

Abstract
We have developed an approach that integrates electroosmotic perfusion of tissue with a substrate-containing solution and online microfluidic analysis of products, in this case thiols. Using this approach we have tracked the metabolism of cystamine, pantethine and CoA in the extracellular space of organotypic hippocampal slice cultures (OHSCs). Currently, little is known about coenzyme A (CoA) biodegradation and even less is known about the regulation and kinetic characteristics for this sequential multi-enzyme reaction. We found that the steady state percentage yields of cysteamine from cystamine and pantethine during the transit through OHSCs were 91% ± 4% (SEM) and 0.01%–0.03%, respectively. The large difference in the yields of cysteamine can be used to explain the drugs’ different toxicities and clinical effectiveness against cystinosis. The kinetic parameters of the enzyme reaction catalyzed by the ectoenzyme pantetheinase are KM,C/α = 4.4 ± 1.1 mM and Vmax,C = 29 ± 3 nM/s, where α is the percentage yield of pantethine to pantetheine through disulfide exchange. We estimate that the percentage yield of pantethine to pantetheine through disulfide exchange is approximately 0.5%. Based on the formation rate of cysteamine in the OHSCs, we obtained the overall apparent Michaelis constant and maximum reaction rate for sequential, extracellular CoA degradation in an in situ environment, which are K′M = 16 ± 4 μM, V′max = 7.1 ± 0.5 nM/s. Kinetic parameters obtained in situ, although difficult to measure, are better representations of the biochemical flux in the living organism than those from isolated enzymes in vitro.
INTRODUCTION
Endogenous cysteamine exists at a relatively low concentration in the tissue.1 It is the terminal metabolite in coenzyme A (CoA) degradation2–4 and also a vital source for taurine.5 Cysteamine and its disulfide, cystamine, are radio-protective and anti-viral agents.6 Several studies, recently reviewed, demonstrated the efficacy of cystamine and cysteamine in animal models of Huntington’s and Parkinson’s diseases.6 Cystamine’s efficacy is likely mediated by cysteamine formed from the former in vivo.7 One significant effect of cysteamine relevant to neurodegenerative conditions in general is to increase the brain derived neurotrophic factor (BDNF).6, 8 Cysteamine bitartrate is now in Phase II clinical trials for Huntington’s disease.6 Cysteamine and cystamine are also used in the treatment of cystinosis, an autosomal recessive genetic disease that causes cystine to accumulate in lysosomes with fatal consequences.9–10 The disulfide form of pantetheine, pantethine, is also capable of reducing accumulated cystine inside the lysosome but with fewer side effects and lower toxicity in comparison to cysteamine or cystamine.10–13 However, it is also less effective in treating nephropathic cystinosis than cystamine.14
CoA is an essential cofactor for about 4% of all known enzymes and is actively involved in more than one hundred different synthetic and degradative reactions.2, 4 CoA participates in the oxidation of major carbon substrates for energy production and the synthesis of many cellular structural components.4 In brain, CoA also plays an essential role in the production of the neurotransmitter acetylcholine.4 The biosynthesis of CoA is accomplished in cytosol through five well-established steps with the aid of enzymes starting from three substrates, pantothenic acid (also known as vitamin B5), ATP, and cysteine.2, 4, 15 However, very little is known about CoA degradation and even less is known about the regulation of this process, its turnover rate or the net taurine production through this pathway.2–4, 16 Based on the current understanding of enzymes that might catalyze this process, there are several proposed routes for CoA biodegradation summarized in Figure 1. In route I, CoA is first dephosphorylated to dephospho-CoA by acid phosphatase (EC 3.1.3.2) located inside the lysosome and then degraded to 4′-phosphopantetheine by the ecto-nucleotide diphosphatase (EC 3.6.1.9) located on the plasma membrane.2, 4, 17 CoA has also been reported to be a direct substrate of ecto-nucleotide diphosphatase with the products of 3′, 5′-ADP and 4′-phosphopantetheine through route II.2, 4, 18 In route III, a peroxisomal nudix hydrolase (EC 3.6.1.13) is capable of converting CoA to 4′-phosphopantetheine and shows very low activity with dephospho-CoA as the substrate.2, 18–19 After further dephosphorylation of 4′-phosphopantetheine by acid phosphatase, the product pantetheine is then degraded to pantothenic acid and cysteamine, which is catalyzed by the ectoenzyme pantetheinase (EC 3.5.1.92).2–4 All of these pathways mentioned above for CoA degradation involve transfer of CoA and its downstream metabolites across the plasma membrane. It is difficult to imagine such a process in in vivo.
Figure 1.
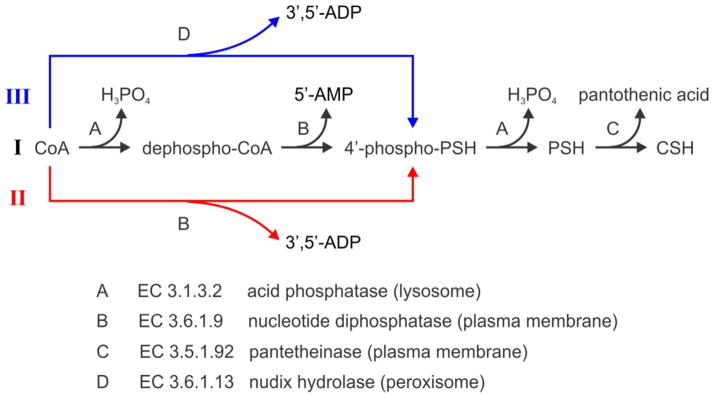
Proposed CoA biodegradation pathways. Note that H2O participating in each step is omitted. PSH and CSH represent pantetheine and cysteamine, respectively.
Currently, there is little knowledge about the rate of cysteamine formation in mammalian tissues due to the relatively low endogenous concentration of this molecule in tissues and also the technical difficulty in measuring it.1, 3 We have reported integrating electroosmotic perfusion with a microfluidic chip for online analysis of fluid perfused through the extracellular space of organotypic hippocampal slice cultures (OHSCs).20 OHSCs have the well-organized 3D architecture of the hippocampus in vivo. They provide a means to study biochemical and neurochemical events in the hippocampus.21 Conveniently, under the influence of an electric field, extracellular fluid in OHSCs can be drawn into a capillary by the same natural driving force, electroosmotic flow, as that used in the microfluidic chip. The coupling of electroosmotic perfusion, sample transport and quantitative analyses solves many practical problems. Samples transported directly to a microfluidic device for analysis are more representative of the fluid in the tissue being sampled than samples that undergo several handling steps. This is especially true when the sample volumes are nanoliters or smaller.
The integration of a tissue culture with a micro-analytical system can provide insight not currently possible. In intact biological systems, numerous enzyme reactions occur simultaneously with complex inhibiting and activating regulation and feedback among them.22 Therefore, analyzing a metabolic process in an in situ model provides a representation of the process that is distinct from and complementary to the in vitro determination of the physicochemical properties of the enzymes involved in the process. Analysis of a metabolic process in situ is also distinct from and complementary to whole animal studies of metabolism and clearance of drugs or endogenous compounds. Enzymatic activity and metabolism of endogenous compounds have been reported using integrated micro-analytical systems. For example, alkaline phosphatase and sphingosine kinase inside in a single cell have been evaluated in a microfluidic delivery device and capillary electrophoresis system respectively.23–24 Peptide degradation in a single cell has also been analyzed with and without the treatment of a drug compound on a microfluidic platform.25 Tissue cultures are more complex than single-cell models. The brain has more than one type of cell (neurons, glia). Metabolic exchange and dynamic signaling between and among different cell types create a dynamic environment.26–27 Integrated sampling/measurement methods, especially for environmentally sensitive compounds like thiols, have the capability of promoting greater understanding of such dynamic, metabolically active environments.
The hippocampal formation plays an important role in memory and spatial navigation.28 Hippocampal dysfunction along with damage in other brain regions occurs in Huntington’s disease.28 Metabolism involving CoA occurs in all cells.2 Thus, the OHSC is an appropriate target for investigations into metabolism related to cysteamine. In this work, we perfused OHSCs with two drug compounds used to treat cystinosis, cystamine and pantethine, to determine the extent of generation of the active compound, cysteamine, from each of them. The differences in the metabolism of the two drugs correlate with their clinical effectiveness and tendency to produce side effects. We also monitored the changes in the concentrations of cysteamine, pantetheine, homocysteine and cysteine in the extracellular space of the CA3 region in the OHSCs while perfusing with CoA. Direct evidence for degradation of CoA to cysteamine in the extracellular space of OHSCs was observed, based on which the in situ overall kinetic parameters were calculated. To the best of our knowledge, this is the first in situ study that directly measures production rate of cysteamine during extracellular CoA degradation in tissues.
EXPERIMENTAL SECTION
Chemicals and Reagents
Artificial cerebrospinal fluid (ACSF) was composed of (mM), NaCl (128), KCl (3), CaCl2 (2), MgSO4 (1.2), KH2PO4 (0.4), NaHCO3 (25), D-(+)-glucose (10). The Gly-Gly modified ACSF (ggACSF) was prepared by replacing 75 mM NaCl in the ACSF with 125 mM Gly-Gly with all other components unchanged. Both solutions have similar osmolarity (~310 mOsm/L) and pH value (pH = 7.4). NaCl and KCl were obtained from Avantor Performance Materials (Center Valley, PA). CaCl2 and KH2PO4 were from EM Science (Gibbstown, NJ). MgSO4 and NaHCO3 were purchased from EMD Millipore (Billerica, MA). Gly-Gly and D-(+)-glucose were from Sigma-Aldrich Chemical Co. (St. Louis, MO).
The running buffer for microfluidic electrophoresis consisted of 40 mM bis-tris propane with 15 mM NaCl at pH 8.5. On-chip thiol derivatizing buffer was 20 mM Tris-HCl at pH 7.5. The buffer preloaded in the sampling capillary and the auxiliary reservoir was composed of 20 mM Tris-HCl and 60 mM NaCl at pH 7.5. All buffers were filtered through 0.1 μm polyethersulfone membrane (EMD Millipore) and degassed by ultrasonication prior to use. The derivatizing stock solution was made by dissolving ThioGlo-1(EMD Millipore) to anhydrous DMSO at a final concentration of 2.5 mM and stored in freezer. The stock solutions of different thiol/disulfide, CoA (CoA, ~95%) and cysteamine, DL-homocysteine, L-cysteine and D-pantethine, cystamine were prepared by dissolving the solids to degassed Milli-Q water (EMD Millipore) to final concentrations of 7.1 mM, 272 mM, 69.1 mM, 49.7 mM, 183 mM, 139 mM respectively. These solutions were stored at −20 °C in aliquots and diluted serially to desired concentrations before use. The pantetheinases inhibitor stock solution, 8-cyclopentyl-1, 3-dipropylxanthine (DPCPX), was prepared with DMSO to a final concentration of 26 mM. All these chemicals were from Sigma-Aldrich.
Preparation of OHSCs
The preparation of OHSCs was modified from Gogolla’s protocol21 and has been described elsewhere.20 The protocol has been approved by the IACUC of the University of Pittsburgh.
Microfluidic Chip Design and Instrumentation
The design of the microfluidic chip, illustrated in Figure 2, is the same as used in a previous paper.20 It was fabricated using standard lithographic techniques as described earlier29 with a 30 μm mask width for all channels. The etching depth is 20 μm and the channel length (solid line) is drawn to scale. The total length of the reaction channel is ~ 46 mm and the distance between the detection point and injection cross is 23 mm. The electrical connection to all liquid access holes, the derivatization (+HV), the sample input (+HV), the auxiliary (ground), the gate (switched between ground and floating through a HV relay) and the waste (−HV) are represented by the short dashed lines. Microfluidic reservoirs were bought from IDEX Health & Science LLC (Oak Harbor, WA) and adapted to accommodate ~500 μL reagents each. An 11 cm (length) × 50 μm (ID)/360 μm (OD) fused silica capillary (long dashed line, Polymicro Technologies, Phoenix, AZ) are connected to the microfluidic chip via a NanoPort™ assembly (IDEX Health & Science LLC) with a minimized dead volume. Shorter capillaries, while desirable, cannot be used because of the need to bend them. The detection of ThioGlo-1 derivatized thiol compounds was accomplished by a home-made confocal laser induced fluorescence (LIF) detection system, which together with two four-channel high voltage power supplies (UltraVolt, Inc., Ronkonkoma, NY) were controlled by a locally written program in LabVIEW 8.2 (National Instruments, Austin, TX).29
Figure 2.
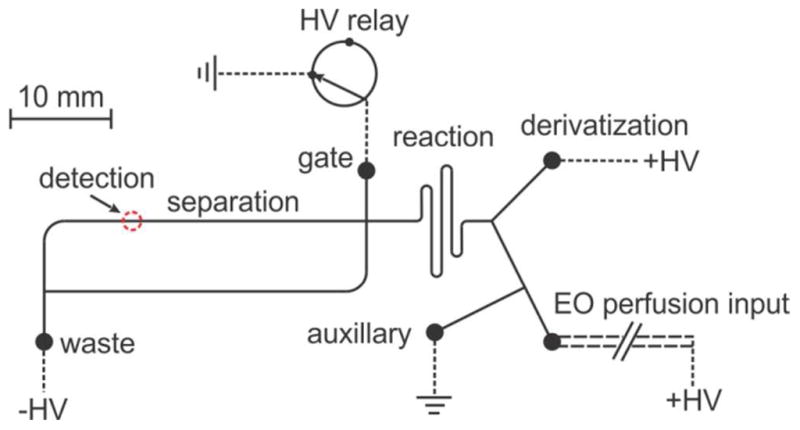
Sketch of the microfluidic device integrated with online electroosmotic perfusion. Channel (solid line) lengths are drawn to scale and all channels are 20 μm in depth. The short dashed line represents the electrical connection in each reservoir. Long dashed line labeled with electroosmotic (EO) perfusion input stands for the fused silica sampling capillary.
Online Electroosmotic Perfusion
At the beginning of the experiment, an OHSC was transferred to a Petri dish with 1.2 mL pre-warmed and gassed GBSS and then to another Petri dish containing 1.2 mL of the ggACSF alone or augmented with a substrate, inhibitor, drug compound or analyte standard at a desired concentration. Microfluidic chips and sampling capillaries were preconditioned with 1 M NaOH or 0.1 M NaOH, followed by deionized water and the experimental buffers for 5 minutes each. The auxiliary reservoir and the sampling capillary were preloaded with Tris buffer augmented with NaCl. The gate and waste reservoirs were filled with running buffer. The derivatization reservoir contained 2.7 μM ThioGlo-1 in derivatizing buffer. The liquid volume at each reservoir was 300–310 μL. The electroosmotic perfusion setup and voltage settings were described in another paper.20 Briefly, for electroosmotic perfusion, the end of the sampling capillary was lowered perpendicularly towards the OHSC by a Sutter MP-285 micromanipulator (Sutter Instrument Company, Novato, CA) until the tip made contact with the thin layer of liquid on the tissue surface in the CA3 region and then was lifted up 20 μm while remaining in contact with the tissue through the thin layer of liquid. There was a pre-perfusion step prior to each experiment. In this step, +3000 V and “ground” were applied to the Petri dish and auxiliary reservoir, respectively for 6 min. The resulting electric field in the capillary was about 240 V/cm. All other reservoirs were “floating”. After this step, +300 V and −4500 V were applied to derivatization and waste reservoirs, respectively. The gate reservoir was switched between sample loading (floating, 0.5 s) and separation modes (ground, 24.5 s) alternately using a high voltage relay. Electrical settings at the auxiliary reservoir and the Petri dish were kept unchanged from the pre-perfusion step. The resulting electric field in the separation channel in the separation mode was about 800 V/cm. Extracellular fluid collected by the electroosmotic perfusion in the CA3 region of the hippocampal slice was analyzed online with the microfluidic chip with the confocal LIF detector through continuous gated injections. There were about 30–40 repeated injections per run. Experiments were carried out at room temperature.
RESULTS AND DISCUSSION
Calibration and quantitation
In the process of electroosmotic perfusion of the extracellular space of an OHSC with a natural enzyme substrate, the substrate will be metabolized. The conditions used for the perfusion by electroosmosis are always the same, thus the residence time of a particular substrate in the tissue is constant. The fluid velocity is governed by the electric field in the tissue (and in the sampling capillary). Because the electric field in the tissue is proportional to the current density and the current density decreases away from the opening of the sampling capillary, the fluid velocity is highest near the sampling capillary (Figure S-2). As a result of this, substrate conversion to product will be highest far from the sampling capillary. We estimate that the residence time of the substrates, tS, is approximately 55 s (see SI).
As described in our previous work.20, 29 solutes from the tissue arrive at the chip after a short delay time. Peak heights increase over a short time and reach a steady state. Plots of peak height vs. time describe a symmetrical sigmoidal curve. We use the steady state peak heights for measurements with one exception. Pantetheine has calculated logP values of −0.92 ± 0.72 (ACD/Labs 12.0, Advanced Chemistry Development) and −0.10 (ALOGPS 2.1, http://146.107.217.178/lab/alogps/index.html). Thus, it can pass across the cell membrane by passive diffusion with the result that the volume accessible to pantetheine is greater than the volume accessible to charged solutes. The peak height from pantetheine changes more slowly over time as it reaches a steady state in comparison to, e.g. cysteamine. In fact, because the time for a single measurement is restricted to about fifteen minutes, in some cases a clear steady state is not reached. In these latter cases, we use the slope at the inflection point of the sigmoidal curve (or the peak height growth rate around the inflection point of the sigmoid progress curve, P) as the measurement as this slope is directly proportional to the plateau height (See Figure S-3).
Many of our inferences are based on quantitative determinations of cysteamine in the perfusate. We have determined earlier20 that the basal concentration of cysteamine in OHSCs is 10.6 ± 1.0 nM. Errors are the standard error of the mean (SEM) and data points in the plots are shown as mean ± standard deviation (SD). The calibration procedure is to introduce cysteamine into the perfusate at various concentrations and establish a linear relationship between the signal and the perfusate concentration. It is important to note that this process provides a measurement that is not sensitive to the presence of biochemical processes that remove cysteamine from the extracellular space as long as the rates of the processes are constant and first order in cysteamine (see SI). Thus, even though the analyte cysteamine may also be a substrate in one or more enzyme reactions or transport processes, the calibration will be accurate. If the processes removing cysteamine are significant and non-first order, the calibration graph would not be linear. Cysteamine calibrations are linear over a large concentration range (Figure S-4).
Other inferences are based on measurements of changes in pantetheine concentrations. Pantetheine is neutral under our experimental conditions. Neutral thiols can in principle be determined by our measurement approach. However, because the separation is based on electrophoresis, neutral molecules are not separated from each other. As a result, a peak in the electropherogram can only be used for relative quantitation of a neutral compound when it can be certain that under the specific conditions used the change in the peak height of the neutral peak correlates with changes in the concentration of the sought for species. We have three observations that allow us to use the change in the height of the neutral peak to infer changes in pantetheine. Details are discussed below, but a statement of the observations here will support our use of changes in the neutral peak height to infer changes in pantetheine concentration in the following discussion. (I) Perfusion with the disulfide pantethine leads to an increase in the neutral peak height that is linearly related to the pantethine concentration in the perfusate. This is consistent with the expectation that pantethine engages in disulfide exchange reactions. (II) Perfusion with CoA leads to an increase in the neutral peak height. Thiol products other than pantetheine in the proposed catabolic pathway (Figure 1) are charged, thus they cannot augment the neutral peak height. This observation is consistent with the expectation that pantetheine is a product of CoA degradation. (III) Coperfusion of CoA and the pantetheinase inhibitor 8-cyclopentyl-1, 3-dipropylxanthine leads to an increase in the neutral peak height with a corresponding decrease in the cysteamine peak height (Figure S-5). These observations permit us to use changes in the neutral peak height to infer changes in pantetheine concentration.
Cystamine is converted more rapidly than pantethine to cysteamine
Many bioactive functions of the disulfides cystamine and pantethine rely on their reduction to the corresponding thiols through disulfide exchange with endogenous thiols and an additional metabolic step for pantetheine with the help of pantetheinase to generate cysteamine.10, 30 Cysteamine is the active molecule in the treatment of cystinosis. Cystine trapped inside the lysosome reacts with cysteamine following the latter’s transport across the lysosomal membrane by a specific membrane transporter to form the cysteamine-cysteine mixed disulfide, which can leave the lysosome with the help of lysosomal lysine transporter.31 However cysteamine is cytotoxic at high concentration; the LC50 of cysteamine to the human leukemia cell line B4D is 52 μM.13, 32 Cysteamine precursors cystamine and pantethine have also been used as drugs against cystinosis.10–11, 14 Differences in the conversion rates of the two precursors to cysteamine in the OHSCs may well explain their distinct clinical profiles.
Cysteamine was determined in perfusate containing cystamine or pantethine. Figure 3 demonstrates the electropherograms from all of the injections in a single measurement. ThioGlo-1, which is neutral and weakly fluorescent, has a small peak with a migration time 9.4 s (Figure 3A). Because the source of the reagent is on the chip, it appears in the 5th injection, which is earlier than the arrival time of thiols coming from the tissue. Peaks from the latter compounds were not detected until the 16th injection. This is attributed to the time spent in the sampling capillary. Although the maleimide reagent (ThioGlo-1) is intended to react with thiols, it also reacts with amines at a much lower rate.33 Due to the relatively high concentration of Gly-Gly in ggACSF, we also observed the derivatized Gly-Gly peak in the electropherogram most easily seen in Figure 3C and Figure 3D with a migration time near 21 s. Figure 3A and Figure 3B show that cystamine is converted to cysteamine, while Figure 3C and Figure 3D show that pantethine is converted to pantetheine (increase in the neutral peak). Figure 4A shows that the dependence of measured cysteamine concentration on that of the initial cystamine concentration (Δ[CSH]/[CSSC]0) in the extracellular space is linear. The conversion rate is high (91% ± 4% with the assumption that one cysteamine is formed from a disulfide exchange reaction with each cystamine). The in situ measurement on intact tissue reflects a biochemically relevant process for extracellular conversion. To determine the effect of added pantethine on pantetheine concentration, we use the peak height growth rate around the inflection point of the sigmoid progress curve, P, which is directly proportional to Δ[PSH] (see Figure S-3). Linear regression of P vs. the added concentration of pantethine yields Figure 4B. We find that the production of pantetheine is linearly related to the concentration of pantethine introduced. This is consistent with an uncatalyzed disulfide exchange reaction being responsible for the conversion.
Figure 3.

Electropherograms obtained from experiments performed on the microfluidic chip coupled with online electroosmotic perfusion in the CA3 region of the OHSCs. Electropherograms in each plot are obtained from a single run. The peak labeled “CSH” is from the adduct of CSH with ThioGlo-1. Neutral compounds (unreacted ThioGlo-1 and pantetheine) are eluted as a single neutral peak. Separation conditions are described in experimental section. Initial cystamine or pantethine concentrations in the perfusate are (A) cystamine: 3.6 μM; (B) cystamine: 12 μM; (C) pantethine: 1.0 mM; (D) pantethine: 8.0 mM.
Figure 4.

The conversion of disulfides pantethine and cystamine in the CA3 region of the OHSCs during electroosmotic perfusion. The conversion of (A) cystamine to cysteamine; (B) pantethine to pantetheine and (C) pantethine to cysteamine as a function of drug concentration. Curves and lines (red) are from nonlinear and linear regression respectively. CSH: cysteamine; PSSP: pantethine. Each point is the mean ± SD (n ≥ 4).
In Figure 4C, the concentration of cysteamine is plotted against the initial concentration of pantethine. The relationship is nonlinear implying the presence of an enzyme-catalyzed step. The dependence of measured cysteamine concentration on that of the initial pantethine concentration (Δ[CSH]/[PSSP]0) in the extracellular space is very low, from 0.03% to 0.01% when the concentration of pantethine varies from 1.1 mM to 8.0 mM. Pantetheinase, the ectoenzyme that creates cysteamine from pantetheine, is inhibited by disulfides. An uncompetitive inhibition of pantetheinase by pantethine at pH 8.0 has been observed and the inhibition effect is increased as the incubation time of the pantetheinase and pantethine is prolonged.34 Therefore the rate equation based on the production of cysteamine (CSH) can be written as follows where KM,C, Vmax,C, and Ki,C are the Michaelis constant, the maximum reaction rate, and the inhibitor constant of pantetheinase with pantetheine (PSH) as the substrate and pantethine (PSSP) as the uncompetitive inhibitor, respectively.35
| [1] |
| [2] |
The factor α is the fraction of initial pantethine, [PSSP]0, that is converted with 1:1 stoichiometry to pantetheine. [PSH] and [PSSP] are average concentrations of pantetheine and pantethine in the perfusate, respectively. The results of a nonlinear fit of Eq. 1 to Δ[CSH] as a function of [PSSP]0 indicates that the last term of the denominator, [PSSP]02(1−α)/Ki,C, is negligible compared to other two terms. By omitting this term, Eq. 1 is reduced to Eq. 2. A nonlinear fit of Eq. 2 is plotted in Figure 4C, which gives KM,C/α = 4.4 ± 1.1 mM and Vmax,C = 29 ± 3 nM/s. Reported KM,C values for pantetheinase in in vitro assays using pantetheine or its analogs as substrate are in the range of ~20 μM 36–38. Based on that, we can estimate that the percentage yield, α, for pantethine to pantetheine to be approximately 0.5%, which is much lower than that of cystamine to cysteamine.
The generation rate of cysteamine from precursors, cystamine or pantethine, will determine the concentration of active substance in the treatment of cystinosis. Cystamine is as effective as cysteamine in lowering cystine levels in cystinosis,10 however, pantethine is less effective.14 This is consistent with our observation that the production rate of cysteamine from pantethine is significantly lower than the production rate from cystamine. Side effects and toxicity are common in the treatment with cysteamine or cystamine.10–12 To the contrary, pantethine has no or limited side effects even at high doses.14, 39 The same rationale mentioned above can be used to explain this difference. Moreover, other beneficial effects of pantethine, such as its anti-catabolic effect and the acceleration of fatty acid oxidation, are related to its promotion of CoA concentration due to the biological conversion of pantethine to pantetheine, and the following metabolism of pantetheine to 4′-phosphopantetheine and finally to CoA, which bypasses the first three steps in the CoA biosynthesis.11, 14, 40 The comparison of cystamine and pantethine metabolism as they pass through the extracellular space of OHSCs demonstrates the value of integration of a tissue culture and a micro-analytical system. We are measuring low concentrations of highly reactive compounds with high spatial resolution in intact tissue. The analytical result is consistent with clinical observations.
Enzymatic degradation of CoA---cysteamine
Endogenous cysteamine, homocysteine and cysteine in the extracellular space of the OHSCs can be observed under the conditions used (Figure 5A). In this figure, cysteamine was first detected in the 17th electropherogram and its height quickly formed a plateau. To monitor the metabolism of CoA in OHSCs, CoA was added to the perfusate. Adding CoA resulted in an increase in the cysteamine peak height (Figure 5B). Representative electropherograms from online electroosmotic perfusion experiments with different CoA concentrations in ggACSF are given in Figure S-6. As the plateau peak height is directly proportional to the enzyme-catalyzed production rate of cysteamine from CoA, we use it in determining the effect of CoA concentration on the cysteamine production rate. Thus by measuring the cysteamine concentration as a function of CoA concentration, we should observe a nonlinear dependence. The plateau concentration, Δ[CSH] was obtained from the calibration curve based on the plateau peak height of the analyte. Δ[CSH] is plotted against the initial [CoA]0 concentration in Figure 6A.
Figure 5.

Series of electropherograms obtained in two experiments performed on the microfluidic chip coupled with online electroosmotic perfusion in the CA3 region of the OHSCs. Initial CoA concentrations in the perfusate ggACSF are (A) 0 mM; (B) 0.12 mM. Peaks labeled with cysteamine (CSH), pantetheine (PSH), homocysteine (Hcy), cysteine (Cys) and glycylglycine (Gly-Gly) on the plots are all products from derivatizing reaction with ThioGlo-1. Peaks marked with ‘*’ are impurity in the dye. Separation conditions are described in experimental section.
Figure 6.
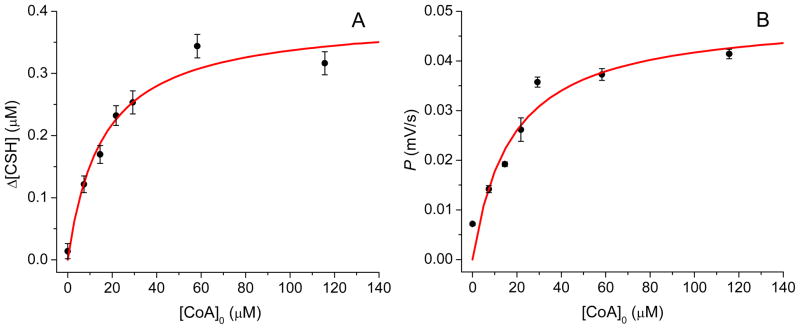
Nonlinear fits based on the conversion of CoA to cysteamine and pantetheine. (A) Δ[CSH] vs. [CoA]0 (B) peak height growth rate, P, vs. [CoA]0, were obtained by electroosmotic perfusion in the CA3 region of OHSCs. CSH: cysteamine. Each point is the mean ± SD (n ≥ 7).
As discussed above, it is appropriate to assume that cysteamine is the last product of a multi-enzyme reaction with CoA as the initial substance. Therefore, the overall initial reaction rate of a multi-enzyme reaction such as one of the paths shown in Figure 1 can be obtained by noting the change in cysteamine concentration over the residence time of the substrate in the tissue, ts. According to Michaelis-Menten kinetics, the overall reaction rate of the system d[CSH]/dt is given by35
| [3] |
| [4] |
where Δ[CSH] is the measured cysteamine concentration; [CoA]0 represents the initial CoA concentration in the perfusate, K′M and V′max and are the apparent Michaelis constant and apparent maximum reaction rate for the multi-enzyme reaction with CoA as the substrate, respectively. A nonlinear fit of Eq. 4 to the data shown in Figure 6A gave K′M = 16 ± 4 μM and V′max = 7.1 ± 0.5 nM/s, respectively.
Enzymatic degradation of CoA---pantetheine
Pantetheine is an intermediate molecule in the metabolic pathway of CoA (Figure 1) and is expected to show up as a neutral molecule on the electropherogram. When CoA is added in the perfusate, the height of the neutral peak stops decreasing around the 19th injection and starts to increase in Figure 5B, which suggests that pantetheine elutes together with the unreacted ThioGlo-1 as expected. If the measured pantetheine signal is used to estimate the multi-enzyme reaction rate, a relationship analogous to Eq. 4 can be established. This relationship, Eq. 5, has the same form as the Michaelis-Menten equation, where K′M,p and V′max,p are the apparent Michaelis constant and the apparent maximum reaction rate based on the production of pantetheine. As discussed above, peak height growth rate, P, is directly proportional to Δ[PSH].
| [5] |
Here q is a constant that connects the steady state concentration of pantetheine with the pantetheine peak height growth rate. Figure 6B shows P for pantetheine plotted against [CoA]0 with a nonlinear fit of Eq. 5. From the fit, we find that K′M,p is 18 ± 6 μM. K′M,p is statistically indistinguishable from the K′M obtained above based on cysteamine concentration. The fact that this constant is the same for the ultimate product formation rate, Δ[CSH]/tS and the penultimate product formation rate, Δ[PSH]/tS, suggests that the final enzyme, pantetheinase, is not limiting and is operating in the first-order regime.
The electroosmotic perfusion technique used here is capable of collecting perfusate under conditions that only cause minimal damage to the tissue.20, 41 The damage of tissue is mainly located in the region near the capillary lumen, where the fluid velocity in tissue is the highest and the substrate conversion to product is minimal. Therefore the damaged tissue will exert negligible effect on the total enzyme reaction. In addition, CoA and its metabolite 4′-phosphopantetheine are negatively charged. These molecules and their derivatives do not cross the cell membrane found in E. coli, rat liver and neuronal cells.40, 42–43 Together with these facts, our results directly show that CoA can be degraded to cysteamine in the extracellular space. The proposed CoA metabolic routes I, II and III (Figure 1) involve lysosomal acid phosphatase which is in lysosomes and nucleotide diphosphatase and pantetheinase which are ectoenzymes on the plasma membrane and nudix hydrolase which is in peroxisomes.2–4 Assuming that the in vivo degradation of CoA is carried out through one of these routes, there would be extensive trafficking of CoA and its metabolites between lysosomes or peroxisomes and outer surface of the plasma membrane. Due to the transport obstacles for intermediate metabolites to access these enzymes in sequential reactions, it is quite difficult to envision these pathways in practice and to explain the efficient degradation of CoA found in rat liver.4 Our results demonstrate that extracellular CoA is very likely to be degraded in a pathway similar to route I or III, but with all the dephosphorylation process taking place in the extracellular space. It is also possible that dephospho-CoA in lysosomes produced by lysosomal acid phosphatase is released to the extracellular space through lysosomal exocytosis44–45 and is then metabolized outside the cells similar to route II. The nucleotide diphosphatase involved in this process prefers dephospho-CoA as a substrate over CoA.4, 46
CONCLUSIONS
We have demonstrated here the successful integration of electroosmotic perfusion with online microfluidic analysis to study the metabolism of two drug compounds and CoA when these analytes passed through the extracellular space of OHSCs. The much higher percentage yields of cysteamine from cystamine than that from pantethine are directly correlated with the differences of these drug compounds in their effectiveness and toxicity observed clinically. The detection of cysteamine and pantetheine while perfusing CoA directly demonstrate the degradation of CoA to cysteamine in the extracellular space of OHSCs. Unlike other proposed pathways for CoA degradation, the route proposed here is easier to visualize in vivo as it does not involve extensive trafficking of CoA and related metabolites. The products of CoA biodegradation in the extracellular space, namely pantetheine, pantothenic acid and cysteamine can each be retaken by cells and reenter the CoA biosynthesis pathway or other metabolic pathways. The kinetic parameters of the overall sequential multi-enzyme reaction of CoA biodegradation obtained by measuring the cysteamine formation rates in the OSHCs at different CoA concentrations are useful for evaluating the taurine flux through cysteamine pathway and for understanding and solving the challenges remaining in the mechanism, regulation and turnover rate of CoA degradation.
Supplementary Material
Acknowledgments
The authors would like to thank the National Institutes of Health (R01 GM066018) for supporting this work. The authors are gratefully to Dr. Francisco Lara Vargas, Dr. Jerome P. Ferrance, and Dr. James P. Landers at the University of Virginia for making glass chips. We thank Prof. Xiang-Qun (Sean) Xie, University of Pittsburgh for advice and calculations of logD.
Footnotes
Supporting Information Available:
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pinto JT, Khomenko T, Szabo S, McLaren GD, Denton TT, Krasnikov BF, Jeitner TM, Cooper AJL. J Chromatogr B: Anal Technol Biomed Life Sci. 2009;877:3434–3441. doi: 10.1016/j.jchromb.2009.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leonardi R, Zhang YM, Rock CO, Jackowski S. Prog Lipid Res. 2005;44:125–153. doi: 10.1016/j.plipres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Stipanuk MH, Dominy JE, Lee JI, Coloso RM. J Nutr. 2006;136:1652S–1659S. doi: 10.1093/jn/136.6.1652S. [DOI] [PubMed] [Google Scholar]
- 4.Robishaw JD, Neely JR. Am J Physiol Endocrinol Metabol. 1985;248:E1–E9. doi: 10.1152/ajpendo.1985.248.1.E1. [DOI] [PubMed] [Google Scholar]
- 5.Dominy J, Simmons CR, Hirschberger LL, Hwang J, Coloso RM, Stipanuk MH. J Biol Chem. 2007;282:25189–25198. doi: 10.1074/jbc.M703089200. [DOI] [PubMed] [Google Scholar]
- 6.Gibrat C, Cicchetti F. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011;35:380–389. doi: 10.1016/j.pnpbp.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 7.Bousquet M, Gibrat C, Ouellet M, Rouillard C, Calon F, Cicchetti F. J Neurochem. 2010;114:1651–1658. doi: 10.1111/j.1471-4159.2010.06874.x. [DOI] [PubMed] [Google Scholar]
- 8.Pillai A, Veeranan-Karmegam R, Dhandapani KM, Mahadik SP. J Neurochem. 2008;107:941–951. doi: 10.1111/j.1471-4159.2008.05665.x. [DOI] [PubMed] [Google Scholar]
- 9.Jézégou A, Llinares E, Anne C, Kieffer-Jaquinod S, O’Regan S, Aupetit J, Chabli A, Sagné C, Debacker C, Chadefaux-Vekemans B, Journet A, André B, Gasnier B. Proc Natl Acad Sci USA. 2012;109:E3434–E3443. doi: 10.1073/pnas.1211198109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butler JD, Zatz M. J Clin Invest. 1984;74:411–416. doi: 10.1172/JCI111436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horvath Z, Vecsei L. Ideggyogy Sz (Clin Neurosci) 2009;62:220–229. [PubMed] [Google Scholar]
- 12.Kuna P, Dostal M, Neruda O, Knajfl J, Petyrek P, Podzimek F, Severa J, Svoboda V, Simsa J, Spelda S. J Appl Biomed. 2004;2:43–49. [Google Scholar]
- 13.Jeitner TM, Delikatny EJ, Bartier WA, Capper HR, Hunt NH. Biochem Pharmacol. 1998;55:793–802. doi: 10.1016/s0006-2952(97)00575-3. [DOI] [PubMed] [Google Scholar]
- 14.Wittwer CT, Gahl WA, Butler JD, Zatz M, Thoene JG. J Clin Invest. 1985;76:1665–1672. doi: 10.1172/JCI112152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spry C, Kirk K, Saliba KJ. FEMS Microbiol Rev. 2008;32:56–106. doi: 10.1111/j.1574-6976.2007.00093.x. [DOI] [PubMed] [Google Scholar]
- 16.Stipanuk MH. Annu Rev Nutr. 2004;24:539–577. doi: 10.1146/annurev.nutr.24.012003.132418. [DOI] [PubMed] [Google Scholar]
- 17.Goding JW, Grobben B, Slegers H. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2003;1638:1–19. doi: 10.1016/s0925-4439(03)00058-9. [DOI] [PubMed] [Google Scholar]
- 18.Strauss E. In: Comprehensive Natural Products II: Chemistry and Biology. 1. Mander L, Liu H-W, editors. Vol. 7.11. Elsevier Ltd; Oxford: 2010. pp. 351–410. [Google Scholar]
- 19.Cartwright JL, Gasmi L, Spiller DG, McLennan AG. J Biol Chem. 2000;275:32925–32930. doi: 10.1074/jbc.M005015200. [DOI] [PubMed] [Google Scholar]
- 20.Wu J, Xu K, Landers JP, Weber SG. Anal Chem. 2013;85:3095–3103. doi: 10.1021/ac302676q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gogolla N, Galimberti I, DePaola V, Caroni P. Nat Protoc. 2006;1:1165–1171. doi: 10.1038/nprot.2006.168. [DOI] [PubMed] [Google Scholar]
- 22.Schoffelen S, van Hest JCM. Soft Matter. 2012;8:1736–1746. [Google Scholar]
- 23.Olofsson J, Xu S, Jeffries GDM, Jesorka A, Bridle H, Isaksson I, Weber SG, Orwar O. Anal Chem. 2013;85:10126–10133. doi: 10.1021/ac4013122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dickinson AJ, Armistead PM, Allbritton NL. Anal Chem. 2013;85:4797–4804. doi: 10.1021/ac4005887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovarik ML, Shah PK, Armistead PM, Allbritton NL. Anal Chem. 2013;85:4991–4997. doi: 10.1021/ac4002029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Araque A, Carmignoto G, Haydon PG. Annu Rev Physiol. 2001;63:795–813. doi: 10.1146/annurev.physiol.63.1.795. [DOI] [PubMed] [Google Scholar]
- 27.Dringen R, Hirrlinger J. Biol Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 28.Ransome MI, Renoir T, Hannan AJ. Neural Plast. 2012;2012:874387. doi: 10.1155/2012/874387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J, Ferrance JP, Landers JP, Weber SG. Anal Chem. 2010;82:7267–7273. doi: 10.1021/ac101182r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wood PL, Khan MA, Moskal JR. Brain Res. 2007;1158:158–163. doi: 10.1016/j.brainres.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Eskelinen EL, Tanaka Y, Saftig P. Trends Cell Biol. 2003;13:137–145. doi: 10.1016/s0962-8924(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 32.Jeitner TM, Lawrence DA. Toxicol Sci. 2001;63:57–64. doi: 10.1093/toxsci/63.1.57. [DOI] [PubMed] [Google Scholar]
- 33.Sharpless NE, Flavin M. Biochemistry. 1966;5:2963–2971. doi: 10.1021/bi00873a028. [DOI] [PubMed] [Google Scholar]
- 34.Pitari G, Maurizi G, Ascenzi P, Ricci G, Dupre S. Eur J Biochem. 1994;226:81–86. doi: 10.1111/j.1432-1033.1994.tb20028.x. [DOI] [PubMed] [Google Scholar]
- 35.Segel IH, editor. 1. John Wiley & Sons; New York: 1975. pp. 1–29.pp. 136–143. [Google Scholar]
- 36.Wittwer C, Wyse B, Hansen RG. Anal Biochem. 1982;122:213–222. doi: 10.1016/0003-2697(82)90273-1. [DOI] [PubMed] [Google Scholar]
- 37.Dupre S, Chiaraluce R, Nardini M, Cannella C, Ricci G, Cavallini D. Anal Biochem. 1984;142:175–181. doi: 10.1016/0003-2697(84)90534-7. [DOI] [PubMed] [Google Scholar]
- 38.Ruan BH, Cole DC, Wu P, Quazi A, Page K, Wright JF, Huang N, Stock JR, Nocka K, Aulabaugh A, Krykbaev R, Fitz LJ, Wolfman NM, Fleming ML. Anal Biochem. 2010;399:284–292. doi: 10.1016/j.ab.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 39.Penet MF, Abou-Hamdan M, Coltel N, Cornille E, Grau GE, de Reggi M, Gharib B. Proc Natl Acad Sci USA. 2008;105:1321–1326. doi: 10.1073/pnas.0706867105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balibar CJ, Hollis-Symynkywicz MF, Tao JS. J Bacteriol. 2011;193:3304–3312. doi: 10.1128/JB.00334-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamsher AE, Xu HJ, Guy Y, Sandberg M, Weber SG. Anal Chem. 2010;82:6370–6376. doi: 10.1021/ac101271r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Domschke W, Liersch N, Decker K. Hoppe-Seyler’s Z Physiol Chem. 1971;253:85–88. doi: 10.1515/bchm2.1971.352.1.85. [DOI] [PubMed] [Google Scholar]
- 43.Kropf M, Rey G, Glauser L, Kulangara K, Johnsson K, Hirling H. Eur J Cell Biol. 2008;87:763–778. doi: 10.1016/j.ejcb.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 44.Reddy A, Caler EV, Andrews NW. Cell. 2001;106:157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 45.Zhang ZJ, Chen G, Zhou W, Song AH, Xu T, Luo QM, Wang W, Gu XS, Duan SM. Nat Cell Biol. 2007;9:945–953. doi: 10.1038/ncb1620. [DOI] [PubMed] [Google Scholar]
- 46.Skrede S. Eur J Biochem. 1973;38:401–407. doi: 10.1111/j.1432-1033.1973.tb03073.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.