugs (Fig. 1) [10].
However, significant progress is lacking and needs to be made. The first step would be the confirmation of these smaller studies with large, multicenter, controlled trials. In doing so, many questions need to be addressed some of which include the following: What is the appropriate duration of the trial? What is the natural course of TD? What are the most appropriate measures of TD for trials? The AIMS and St Hans Rating scale have been validated [112, 113], but others have also been utilized [91]. Will patients remaining on neuroleptics respond differently to those who can come off them? Do patients with psychiatric diseases respond differently to medical interventions than nonpsychiatric patients? Should these trials involve multiple agents for comparative purposes? What is role of the type of TD (e.g., classical vs dystonia) on response? There is some suggestion that different types of TD will respond differently to agents, for example anticholinergics are helpful in dystonia [114], but can worsen classical oral–buccal–lingual dyskinesia [55]. Can the responsiveness to medications in TD be predicted by studies of genetic polymorphisms known to influence the risk and severity of TD [10]? For instance, in the EGb-761 trial there was greater improvement in the AIMS score in those with the valine/valine allele of the brain-derived neurotrophic factor gene and less improvement with the methionine/methionine allele [115].
This significant effort requires collaboration between basic, translational, and clinical scientists across the disciplines of psychiatry and neurology. While the agents discussed here represent a good first step, a multifaceted approach to drug discovery going beyond these agents will be necessary. This will require more detailed examination of the pathogenesis so that rational drug development becomes possible, as well as the use of well-designed clinical trials if we wish to significantly alter the trajectory of TD.
Electronic supplementary material
(PDF 1225 kb)
Acknowledgments
This work was supported by the Vance Lanier Endowed Chair in Neurology. The funding source had no input into the contents of this article. We would like to thank Thomas Wichmann, MD for his editorial advice. Full conflict of interest disclosures are available in the electronic supplementary material for this article.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Financial Disclosures
Dr. Factor has the following disclosures: honoraria—Scientiae for CME program, University of Florida speaker program, Current Neurology and Neuroscience section editor, Merz, Chelsea Therapeutics, ADAMAS; grants —Ceregene, TEVA, Ipsen, Allergan, Medtronics, Michael J. Fox Foundation, NIH; royalties—Demos, Blackwell Futura for textbooks. Dr. Zutshi had travel paid by UCB.
References
- 1.Soares-Weiser K, Fernandez HH. Tardive dyskinesia. Semin Neurol. 2007;27:159–169. doi: 10.1055/s-2007-971169. [DOI] [PubMed] [Google Scholar]
- 2.Tarsy D, Baldessarini RJ. Epidemiology of tardive dyskinesia: is risk declining with modern antipsychotics? Mov Disord. 2006;21:589–598. doi: 10.1002/mds.20823. [DOI] [PubMed] [Google Scholar]
- 3.Li CR, Chung YC, Park TW, et al. Clozapine-induced tardive dyskinesia in schizophrenic patients taking clozapine as a first-line antipsychotic drug. World J Biol Psychiatry. 2009;10:919–924. doi: 10.1080/15622970802481895. [DOI] [PubMed] [Google Scholar]
- 4.Molho ES, Factor SA. Possible tardive dystonia resulting from clozapine therapy. Mov Disord. 1999;14:873–874. doi: 10.1002/1531-8257(199909)14:5<873::AID-MDS1027>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 5.Pasricha PJ, Pehlivanov N, Sugumar A, Jankovic J. Drug Insight: from disturbed motility to disordered movement–a review of the clinical benefits and medicolegal risks of metoclopramide. Nat Clin Pract Gastroenterol Hepatol. 2006;3:138–148. doi: 10.1038/ncpgasthep0442. [DOI] [PubMed] [Google Scholar]
- 6.Waln O, Jankovic J. An update on tardive dyskinesia: from phenomenology to treatment. Tremor Other Hyperkinet Mov 2013;3. [DOI] [PMC free article] [PubMed]
- 7.Bhidayasiri R, Fahn S, Weiner WJ, Gronseth GS, Sullivan KL, Zesiewicz TA. Evidence-based guideline: Treatment of tardive syndromes: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;8:463–469. doi: 10.1212/WNL.0b013e31829d86b6. [DOI] [PubMed] [Google Scholar]
- 8.Faurbye A, Rasch PJ, Petersen PB, Brandborg G, Pakkenberg H. Neurological symptoms in pharmacotherapy of psychoses. Acta Psychiatr Scand. 1964;40:10–27. doi: 10.1111/j.1600-0447.1964.tb05731.x. [DOI] [PubMed] [Google Scholar]
- 9.Burke RE, Fahn S, Jankovic J, Marsden CD, Lang AE, Gollomp S, et al. Tardive dystonia: late-onset and persistent dystonia caused by antipsychotic drugs. Neurology. 1982;32:1335–1346. doi: 10.1212/WNL.32.12.1335. [DOI] [PubMed] [Google Scholar]
- 10.Revuelta GJ, Cloud L, Aia PG, Factor SA. Tardive dyskinesias. In: Albanese AJ, editor. Hyperkinetic movement disorders: differential diagnosis and treatment. Chichester: Wiley-Blackwell; 2012. pp. 331–352. [Google Scholar]
- 11.Samie MR, Dannenhoffer MA, Rozek S. Life-threatening tardive dyskinesia caused by metoclopramide. Mov Disord. 1987;2:125–129. doi: 10.1002/mds.870020207. [DOI] [PubMed] [Google Scholar]
- 12.Skidmore FW, Weiner WJ, Burke R. Neuroleptic-Induced tardive dyskinesia variants. In: Factor SA, Lang AE, Weiner WJ, editors. Drug induced movement disorders. Malden, MA: Blackwell Futura; 2005. pp. 257–284. [Google Scholar]
- 13.Yassa R, Nastase C, Dupont D, Thibeau M. Tardive dyskinesia in elderly psychiatric patients: a 5-year study. Am J Psychiatry. 1992;149:1206–1211. doi: 10.1176/ajp.149.9.1206. [DOI] [PubMed] [Google Scholar]
- 14.Jeste DV, Caligiuri MP, Paulsen JS, et al. Risk of tardive dyskinesia in older patients. A prospective longitudinal study of 266 outpatients. Arch Gen Psychiatry. 1995;52:756–765. doi: 10.1001/archpsyc.1995.03950210050010. [DOI] [PubMed] [Google Scholar]
- 15.Tenback DE, van Harten PN, van Os J. Non-therapeutic risk factors for onset of tardive dyskinesia in schizophrenia: a meta-analysis. Mov Disord. 2009;24:2309–2315. doi: 10.1002/mds.22707. [DOI] [PubMed] [Google Scholar]
- 16.Wonodi I, Adami HM, Cassady SL, Sherr JD, Avila MT, Thaker GK. Ethnicity and the course of tardive dyskinesia in outpatients presenting to the motor disorders clinic at the Maryland psychiatric research center. J Clin Psychopharmacol. 2004;24:592–598. doi: 10.1097/01.jcp.0000144888.43449.54. [DOI] [PubMed] [Google Scholar]
- 17.Woerner MG, Saltz BL, Kane JM, Lieberman JA, Alvir JM. Diabetes and development of tardive dyskinesia. Am J Psychiatry. 1993;150:966–968. doi: 10.1176/ajp.150.6.966. [DOI] [PubMed] [Google Scholar]
- 18.Ganzini L, Heintz RT, Hoffman WF, Casey DE. The prevalence of tardive dyskinesia in neuroleptic-treated diabetics. A controlled study. Arch Gen Psychiatry. 1991;48:259–263. doi: 10.1001/archpsyc.1991.01810270071010. [DOI] [PubMed] [Google Scholar]
- 19.Woerner MG, Alvir JM, Saltz BL, Lieberman JA, Kane JM. Prospective study of tardive dyskinesia in the elderly: rates and risk factors. Am J Psychiatry. 1998;155:1521–1528. doi: 10.1176/ajp.155.11.1521. [DOI] [PubMed] [Google Scholar]
- 20.Kane JM, Woerner M, Borenstein M, Wegner J, Lieberman J. Integrating incidence and prevalence of tardive dyskinesia. Psychopharmacol Bull. 1986;22:254–258. [PubMed] [Google Scholar]
- 21.Hyde TM, Apud JA, Fisher WC, Egan MF. Tardive dyskinesia. In: Factor SA, Lang AE, Weiner WJ, editors. Drug induced movement disorders. Malden, MA: Blackwell Futura; 2005. pp. 213–256. [Google Scholar]
- 22.Chatterjee A, Chakos M, Koreen A, et al. Prevalence and clinical correlates of extrapyramidal signs and spontaneous dyskinesia in never-medicated schizophrenic patients. Am J Psychiatry. 1995;152:1724–1729. doi: 10.1176/ajp.152.12.1724. [DOI] [PubMed] [Google Scholar]
- 23.Delwaide PJ, Desseilles M. Spontaneous buccolinguofacial dyskinesia in the elderly. Acta Neurol Scand. 1977;56:256–262. doi: 10.1111/j.1600-0404.1977.tb01431.x. [DOI] [PubMed] [Google Scholar]
- 24.Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27:95–99. doi: 10.1001/archpsyc.1972.01750250081011. [DOI] [PubMed] [Google Scholar]
- 25.Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. II. Short-term efficacy of dopamine-blocking agents haloperidol and thiopropazate. Arch Gen Psychiatry. 1972;27:100–103. doi: 10.1001/archpsyc.1972.01750250086012. [DOI] [PubMed] [Google Scholar]
- 26.Remington G. Tardive dyskinesia: eliminated, forgotten, or overshadowed? Curr Opin Psychiatry. 2007;20:131–137. doi: 10.1097/YCO.0b013e328017f6b1. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan S. Metoclopramide: drug use data review. memorandum. Food and Drug Administration, Center for Drug Evaluation and Research, 2005.
- 28.Kane JM, Smith JM. Tardive dyskinesia: prevalence and risk factors, 1959 to 1979. Arch Gen Psychiatry. 1982;39:473–481. doi: 10.1001/archpsyc.1982.04290040069010. [DOI] [PubMed] [Google Scholar]
- 29.Yassa R, Jeste DV. Gender differences in tardive dyskinesia: a critical review of the literature. Schizophr Bull. 1992;18:701–715. doi: 10.1093/schbul/18.4.701. [DOI] [PubMed] [Google Scholar]
- 30.Kane JM, Woerner M, Lieberman J. Tardive dyskinesia: prevalence, incidence, and risk factors. J Clin Psychopharmacol. 1988;8:52S–56S. doi: 10.1097/00004714-198808001-00010. [DOI] [PubMed] [Google Scholar]
- 31.Glazer WM, Morgenstern H, Doucette JT. Predicting the long-term risk of tardive dyskinesia in outpatients maintained on neuroleptic medications. J Clin Psychiatry. 1993;54:133–139. [PubMed] [Google Scholar]
- 32.Chakos MH, Alvir JM, Woerner MG, et al. Incidence and correlates of tardive dyskinesia in first episode of schizophrenia. Arch Gen Psychiatry. 1996;53:313–319. doi: 10.1001/archpsyc.1996.01830040049009. [DOI] [PubMed] [Google Scholar]
- 33.Factor SA, Friedman JH. The emerging role of clozapine in the treatment of movement disorders. Mov Disord. 1997;12:483–496. doi: 10.1002/mds.870120403. [DOI] [PubMed] [Google Scholar]
- 34.Friedman RA. A call for caution in the use of antipsychotic drugs. New York Times, 2012.
- 35.Olfson M, Blanco C, Liu SM, Wang S, Correll CU. National trends in the office-based treatment of children, adolescents, and adults with antipsychotics. Arch Gen Psychiatry. 2012;69:1247–1256. doi: 10.1001/archgenpsychiatry.2012.647. [DOI] [PubMed] [Google Scholar]
- 36.Correll CU, Schenk EM. Tardive dyskinesia and new antipsychotics. Curr Opin Psychiatry. 2008;21:151–156. doi: 10.1097/YCO.0b013e3282f53132. [DOI] [PubMed] [Google Scholar]
- 37.Bakker PR, de Groot IW, van Os J, van Harten PN. Long-stay psychiatric patients: a prospective study revealing persistent antipsychotic-induced movement disorder. PLoS One. 2011;6:e25588. doi: 10.1371/journal.pone.0025588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193:279–288. doi: 10.1192/bjp.bp.108.050088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones PB, Barnes TR, Davies L, et al. Randomized controlled trial of the effect on Quality of Life of second- vs first-generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1) Arch Gen Psychiatry. 2006;63:1079–1087. doi: 10.1001/archpsyc.63.10.1079. [DOI] [PubMed] [Google Scholar]
- 40.Woods SW, Morgenstern H, Saksa JR, et al. Incidence of tardive dyskinesia with atypical versus conventional antipsychotic medications: a prospective cohort study. J Clin Psychiatry. 2010;71:463–474. doi: 10.4088/JCP.07m03890yel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kane JM, Woerner MG, Pollack S, Safferman AZ, Lieberman JA. Does clozapine cause tardive dyskinesia? J Clin Psychiatry. 1993;54:327–330. [PubMed] [Google Scholar]
- 42.Gasquet I, Gury C, Tcherny-Lessenot S, Quesnot A, Gaudebout P. Patterns of prescription of four major antipsychotics: a retrospective study based on medical records of psychiatric inpatients. Pharmacoepidemiol Drug Saf. 2005;14:805–811. doi: 10.1002/pds.1122. [DOI] [PubMed] [Google Scholar]
- 43.Bergen JA, Eyland EA, Campbell JA, et al. The course of tardive dyskinesia in patients on long-term neuroleptics. Br J Psychiatry. 1989;154:523–528. doi: 10.1192/bjp.154.4.523. [DOI] [PubMed] [Google Scholar]
- 44.Casey DE. Tardive dyskinesia: reversible and irreversible. Psychopharmacology Suppl. 1985;2:88–97. doi: 10.1007/978-3-642-70140-5_11. [DOI] [PubMed] [Google Scholar]
- 45.Fernandez HH, Krupp B, Friedman JH. The course of tardive dyskinesia and parkinsonism in psychiatric inpatients: 14-year follow-up. Neurology. 2001;56:805–807. doi: 10.1212/WNL.56.6.805. [DOI] [PubMed] [Google Scholar]
- 46.Glazer WM, Morgenstern H, Schooler N, Berkman CS, Moore DC. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585–592. doi: 10.1192/bjp.157.4.585. [DOI] [PubMed] [Google Scholar]
- 47.Klawans HL, Tanner CM, Barr A. The reversibility of “permanent” tardive dyskinesia. Clin Neuropharmacol. 1984;7:153–159. doi: 10.1097/00002826-198406000-00006. [DOI] [PubMed] [Google Scholar]
- 48.Modestin J, Wehrli MV, Stephan PL, Agarwalla P. Evolution of neuroleptic-induced extrapyramidal syndromes under long-term neuroleptic treatment. Schizophr Res. 2008;100:97–107. doi: 10.1016/j.schres.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 49.Zutshi D, Cloud L, Factor S. Reversibility of tardive syndromes. 64th Annual Meeting of the American Academy of Neurology, 2012, New Orleans, LA. Abstract.
- 50.Smith JM, Baldessarini RJ. Changes in prevalence, severity, and recovery in tardive dyskinesia with age. Arch Gen Psychiatry. 1980;37:1368–1373. doi: 10.1001/archpsyc.1980.01780250054006. [DOI] [PubMed] [Google Scholar]
- 51.Glazer WM, Morgenstern H, Doucette JT. The prediction of chronic persistent versus intermittent tardive dyskinesia. A retrospective follow-up study. Br J Psychiatry. 1991;158:822–828. doi: 10.1192/bjp.158.6.822. [DOI] [PubMed] [Google Scholar]
- 52.Yassa R, Nair NP. A 10-year follow-up study of tardive dyskinesia. Acta Psychiatr Scand. 1992;86:262–266. doi: 10.1111/j.1600-0447.1992.tb03264.x. [DOI] [PubMed] [Google Scholar]
- 53.Bergen J, Kitchin R, Berry G. Predictors of the course of tardive dyskinesia in patients receiving neuroleptics. Biol Psychiatry. 1992;32:580–594. doi: 10.1016/0006-3223(92)90071-7. [DOI] [PubMed] [Google Scholar]
- 54.Goetz CG, Weiner WJ, Nausieda PA, Klawans HL. Tardive dyskinesia: pharmacology and clinical implications. Clin Neuropharmacol. 1982;5:3–22. doi: 10.1097/00002826-198205010-00002. [DOI] [PubMed] [Google Scholar]
- 55.Gardos G, Cole JO, Rapkin RM, et al. Anticholinergic challenge and neuroleptic withdrawal. Changes in dyskinesia and symptom measures. Arch Gen Psychiatry. 1984;41:1030–1035. doi: 10.1001/archpsyc.1983.01790220020003. [DOI] [PubMed] [Google Scholar]
- 56.Shenoy RS, Sadler AG, Goldberg SC, Hamer RM, Ross B. Effects of a six-week drug holiday on symptom status, relapse, and tardive dyskinesia in chronic schizophrenics. J Clin Psychopharmacol. 1981;1:141–145. doi: 10.1097/00004714-198105000-00005. [DOI] [PubMed] [Google Scholar]
- 57.Carpenter WT, Rey AG, Stephens JH. Covert dyskinesia in ambulatory schizophrenia. Lancet. 1980;2:212–213. doi: 10.1016/S0140-6736(80)90107-5. [DOI] [PubMed] [Google Scholar]
- 58.Branchey MH, Branehey LB, Richardson MA. Effects of gradual decrease and discontinuation of neuroleptics on clinical condition and tardive dyskinesia [proceedings] Psychopharmacol Bull. 1981;17:118–120. [PubMed] [Google Scholar]
- 59.Glazer WM, Hafez HM, Benarroche CL. Molindone and haloperidol in tardive dyskinesia. J Clin Psychiatry. 1985;46:4–7. [PubMed] [Google Scholar]
- 60.Chouinard G. Effects of risperidone in tardive dyskinesia: an analysis of the Canadian multicenter risperidone study. J Clin Psychopharmacol. 1995;15:36S–44S. doi: 10.1097/00004714-199502001-00007. [DOI] [PubMed] [Google Scholar]
- 61.Bai YM, Yu SC, Lin CC. Risperidone for severe tardive dyskinesia: a 12-week randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2003;64:1342–1348. doi: 10.4088/JCP.v64n1110. [DOI] [PubMed] [Google Scholar]
- 62.Chan HY, Chiang SC, Chang CJ, et al. A randomized controlled trial of risperidone and olanzapine for schizophrenic patients with neuroleptic-induced tardive dyskinesia. J Clin Psychiatry. 2010;71:1226–1233. doi: 10.4088/JCP.09m05155yel. [DOI] [PubMed] [Google Scholar]
- 63.Ono S, Suzuki Y, Shindo M, et al. Improvement of tardive dyskinesia and dystonia associated with aripiprazole following a switch to quetiapine: case report and review of the literature. J Clin Pharm Ther. 2012;37:370–372. doi: 10.1111/j.1365-2710.2011.01290.x. [DOI] [PubMed] [Google Scholar]
- 64.Lieberman JA, Saltz BL, Johns CA, Pollack S, Borenstein M, Kane J. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503–510. doi: 10.1192/bjp.158.4.503. [DOI] [PubMed] [Google Scholar]
- 65.Aia PG, Revuelta GJ, Cloud LJ, Factor SA. Tardive dyskinesia. Curr Treat Options Neurol. 2011;13:231–241. doi: 10.1007/s11940-011-0117-x. [DOI] [PubMed] [Google Scholar]
- 66.Huntington Study Group Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006;66:366–372. doi: 10.1212/01.wnl.0000198586.85250.13. [DOI] [PubMed] [Google Scholar]
- 67.Ondo WG, Hanna PA, Jankovic J. Tetrabenazine treatment for tardive dyskinesia: assessment by randomized videotape protocol. Am J Psychiatry. 1999;156:1279–1281. doi: 10.1176/ajp.156.8.1279. [DOI] [PubMed] [Google Scholar]
- 68.Kenney C, Hunter C, Jankovic J. Long-term tolerability of tetrabenazine in the treatment of hyperkinetic movement disorders. Mov Disord. 2007;22:193–197. doi: 10.1002/mds.21222. [DOI] [PubMed] [Google Scholar]
- 69.Jankovic J, Beach J. Long-term effects of tetrabenazine in hyperkinetic movement disorders. Neurology. 1997;48:358–362. doi: 10.1212/WNL.48.2.358. [DOI] [PubMed] [Google Scholar]
- 70.Leung JG, Breden EL. Tetrabenazine for the treatment of tardive dyskinesia. Ann Pharmacother. 2011;45:525–531. doi: 10.1345/aph.1P312. [DOI] [PubMed] [Google Scholar]
- 71.Huang CC, Wang RI, Hasegawa A, Alverno L. Evaluation of reserpine and alpha-methyldopa in the treatment of tardive dyskinesia. Psychopharmacol Bull. 1980;16:41–43. [PubMed] [Google Scholar]
- 72.Huang CC, Wang RI, Hasegawa A, Alverno L. Reserpine and alpha-methyldopa in the treatment of tardive dyskinesia. Psychopharmacology (Berl) 1981;73:359–362. doi: 10.1007/BF00426466. [DOI] [PubMed] [Google Scholar]
- 73.Kenney C, Jankovic J. Tetrabenazine in the treatment of hyperkinetic movement disorders. Expert Rev Neurother. 2006;6:7–17. doi: 10.1586/14737175.6.1.7. [DOI] [PubMed] [Google Scholar]
- 74.Allen RM. Palliative treatment of tardive dyskinesia with combination of amantadine-neuroleptic administration. Biol Psychiatry. 1982;17:719–727. [PubMed] [Google Scholar]
- 75.Vale S, Espejel MA. Amantadine for dyskinesia tarda. N Engl J Med. 1971;284:673. doi: 10.1056/nejm197103252841216. [DOI] [PubMed] [Google Scholar]
- 76.Decker BL, Davis JM, Jonowsky DS, el-Yousef MK, Sekerke HJ. Amantadine hydrochloride treatment of tardive dyskinesia. N Engl J Med. 1971;285:860. doi: 10.1056/NEJM197110072851516. [DOI] [PubMed] [Google Scholar]
- 77.Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson's disease. Neurology. 1998;50:1323–1326. doi: 10.1212/WNL.50.5.1323. [DOI] [PubMed] [Google Scholar]
- 78.Thomas A, Iacono D, Luciano AL, Armellino K, Di Iorio A, Onofrj M. Duration of amantadine benefit on dyskinesia of severe Parkinson's disease. J Neurol Neurosurg Psychiatry. 2004;75:141–143. [PMC free article] [PubMed] [Google Scholar]
- 79.Snow BJ, Macdonald L, McAuley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson's disease: a double-blind, placebo-controlled study. Clin Neuropharmacol. 2000;23:82–85. doi: 10.1097/00002826-200003000-00004. [DOI] [PubMed] [Google Scholar]
- 80.Jackisch R, Link T, Neufang B, Koch R. Studies on the mechanism of action of the antiparkinsonian drugs memantine and amantadine: no evidence for direct dopaminomimetic or antimuscarinic properties. Arch Int Pharmacodyn Ther. 1992;320:21–42. [PubMed] [Google Scholar]
- 81.Angus S, Sugars J, Boltezar R, Koskewich S, Schneider NM. A controlled trial of amantadine hydrochloride and neuroleptics in the treatment of tardive dyskinesia. J Clin Psychopharmacol. 1997;17:88–91. doi: 10.1097/00004714-199704000-00004. [DOI] [PubMed] [Google Scholar]
- 82.Pappa S, Tsouli S, Apostolou G, Mavreas V, Konitsiotis S. Effects of amantadine on tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Clin Neuropharmacol. 2010;33:271–275. doi: 10.1097/WNF.0b013e3181ffde32. [DOI] [PubMed] [Google Scholar]
- 83.Woods SW, Saksa JR, Baker CB, Cohen SJ, Tek C. Effects of levetiracetam on tardive dyskinesia: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2008;69:546–554. doi: 10.4088/JCP.v69n0405. [DOI] [PubMed] [Google Scholar]
- 84.McGavin CL, John V, Musser WS. Levetiracetam as a treatment for tardive dyskinesia: a case report. Neurology. 2003;61:419. doi: 10.1212/01.WNL.0000073538.60021.50. [DOI] [PubMed] [Google Scholar]
- 85.Bona JR. Treatment of neuroleptic-induced tardive dyskinesia with levetiracetam: a case series. J Clin Psychopharmacol. 2006;26:215–216. doi: 10.1097/01.jcp.0000203823.60603.80. [DOI] [PubMed] [Google Scholar]
- 86.Konitsiotis S, Pappa S, Mantas C, Mavreas V. Levetiracetam in tardive dyskinesia: an open label study. Mov Disord. 2006;21:1219–1221. doi: 10.1002/mds.20835. [DOI] [PubMed] [Google Scholar]
- 87.Meco G, Fabrizio E, Epifanio A, Morgante F, Valente M, Vanacore N, et al. Levetiracetam in tardive dyskinesia. Clin Neuropharmacol. 2006;29:265–268. doi: 10.1097/01.WNF.0000228807.49044.7D. [DOI] [PubMed] [Google Scholar]
- 88.Wolz M, Lohle M, Strecker K, et al. Levetiracetam for levodopa-induced dyskinesia in Parkinson's disease: a randomized, double-blind, placebo-controlled trial. J Neural Transm. 2010;117:1279–1286. doi: 10.1007/s00702-010-0472-x. [DOI] [PubMed] [Google Scholar]
- 89.Kabes J, Sikora J, Pisvejc J, Hanzlicek L, Skondia V. Effect of piracetam on extrapyramidal side effects induced by neuroleptic drugs. Int Pharmacopsychiatry. 1982;17:185–192. [PubMed] [Google Scholar]
- 90.Libov I, Miodownik C, Bersudsky Y, Dwolatzky T, Lerner V. Efficacy of piracetam in the treatment of tardive dyskinesia in schizophrenic patients: a randomized, double-blind, placebo-controlled crossover study. J Clin Psychiatry. 2007;68:1031–1037. doi: 10.4088/JCP.v68n0709. [DOI] [PubMed] [Google Scholar]
- 91.Thaker GK, Nguyen JA, Strauss ME, Jacobson R, Kaup BA, Tamminga CA. Clonazepam treatment of tardive dyskinesia: a practical GABAmimetic strategy. Am J Psychiatry. 1990;147:445–451. doi: 10.1176/ajp.147.4.445. [DOI] [PubMed] [Google Scholar]
- 92.Factor SA. Propranolol therapy for tardive dyskinesia revisited. Mov Disord. 2012;27:1703. doi: 10.1002/mds.25231. [DOI] [PubMed] [Google Scholar]
- 93.Bacher NM, Lewis HA. Low-dose propranolol in tardive dyskinesia. Am J Psychiatry. 1980;137:495–497. doi: 10.1176/ajp.137.4.495. [DOI] [PubMed] [Google Scholar]
- 94.Perenyi A, Farkas A. Propranolol in the treatment of tardive-dyskinesia. Biol Psychiatry. 1983;18:391–394. [PubMed] [Google Scholar]
- 95.Schrodt GR, Wright JH, Simpson R, Moore DP, Chase S. Treatment of tardive-dyskinesia with propranolol. J Clin Psychiatry. 1982;43:328–331. [PubMed] [Google Scholar]
- 96.Silver JM, Yudofsky SC, Kogan M, Katz BL. Elevation of thioridazine plasma levels by propranolol. Am J Psychiatry. 1986;143:1290–1292. doi: 10.1176/ajp.143.10.1290. [DOI] [PubMed] [Google Scholar]
- 97.Vestal RE, Kornhauser DM, Hollifield JW, Shand DG. Inhibition of propranolol metabolism by chlorpromazine. Clin Pharmacol Ther. 1979;25:19–24. doi: 10.1002/cpt197925119. [DOI] [PubMed] [Google Scholar]
- 98.Lerner V, Kaptsan A, Miodownik C, Kotler M. Vitamin B6 in treatment of tardive dyskinesia: a preliminary case series study. Clin Neuropharmacol. 1999;22:241–243. [PubMed] [Google Scholar]
- 99.Lerner V, Miodownik C, Kaptsan A, et al. Vitamin B6 treatment for tardive dyskinesia: a randomized, double-blind, placebo-controlled, crossover study. J Clin Psychiatry. 2007;68:1648–1654. doi: 10.4088/JCP.v68n1103. [DOI] [PubMed] [Google Scholar]
- 100.Zhang WF, Tan YL, Zhang XY, Chan RC, Wu HR, Zhou DF. Extract of Ginkgo biloba treatment for tardive dyskinesia in schizophrenia: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2011;72:615–621. doi: 10.4088/JCP.09m05125yel. [DOI] [PubMed] [Google Scholar]
- 101.Richardson MA, Bevans ML, Weber JB, et al. Branched chain amino acids decrease tardive dyskinesia symptoms. Psychopharmacology (Berl) 1999;143:358–364. doi: 10.1007/s002130050959. [DOI] [PubMed] [Google Scholar]
- 102.Richardson MA, Small AM, Read LL, Chao HM, Clelland JD. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65:92–96. doi: 10.4088/JCP.v65n0116. [DOI] [PubMed] [Google Scholar]
- 103.Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160:1117–1124. doi: 10.1176/appi.ajp.160.6.1117. [DOI] [PubMed] [Google Scholar]
- 104.Esper CD, Freeman A, Factor SA. Lingual protrusion dystonia: frequency etiology and botulinum toxin therapy. Park Rel Disord. 2010;16:438–441. doi: 10.1016/j.parkreldis.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 105.Tschopp L, Salazar Z, Micheli F. Botulinum toxin in painful tardive dyskinesia. Clin Neuropharmacol. 2009;32:165–166. doi: 10.1097/WNF.0b013e31818ddbc4. [DOI] [PubMed] [Google Scholar]
- 106.Hennings JM, Krause E, Botzel K, Wetter TC. Successful treatment of tardive lingual dystonia with botulinum toxin: case report and review of the literature. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1167–1171. doi: 10.1016/j.pnpbp.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 107.Slotema CW, van Harten PN, Bruggeman R, Hoek HW. Botulinum toxin in the treatment of orofacial tardive dyskinesia: a single blind study. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:507–509. doi: 10.1016/j.pnpbp.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 108.Kefalopoulou Z, Paschali A, Markaki E, Vassilakos P, Ellul J, Constantoyannis C. A double-blind study on a patient with tardive dyskinesia treated with pallidal deep brain stimulation. Acta Neurol Scand. 2009;119:269–273. doi: 10.1111/j.1600-0404.2008.01115.x. [DOI] [PubMed] [Google Scholar]
- 109.Damier P, Thobois S, Witjas T, et al. Bilateral deep brain stimulation of the globus pallidus to treat tardive dyskinesia. Arch Gen Psychiatry. 2007;64:170–176. doi: 10.1001/archpsyc.64.2.170. [DOI] [PubMed] [Google Scholar]
- 110.Schrader C, Peschel T, Petermeyer M, Dengler R, Hellwig D. Unilateral deep brain stimulation of the internal globus pallidus alleviates tardive dyskinesia. Mov Disord. 2004;19:583–585. doi: 10.1002/mds.10705. [DOI] [PubMed] [Google Scholar]
- 111.Zhang JG, Zhang K, Wang ZC. Deep brain stimulation in the treatment of tardive dystonia. Chin Med J (Engl) 2006;119:789–792. [PubMed] [Google Scholar]
- 112.Lane RD, Glazer WM, Hansen TE, Berman WH, Kramer SI. Assessment of tardive dyskinesia using the Abnormal Involuntary Movement Scale. J Nerv Ment Dis. 1985;173:353–357. doi: 10.1097/00005053-198506000-00005. [DOI] [PubMed] [Google Scholar]
- 113.Gerlach J, Korsgaard S, Clemmesen P, et al. The St. Hans Rating Scale for extrapyramidal syndromes: reliability and validity. Acta Psychiatr Scand. 1993;87:244–252. doi: 10.1111/j.1600-0447.1993.tb03366.x. [DOI] [PubMed] [Google Scholar]
- 114.Kang UJ, Burke RE, Fahn S. Natural history and treatment of tardive dystonia. Mov Disord. 1986;1:193–208. doi: 10.1002/mds.870010305. [DOI] [PubMed] [Google Scholar]
- 115.Zhang XY, Zhang WF, Zhou DF, et al. Brain-derived neurotrophic factor levels and its Val66Met gene polymorphism predict tardive dyskinesia treatment response to Ginkgo biloba. Biol Psychiatry. 2012;72:700–706. doi: 10.1016/j.biopsych.2012.04.032. [DOI] [PubMed] [Google Scholar]
- 116.Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry 2000;61(Suppl.)4:5–9. [PubMed]
- 117.Margolese HC, Chouinard G, Kolivakis TT, Beauclair L, Miller R. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 1: pathophysiology and mechanisms of induction. Can J Psychiatry. 2005;50:541–547. doi: 10.1177/070674370505000907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1225 kb)