

Abstract
Acetaminophen (APAP) hepatotoxicity is the main cause of acute liver failure in humans. Although mitochondrial oxidant stress and induction of the mitochondrial permeability transition (MPT) have been implicated in APAP-induced hepatotoxicity, the link between these events is unclear. To investigate this, this study evaluated APAP hepatotoxicity in mice deficient of cyclophilin D, a protein component of the MPT. Treatment of wild type mice with APAP resulted in focal centrilobular necrosis, nuclear DNA fragmentation and formation of reactive oxygen (elevated glutathione disulphide levels) and peroxynitrite (nitrotyrosine immunostaining) in the liver. CypD-deficient (Ppif−/−) mice were completely protected against APAP-induced liver injury and DNA fragmentation. Oxidant stress and peroxynitrite formation were blunted but not eliminated in CypD-deficient mice. Thus, mitochondrial oxidative stress and induction of the MPT are critical events in APAP hepatotoxicity in vivo and at least part of the APAP-induced oxidant stress and peroxynitrite formation occurs downstream of the MPT.
Keywords: Acetaminophen, cyclophilin D, mitochondrial permeability transition, oxidative stress, hepatocytes, necrosis
Introduction
Acetaminophen (APAP) is currently one of the most widely consumed drugs in the world. Although safe when used at therapeutics doses, intentional or unintentional over-dosing can cause severe liver injury and even liver failure [1]. During the last decade APAP hepatotoxicity became the single most frequent cause of acute liver failure in the US and many other countries [1]. Early animal studies indicated that the toxicity is dependent on the metabolic activation of APAP by the cytochrome P450 system, which generates the reactive metabolite N-acetyl-p-benzoquinoneimine (NAPQI) [2]. NAPQI can be scavenged by glutathione; the majority of the glutathione adducts is eliminated as a mercapturic acid conjugate via urine [2]. However, when NAPQI exhausts the cellular glutathione pools, NAPQI will covalently bind to cellular proteins [2,3]. For many years research focused on identifying cellular targets for NAPQI [4] and, although many protein adducts were identified, none could readily explain cellular toxicity [5,6].
A more recently emerging concept is that the formation of protein adducts is a critical initiating signal that requires amplification in order to cause cell death [6,7]. Central to this amplification concept of APAP-induced cell death is the mitochondria [6,7]. Early studies showed that there is inhibition of mitochondrial respiration [8,9] leading to cellular ATP depletion [10,11] during APAP hepatotoxicity. Although some of the mitochondrial dysfunction is partially reversible by supplying mitochondrial energy substrates [12], NAPQI binding to mitochondrial proteins may also contribute to the mitochondrial damage [13,14]. The amplification of the original protein binding signal is caused by the formation of reactive oxygen species inside the mitochondria, as indicated by the selective increase of mitochondrial glutathione disulphide (GSSG) [10,15] and the formation of peroxynitrite in the mitochondria [16]. This oxidant and nitrosative stress leads to damage of mitochondrial DNA [16] and release of inter-membrane proteins including cyto-chrome c, second mitochondria activator of caspases (Smac), endonuclease G and apoptosis-inducing factor (AIF) [7,17–21]. Although declining ATP levels may prevent induction of caspase activation and apoptosis by the released cytochrome c [22,23], endonuclease G and AIF translocate to the nucleus and are involved in nuclear DNA fragmentation [20]. Interestingly, the initial release of the mitochondrial inter-membrane proteins is facilitated by formation of bax pores in the outer membrane [21]. However, at later times, the effect is independent of bax and most likely related to mitochondrial damage due to the continued oxidant stress and peroxynitrite formation [21].
The formation of the mitochondrial permeability transition pore (MPT) is a critical event in cell death [24]. The MPT is thought to involve proteins such as the voltage-dependent anion channel (VDAC) from the outer membrane, adenine nucleotide translocator from the inner membrane, cyclophilin D (CypD) from the matrix and other proteins such as peripheral benzodiazepine receptor, hexokinase and creatine kinase [24]. Although the role of VDAC in the MPT has been questioned [25], the fact that cyclophilin D-deficient cells are protected against necrotic cell death suggests that this protein is essential for the MPT pore [26]. However, it has been postulated that under conditions of severe stress, unregulated MPT pores may be formed by clustering of misfolded proteins [27]. Cyclosporine A, which can inhibit the MPT pore formation by binding to cyclophilin D, was shown to protect against APAP hepatotoxicity in vivo, suggesting a critical role of the MPT in the pathophysiology [28,29]. However, APAP-induced cell death in cultured hepatocytes appears to involve the regulated, cyclosporine A-dependent MPT and the unregulated MPT [30]. Because pharmacological interventions may have off-target effects, the main goal of this investigation was to evaluate the role of cyclophilin D in APAP-induced oxidant stress and cell death using a mouse model with genetic deficiency of cyclophilin D.
Materials and methods
Animals
Male cyclophilin D-deficient mice (Ppif−/− mice), which are on a B6129SF2/J background, and their age-matched control littermates (B6129SF2/J) with an average weight of 18–20 g were used in this study [26]. All animals were housed in an environmentally controlled room with 12 h light/dark cycle and allowed free access to food and water. The experimental protocols were approved by the Institutional Animal Care and Use Committees of the University of Kansas Medical Center and University of Missouri at Columbia and followed the criteria of the National Research Council for the care and use of laboratory animals in research. All chemicals were purchased from Sigma Chemical Co. (St Louis, MO) unless stated otherwise.
Experimental design
Mice were injected intraperitoneally with 200 mg/kg APAP (dissolved in warm saline) after overnight fasting. The animals were killed 30 min or 6 h after APAP treatment and blood was withdrawn from the vena cava into a heparinized syringe for measurement of alanine aminotransferase (ALT) activities (Kinetic Test Kit 68-B, Biotron Diagnostics, Inc., Hernet, CA). The liver was removed and was rinsed in saline; liver sections were fixed in 10% phosphate-buffered formalin for histological analyses. The remaining liver was snap-frozen in liquid nitrogen and stored at −80°C for measurement of glutathione (GSH) and glutathione disulphide (GSSG) as described [31,32].
Histology and immunohistochemistry
Formalin-fixed tissue samples were embedded in paraffin and 4 μm sections were cut. Replicate sections were stained with haematoxylin and eosin (H&E) for evaluation of necrosis [23]. Sections were also stained for nitrotyrosine (NT) protein adducts with the DAKO LSAB peroxidase Kit (K684) (DAKO Corp., Carpinteria, CA) and an anti-nitrotyrosine antibody (Molecular Probes, Eugene, OR) [31]. For the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) assay, sections of liver were stained with the In Situ Cell Death Detection Kit, AP (Roche Diagnostics, Indianapolis, IN), as described in the manufacturer’s instructions [23].
Western blotting and cytochrome P450 activity measurements
Western blotting was carried out as described in detail [33] using the following antibodies: a rabbit anti-phospho-JNK polyclonal antibody (Cell Signaling Technology, Danvers, MA), a rabbit anti-JNK antibody (recognizes only JNK2) (Santa Cruz Biotechnology Inc., Santa Cruz, CA) and a rabbit anti-Cyp2E1 polyclonal antibody (Abcam, Cambridge, MA). For determination of cytochrome P450 activity, the 20 000 × g supernatant from liver homogenates were used in the 7-ethoxy-4-trifluoromethylcoumarin (7EFC) deethylase assay as described [34,35]. Briefly, samples were incubated in 0.1 M phosphate buffer (pH 7.4) with 0.4 mg/ml albumin and 1 mM NADPH. The reaction was initiated by addition of 7EFC (50 μM final concentration) and the increase in fluorescence with time was monitored on a fluorescence plate reader with an excitation at 410 nm and emission at 510 nm.
Statistics
All results were expressed as mean ± SE. Comparisons between multiple groups were performed with one-way ANOVA followed by a post-hoc Bonferroni test. If the data were not normally distributed, we used the Kruskal-Wallis Test (non-parametric ANOVA) followed by Dunn’s Multiple Comparisons Test; p < 0.05 was considered significant.
Results
Lack of cyclophilin D protects against acetaminophen-induced liver injury
To evaluate the potential role of CypD in APAP hepatotoxicity, a moderate overdose of 200 mg/kg was selected. At 6 h, APAP induced significant liver injury as indicated by the increase in plasma ALT activities (Figure 1) and the development of centrilobular necrosis (Figure 2A). However, liver injury in CypD-deficient mice treated with APAP was completely prevented (Figures 1 and 2A). Previous studies demonstrated that nuclear DNA fragmentation as visualized by the TUNEL assay is a consequence of mitochondrial dysfunction [16]. Consistent with these findings, a substantial number of TUNEL-positive cells were observed in the centrilobular regions, which correlated well with the areas of necrosis (Figure 2B). Nuclear DNA fragmentation was also eliminated in CypD-deficient mice (Figure 2B).
Figure 1.
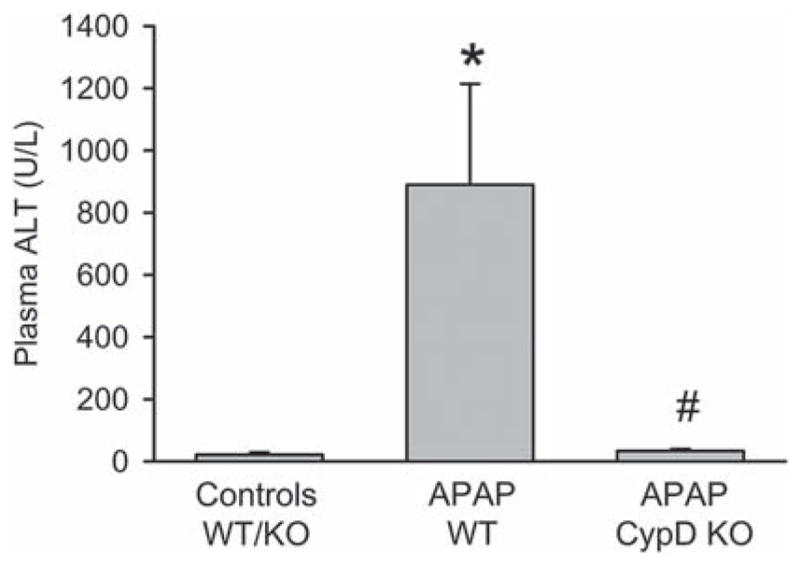
APAP-induced liver injury in CypD KO mice. Plasma alanine aminotransferase (ALT) activities as an indicator for acetaminophen (APAP)-induced hepatotoxicity were measured in B6129SF2/J wild type (WT) and cyclophilin D-deficient (CypD KO) mice 6 h after 200 mg/kg APAP administration. Data represent mean ± SE of n = 5 animals per group. *p < 0.05 (compared to untreated controls), # p < 0.05 (compared to APAP-treated wild type mice).
Figure 2.

(A) Representative liver sections of untreated control mice or animals treated for 6 h with 200 mg/kg acetaminophen (APAP) and stained with H&E. (B) DNA fragmentation as evaluated by the TUNEL assay in mice treated with APAP for 6 h. (C) Nitrotyrosine staining as an indicator of peroxynitrite formation in mice treated with APAP for 6 h. Inserted images of serial liver sections from wild type animals treated with APAP indicate that the centrilubular location of cell necrosis correlates with the area of TUNEL and nitrotyrosine staining.
Acetaminophen-induced oxidative and nitrosative stress
Covalent modification of mitochondrial proteins by NAPQI leads to mitochondrial oxidant stress and peroxynitrite formation [6]. Consistent with these observations, nitrotyrosine protein adducts as a footprint for peroxynitrite formation were detected in the centrilobular areas (Figure 2C). Although there was a trend to less nitrotyrosine staining in CypD-deficient mice, nitrotyrosine adducts were still detectable (Figure 2C). In addition, previous studies documented the selective increase of GSSG levels and the GSSG-to-GSH ratio in mitochondria as an indicator of a mitochondrial oxidant stress [10,15]. At 6 h after APAP injection, GSH levels completely recovered in WT animals (Figure 3A). However, the GSSG levels were increased 6-fold over baseline (Figure 3B), resulting in an elevated GSSG-to-GSH ratio (Figure 3C). In CypD-deficient animals, GSH levels were also fully recovered, but GSSG levels were only increased by 2.5-fold over baseline (Figure 3B). This caused a still significantly elevated GSSG-to-GSH ratio (Figure 3C). Thus, CypD-deficiency attenuated but not eliminated formation of reactive oxygen species and peroxynitrite during APAP hepatotoxicity.
Figure 3.

APAP-induced oxidant stress in CypD KO mice. Liver content of total glutathione (GSH+GSSG) (A), glutathione disulphide (GSSG) (B) and the GSSG-to-GSH ratio (C) in untreated mice or those treated with 200 mg/kg APAP for 6 h. The data represents mean ± SE of n = 5 animals per group. *p < 0.05 (compared to untreated controls), #p < 0.05 (compared to APAP-treated wild type mice).
Because APAP-induced oxidant stress can trigger activation of c-jun-N-terminal kinase (JNK), expression of JNK and its activated form P-JNK were evaluated by western blotting (Figures 4A and B). In WT mice APAP treatment induced JNK activation, which was prevented in CypD-deficient mice (Figures 4A and B). These finding suggest that JNK activation occurs in response to the post-MPT amplification of oxidant stress and cellular damage.
Figure 4.

(A) Western blot analysis of P-JNK and JNK2 protein expression in whole liver homogenates from untreated controls and 6 h after 200 mg/kg APAP in wild type (WT) and cyclophilin D deficient (CypD KO) mice. (B) Densitometric quantitation of P-JNK band intensities from western blot. The data in (B) represent mean ± SE of n = 3 animals per group. (C) Western blot analysis of liver cytochrome P450 2E1 protein expression in untreated controls and 6 h after 200 mg/kg APAP in WT and CypD KO mice. (D) Cytochrome P450-dependent O-deethylase activity. The data in (D) represent mean ± SE of n = 4 animals per group. *p < 0.05 (compared to untreated controls), #p < 0.05 (compared to APAP-treated wild type mice).
Drug metabolism capacity in CypD-deficient mice
To determine if CypD-deficient mice have adequate capacity to metabolize APAP, Cyp2E1 levels were evaluated by western blotting. Cyp2E1 protein levels in CypD-deficient mice were moderately increased over controls and APAP-treated WT animals (Figure 4C). To confirm this finding, P450-dependent O-deethylase activity was determined using the substrate 7-ethoxy-4-trifluoromethylcoumarin (Figure 4D). This reaction can be catalysed by Cyp2E1 [34,35]. Similar to the western blot, there was a higher enzyme activity in CypD-deficient mice compared to controls and APAP-treated WT mice (Figure 4D). Together these data suggests that CypD-deficient mice have an adequate capacity to metabolically activate APAP. To more directly assess NAPQI formation, the initial depletion of hepatic GSH levels was evaluated 30 min after APAP administration [10]. APAP overdose caused > 80% loss of liver GSH levels in livers of both WT and CypD-deficient mice (Figure 5A), suggesting a similar capacity to generate NAPQI. In contrast to later time points (Figure 2), there was a similar decline of GSSG levels (Figure 5B) and, therefore, there was no increase of the GSSG-to-GSH ratio in WT or CypD-deficient mice (Figure 5C).
Figure 5.

APAP-induced glutathione depletion in CypD KO mice. Liver content of total glutathione (GSH+GSSG) (A), glutathione disulphide (GSSG) (B) and the GSSG-to-GSH ratio (C) in untreated mice or those treated with 200 mg/kg APAP for 30 min. The data represents mean ± SE of n = 4 animals per group. *p < 0.05 (compared to untreated controls).
Discussion
Cyclophilin D and APAP-induced cell death
The objective of this investigation was to evaluate the role of cyclophilin D as a critical component of the MPT pore in APAP-induced liver cell injury. Our data demonstrate that liver injury observed in WT animals after a moderate overdose of APAP is completely prevented in CypD-deficient mice. Because it is well established that CypD is essential for Ca2+- and oxidant stress-induced MPT pore formation in mitochondria [26], our data suggest that the initial APAP-induced hepatocellular injury is dependent on the formation of the MPT pore. These findings are consistent with previous reports where a protective effect of cyclosporine A against APAP toxicity was shown in vivo [28,29] and in vitro [30]. However, cyclosporine A has additional effects beyond binding to CypD, e.g. inhibiting calcineurin [36], which may impact the mechanism of APAP toxicity. The current findings provide direct evidence for the importance of CypD in the cell death mechanism after APAP overdose.
Although the molecular composition of the MPT pore is still poorly defined, it is well established that CypD is a critical regulator of this pore [37]. Nevertheless, it was shown that excessive Ca2+ can trigger the MPT in mitochondria of CypD-deficient mice [26]. This suggests that CypD is not part of the actual pore and that under more severe stress conditions an unregulated MPT pore independent of CypD may be formed [27,37]. In fact, cyclosporine A in cultured hepatocytes substantially delayed but did not eliminate cell death after exposure to high (10 mM) doses of APAP [30]. In contrast, cyclosporine A effectively reduced APAP-induced cell death in vivo [28,29]. Our data clearly indicate that CypD is involved in regulating the MPT opening and cell death after a moderate overdose of APAP in vivo. A potential reason for the more limited efficacy of cyclosporine A in vitro might also be the exacerbated APAP-induced oxidant stress under regular cell culture conditions [38]. Thus, more detailed time- and dose-response experiments are needed to evaluate the full extent of the beneficial effects of inhibiting CypD in APAP hepatotoxicity.
An important issue for the interpretation of the data is whether the protective effect was caused by the absence of CypD or potentially by differences in the capacity to produce the reactive metabolite NAPQI. The protein levels of Cyp2E1, which is the most critical cytochrome P450 enzyme for metabolic activation of APAP in vivo [39], were similar if not slightly higher in CypD-deficient mice compared to WT animals. In addition, the P450-dependent O-deethylase activity, which is catalysed at least in part by Cyp2E1 [34,35], was higher in CypD-deficient mice compared to WT mice. Furthermore, the initial depletion of hepatic GSH levels, which is a direct measure of the actual NAPQI formation [10,12], showed no difference between WT and KO mice. Interestingly, there was no increase in GSSG levels or the GSSG-to-GSH ratio as an indicator of oxidant stress during this early metabolism phase. These observations confirm previous findings that the metabolism of APAP is not responsible for the oxidant stress [6]. Together these data suggest that CypD-deficient mice have an adequate capacity to metabolically activate APAP and therefore the protective effect against APAP induced liver injury is unlikely caused by a lack of reactive metabolite formation.
Cyclophilin D and APAP-induced oxidant stress
Oxidant stress is a potent inducer of the MPT pore opening in mitochondria [24,37]. APAP overdose triggers a mitochondrial oxidant stress and peroxynitrite formation as indicated by the selective increase of mitochondrial GSSG in vivo [10,15], selective nitrotyrosine protein adduct formation in mitochondria in vivo [16] and the increased Mitosox Red fluorescence in cultured hepatocytes [38]. The oxidant stress is preceding cell death [40] and the MPT [30] and scavenging reactive oxygen and peroxynitrite caused a profound protection against APAP hepatotoxicity in vivo [31,41] and in vitro [40]. Furthermore, preventing lysosomal iron translocation to the mitochondria prevented the MPT pore opening and cell death [42]. The current findings support the concept of reactive oxygen and peroxynitrite formation during APAP hepatotoxicity and show that CypD deficiency attenuates but not eliminates this oxidant stress. These results are consistent with the hypothesis that at least a part of the oxidant stress occurs upstream of the MPT and is able to trigger the pore opening. On the other hand, the significant reduction of the oxidant stress in CypD-deficient mice indicates that part of the oxidant stress is a consequence of the MPT. These observations are consistent with the concept that the opening of the MPT pore and collapse of the membrane potential may trigger a transient increase in reactive oxygen formation some of which may affect neighbouring mitochondria [43]. This effect could be responsible for the propagation of the injury within the cell.
Although both reactive oxygen species and peroxynitrite are produced in mitochondria during APAP hepatotoxicity [10,11,16], the source of nitric oxide (NO) is controversial. Evidence for [44,45] and against [15,46,47] a contribution of inducible nitric oxide synthase (iNOS) was provided. In addition, involvement of endothelial NOS (eNOS) [48] or neuronal NOS (nNOS) [49] has been suggested. However, given the fact that NO can freely diffuse through membranes, multiple sources can contribute to NO formation as a precursor for peroxynitrite in vivo. Given the critical role of NO for a number of physiological processes, it appears that the increase in mitochondrial superoxide rather than increased NO formation may be the driving force for peroxynitrite generation [15,47].
Previous data indicate that the early mitochondrial oxidant stress triggers JNK activation [47,50] and JNK signalling was shown to be important in APAP hepatotoxicity [47,50,51]. It was suggested that activated JNK translocates to the mitochondria and triggers the MPT [50]. However, the fact that in CypD-deficient mice APAP-induced JNK activation was eliminated suggests that JNK activation can occur downstream of the MPT. The reduced oxidant stress in these mice may have been the reason for the lack of activated JNK. In this respect it needs to be kept in mind that the previous reports showing robust JNK activation and a critical role of JNK in the cell death mechanism used much higher doses of APAP [47,50,51]. Thus, JNK signalling may be more important in the pathophysiology at higher doses, i.e. under more severe stress conditions, than used in the current experiments. This indicates that, dependent on the dose of APAP, intracellular signalling pathways may vary. Nevertheless, mitochondrial dysfunction and the MPT appear to be central for the cell death mechanism independent of the dose.
In summary, our data demonstrate a critical role of CypD in APAP-induced liver injury and to our knowledge is the first report using a genetic model to confirm this in vivo. The protective effect of CypD deficiency was associated with attenuated JNK activation and reduced oxidant stress and peroxynitrite formation suggesting that these events occur at least in part downstream of the MPT. Based on these findings, CypD is a promising therapeutic target to limit APAP hepatotoxicity in vivo.
Acknowledgments
This investigation was supported in part by National Institutes of Health Grants R01 DK070195 and R01 AA12916, and by grants P20 RR016475 and P20 RR021940 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health.
Footnotes
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis. 2007;11:525–548. doi: 10.1016/j.cld.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Nelson SD. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin Liver Dis. 1990;10:267–278. doi: 10.1055/s-2008-1040482. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther. 1973;187:211–217. [PubMed] [Google Scholar]
- 4.Cohen SD, Pumford NR, Khairallah EA, Boekelheide K, Pohl LR, Amouzadeh HR, Hinson JA. Selective protein covalent binding and target organ toxicity. Toxicol Appl Pharmacol. 1997;143:1–12. doi: 10.1006/taap.1996.8074. [DOI] [PubMed] [Google Scholar]
- 5.Pumford NR, Halmes NC. Protein targets of xenobiotic reactive intermediates. Annu Rev Pharmacol Toxicol. 1997;37:91–117. doi: 10.1146/annurev.pharmtox.37.1.91. [DOI] [PubMed] [Google Scholar]
- 6.Jaeschke H, Knight TR, Bajt ML. The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol Lett. 2003;144:279–288. doi: 10.1016/s0378-4274(03)00239-x. [DOI] [PubMed] [Google Scholar]
- 7.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 8.Meyers LL, Beierschmitt WP, Khairallah EA, Cohen SD. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol Appl Pharmacol. 1988;93:378–387. doi: 10.1016/0041-008x(88)90040-3. [DOI] [PubMed] [Google Scholar]
- 9.Ramsay RR, Rashed MS, Nelson SD. In vitro effects of acetaminophen metabolites and analogs on the respiration of mouse liver mitochondria. Arch Biochem Biophys. 1989;273:449–457. doi: 10.1016/0003-9861(89)90504-3. [DOI] [PubMed] [Google Scholar]
- 10.Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther. 1990;255:935–941. [PubMed] [Google Scholar]
- 11.Tirmenstein MA, Nelson SD. Acetaminophen-induced oxidation of protein thiols. Contribution of impaired thiol-metabolizing enzymes and the breakdown of adenine nucleotides. J Biol Chem. 1990;265:3059–3065. [PubMed] [Google Scholar]
- 12.Saito C, Zwingmann C, Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology. 2010;51:246–254. doi: 10.1002/hep.23267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tirmenstein MA, Nelson SD. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3′-hydroxyacetanilide, in mouse liver. J Biol Chem. 1989;264:9814–9819. [PubMed] [Google Scholar]
- 14.Qiu Y, Benet LZ, Burlingame AL. Identification of hepatic protein targets of the reactive metabolites of the non-hepatotoxic regioisomer of acetaminophen, 3′-hydroxyacetanilide, in the mouse in vivo using two-dimensional gel electrophoresis and mass spectrometry. Adv Exp Med Biol. 2001;500:663–673. doi: 10.1007/978-1-4615-0667-6_99. [DOI] [PubMed] [Google Scholar]
- 15.Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol Sci. 2001;62:212–220. doi: 10.1093/toxsci/62.2.212. [DOI] [PubMed] [Google Scholar]
- 16.Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, Jaeschke H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2005;315:879–887. doi: 10.1124/jpet.105.088898. [DOI] [PubMed] [Google Scholar]
- 17.Adams ML, Pierce RH, Vail ME, White CC, Tonge RP, Kavanagh TJ, Fausto N, Nelson SD, Bruschi SA. Enhanced acetaminophen hepatotoxicity in transgenic mice overexpressing BCL-2. Mol Pharmacol. 2001;60:907–915. doi: 10.1124/mol.60.5.907. [DOI] [PubMed] [Google Scholar]
- 18.Knight TR, Jaeschke H. Acetaminophen-induced inhibition of Fas receptor-mediated liver cell apoptosis: mitochondrial dysfunction versus glutathione depletion. Toxicol Appl Pharmacol. 2002;181:133–141. doi: 10.1006/taap.2002.9407. [DOI] [PubMed] [Google Scholar]
- 19.El-Hassan H, Anwar K, Macanas-Pirard P, Crabtree M, Chow SC, Johnson VL, Lee PC, Hinton RH, Price SC, Kass GE. Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: roles of cytochrome c, Bax, Bid, and caspases. Toxicol Appl Pharmacol. 2003;191:118–129. doi: 10.1016/s0041-008x(03)00240-0. [DOI] [PubMed] [Google Scholar]
- 20.Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci. 2006;94:217–225. doi: 10.1093/toxsci/kfl077. [DOI] [PubMed] [Google Scholar]
- 21.Bajt ML, Farhood A, Lemasters JJ, Jaeschke H. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2008;324:8–14. doi: 10.1124/jpet.107.129445. [DOI] [PubMed] [Google Scholar]
- 22.Lawson JA, Fisher MA, Simmons CA, Farhood A, Jaeschke H. Inhibition of Fas receptor (CD95)-induced hepatic caspase activation and apoptosis by acetaminophen in mice. Toxicol Appl Pharmacol. 1999;156:179–186. doi: 10.1006/taap.1999.8635. [DOI] [PubMed] [Google Scholar]
- 23.Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci. 2002;67:322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- 24.Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304:463–470. doi: 10.1016/s0006-291x(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 25.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 27.He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- 28.Masubuchi Y, Suda C, Horie T. Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J Hepatol. 2005;42:110–116. doi: 10.1016/j.jhep.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 29.Haouzi D, Cohen I, Vieira HL, Poncet D, Boya P, Castedo M, Vadrot N, Belzacq AS, Fau D, Brenner C, Feldmann G, Kroemer G. Mitochondrial permeability transition as a novel principle of hepatorenal toxicity in vivo. Apoptosis. 2002;7:395–405. doi: 10.1023/a:1020026923038. [DOI] [PubMed] [Google Scholar]
- 30.Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40:1170–1179. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- 31.Knight TR, Ho YS, Farhood A, Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther. 2002;303:468–475. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- 32.Jaeschke H, Mitchell JR. Use of isolated perfused organs in hypoxia and ischemia/reperfusion oxidant stress. Methods Enzymol. 1990;186:752–759. doi: 10.1016/0076-6879(90)86175-u. [DOI] [PubMed] [Google Scholar]
- 33.Bajt ML, Lawson JA, Vonderfecht SL, Gujral JS, Jaeschke H. Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: evidence for a postmitochondrial processing of caspase-8. Toxicol Sci. 2000;58:109–117. doi: 10.1093/toxsci/58.1.109. [DOI] [PubMed] [Google Scholar]
- 34.Buters JT, Schiller CD, Chou RC. A highly sensitive tool for the assay of cytochrome P450 enzyme activity in rat, dog and man. Direct fluorescence monitoring of the deethylation of 7-ethoxy-4-trifluoromethylcoumarin. Biochem Pharmacol. 1993;46:1577–1584. doi: 10.1016/0006-2952(93)90326-r. [DOI] [PubMed] [Google Scholar]
- 35.Chang TK, Crespi CL, Waxman DJ. Determination of CYP2B6 component of 7-ethoxy-4-trifluoromethylcoumarin O-deethylation activity in human liver microsomes. Methods Mol Biol. 2006;320:97–102. doi: 10.1385/1-59259-998-2:97. [DOI] [PubMed] [Google Scholar]
- 36.Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Curr Med Chem. 2003;10:1485–1506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- 37.Baines CP. The molecular composition of the mitochondrial permeability transition pore. J Mol Cell Cardiol. 2009;46:850–857. doi: 10.1016/j.yjmcc.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan HM, Ramachandran A, Bajt ML, Lemasters JJ, Jaeschke H. The oxygen tension modulates acetaminophen-induced mitochondrial oxidant stress and cell injury in cultured hepatocytes. Toxicol Sci. 2010 doi: 10.1093/toxsci/kfq208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaher H, Buters JT, Ward JM, Bruno MK, Lucas AM, Stern ST, Cohen SD, Gonzalez FJ. Protection against acetaminophen toxicity in CYP1A2 and CYP2E1 double-null mice. Toxicol Appl Pharmacol. 1998;152:193–199. doi: 10.1006/taap.1998.8501. [DOI] [PubMed] [Google Scholar]
- 40.Bajt ML, Knight TR, Lemasters JJ, Jaeschke H. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol Sci. 2004;80:343–349. doi: 10.1093/toxsci/kfh151. [DOI] [PubMed] [Google Scholar]
- 41.James LP, McCullough SS, Lamps LW, Hinson JA. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol Sci. 2003;75:458–467. doi: 10.1093/toxsci/kfg181. [DOI] [PubMed] [Google Scholar]
- 42.Kon K, Kim JS, Uchiyama A, Jaeschke H, Lemasters JJ. Lysosomal iron mobilization and induction of the mitochondrial permeability transition in acetaminophen-induced toxicity to mouse hepatocytes. Toxicol Sci. 2010;117:101–108. doi: 10.1093/toxsci/kfq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 44.Gardner CR, Laskin JD, Dambach DM, Sacco M, Durham SK, Bruno MK, Cohen SD, Gordon MK, Gerecke DR, Zhou P, Laskin DL. Reduced hepatotoxicity of acetaminophen in mice lacking inducible nitric oxide synthase: potential role of tumor necrosis factor-alpha and interleukin-10. Toxicol Appl Pharmacol. 2002;184:27–36. [PubMed] [Google Scholar]
- 45.Hinson JA, Bucci TJ, Irwin LK, Michael SL, Mayeux PR. Effect of inhibitors of nitric oxide synthase on acetaminophen-induced hepatotoxicity in mice. Nitric Oxide. 2002;6:160–167. doi: 10.1006/niox.2001.0404. [DOI] [PubMed] [Google Scholar]
- 46.Knight TR, Fariss MW, Farhood A, Jaeschke H. Role of lipid peroxidation as a mechanism of liver injury after acetaminophen overdose in mice. Toxicol Sci. 2003;76:229–236. doi: 10.1093/toxsci/kfg220. [DOI] [PubMed] [Google Scholar]
- 47.Saito C, Lemasters JJ, Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2010;246:8–17. doi: 10.1016/j.taap.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salhanick SD, Orlow D, Holt DE, Pavlides S, Reenstra W, Buras JA. Endothelially derived nitric oxide affects the severity of early acetaminophen-induced hepatic injury in mice. Acad Emerg Med. 2006;13:479–485. doi: 10.1197/j.aem.2005.11.082. [DOI] [PubMed] [Google Scholar]
- 49.Burke AS, MacMillan-Crow LA, Hinson JA. Reactive nitrogen species in acetaminophen-induced mitochondrial damage and toxicity in mouse hepatocytes. Chem Res Toxicol. 2010;23:1286–1292. doi: 10.1021/tx1001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283:13565–13577. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Henderson NC, Pollock KJ, Frew J, Mackinnon AC, Flavell RA, Davis RJ, Sethi T, Simpson KJ. Critical role of c-jun (NH2) terminal kinase in paracetamol-induced acute liver failure. Gut. 2007;56:982–990. doi: 10.1136/gut.2006.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]