

Summary
Structural plasticity in the adult brain is essential for adaptive behavior. We have found a remarkable anatomical plasticity in the basal ganglia of adult mice that is regulated by dopamine D2 receptors (D2Rs). By modulating neuronal excitability, striatal D2Rs bi-directionally control the density of direct pathway collaterals in the globus pallidus that bridge the direct pathway with the functionally opposing indirect pathway. An increase in bridging collaterals is associated with enhanced inhibition of pallidal neurons in vivo and disrupted locomotor activation after optogenetic stimulation of the direct pathway. Remarkably, chronic blockade with haloperidol, an antipsychotic medication used to treat schizophrenia decreases the extent of bridging collaterals and rescues the locomotor imbalance. These findings identify a role for bridging collaterals in regulating the concerted balance of striatal output, and may have important implications for understanding schizophrenia, a disease involving excessive activation of striatal D2Rs that is treated with D2R blockers.
Introduction
In the classical model of the basal ganglia circuitry, two parallel and distinct neural circuits connecting the input nucleus to the output nuclei have been described (Albin et al., 1989; Gerfen and Surmeier, 2011; Kreitzer and Malenka, 2008; Valjent et al., 2009). The direct (striatonigral) pathway is composed of GABAergic medium spiny neurons (MSNs) that predominantly express dopamine D1 receptors (D1Rs) and projects monosynaptically to the entopenduncular nucleus (EN) and substantia nigra pars reticulata (SNr). In contrast, the indirect (striatopallidal) pathway predominantly expresses D2 receptors (D2Rs) and projects to the external segment of the globus pallidus (GPe) which then relays to the EN and SNr. These two pathways are thought to create a dynamic balance exerting opposing but concerted actions on the control of movement, cognition and motivational processes (Cui et al., 2013; Durieux et al., 2009; Ferguson et al., 2010; Hikida et al., 2010; Hikosaka et al., 2000; Kravitz et al., 2010; Kravitz et al., 2012; Lobo et al., 2010; Mink, 2003; Nambu, 2008; Tai et al., 2012). Accordingly, an imbalance between both pathways has been postulated for several brain disorders including obsessive-compulsive disorder (OCD) and Parkinson’s disease (Albin et al., 1989; Maia and Frank, 2011).
The direct and indirect pathways are often described not only as functionally opposing, but also as anatomically segregated. However, this view has been challenged by single-cell tracing studies in rats and monkeys (Fujiyama et al., 2011; Kawaguchi et al., 1990; Levesque and Parent, 2005; Wu et al., 2000). In the rat, of 120 striatal projection neurons that have been individually labeled in three independent publications (Fujiyama et al., 2011; Kawaguchi et al., 1990; Wu et al., 2000), 37% projected exclusively to the GPe (“pure” indirect pathway), whereas only 3% projected only to the SNr or EN (“pure” direct pathway). 60% of labeled neurons projected to the SNr/EN and possessed collateral terminal fields in the GPe. Because these GPe collaterals may have the ability to bridge the direct and the indirect pathways, we refer to them here as ‘bridging’ collaterals. Regulating the extent of bridging collaterals could be a mechanism by which the direct pathway modulates the indirect pathway thereby affecting the behavioral balance maintained in concert by both pathways.
Neuronal activity represents an important developmental mechanism for regulating axonal projections and establishing proper connectivity between different brain structures (Catalano and Shatz, 1998; De Marco Garcia et al., 2011; Hua et al., 2005). To determine whether similar mechanisms also occur in the basal ganglia circuitry of the adult animal, we took advantage of BAC transgenic mice that express green fluorescent protein (GFP) under the control of the Drd1a or the Drd2 promoter (Drd1a-GFP and Drd2-GFP mice (Gong et al., 2003)). These mice label the direct and the indirect pathways, respectively, and can be used as tracing agents to visualize the axonal projections of both pathways (Valjent et al., 2009).
Using a combination of transgenic mice and viral gene transfer strategies we found that the density of bridging collaterals that connect the direct with the indirect pathway is regulated by neuronal excitability in the adult brain. Specifically, we found that increasing excitability of the indirect pathway is sufficient to induce the growth of bridging collaterals. Chronic up-regulation of D2Rs (D2R-OE mice), which increases excitability of the indirect pathway (Cazorla et al., 2012), similarly leads to a higher density of bridging collaterals, a phenotype that is reversed after viral-mediated decrease in excitability of D2R-OE MSNs. In contrast, genetic D2R downregulation (Drd2−/−) or chronic treatment with the D2R blocker haloperidol decreases the density of bridging collaterals. Increased bridging collaterals are associated with enhanced inhibition of pallidal neurons in vivo and disrupted motor activity after optogenetic stimulation of the direct pathway. Strikingly, both the behavioral and anatomical changes are reversed by chronic treatment with haloperidol. These data show a remarkable degree of structural plasticity in the adult basal ganglia that is regulated by chronic changes in the dopamine system and is sensitive to antipsychotic medication. These findings may have important implications for schizophrenia, in which D2R function is chronically increased in the striatum and for which D2R blockade with antipsychotic medication is still the most effective treatment (Howes et al., 2012).
Results
Striatopallidal MSN excitability regulates the extent of striatonigral bridging collaterals
In analogy to what has been observed during development, we reasoned that manipulating MSN excitability could lead to axonal remodeling of the striatal output pathways in adulthood (De Marco Garcia et al., 2011; Hua et al., 2005). To investigate this hypothesis in the adult animal, we injected the dorsal striatum with an adeno-associated virus (AAV) expressing the transdominant negative Kir2.1AAA potassium channel (AAV-Kir2.1AAA) using Drd1a-GFP and Drd2-GFP BAC transgenic mice to label, respectively, the direct and indirect pathways (Gong et al., 2003; Valjent et al., 2009) (Figure 1A). We previously showed that this strategy efficiently increases MSN excitability by down-regulating endogenous inward rectifying potassium (Kir) channel function (Cazorla et al., 2012). Quantification of Drd1a- and Drd2-GFP-positive terminal fields in AAV-Kir2.1AAA-mice revealed that increasing MSN excitability did not affect the growth of the classical striatonigral and striatopallidal terminal fields (i.e. Drd1a-GFP-positive terminals in SNr and Drd2-GFP-positive terminals in GPe, respectively) but specifically promoted the growth of striatonigral bridging collaterals in the GPe (Figure 1B,C). The overall density of the presynaptic terminal marker synaptophysin was increased in the GPe of AAV-Kir2.1AAA-mice and co-localized with Drd1a-GFP, confirming that Drd1a-GFP-labeled terminals found in the GPe correspond to presynaptic terminals and not to fibers of passage (Figure S1A). Neuronal populations in the GPe have been divided into parvalbumin (PV)-positive and -negative cells (Mallet et al., 2012). Drd1a-GFP-labeled terminals in the GPe of both controls and AAV-Kir2.1AAA-mice were found to contact both of these subpopulations (Figure S1C).
Figure 1. Excitability of the indirect pathway regulates the local growth of direct pathway collaterals into the adult GPe.
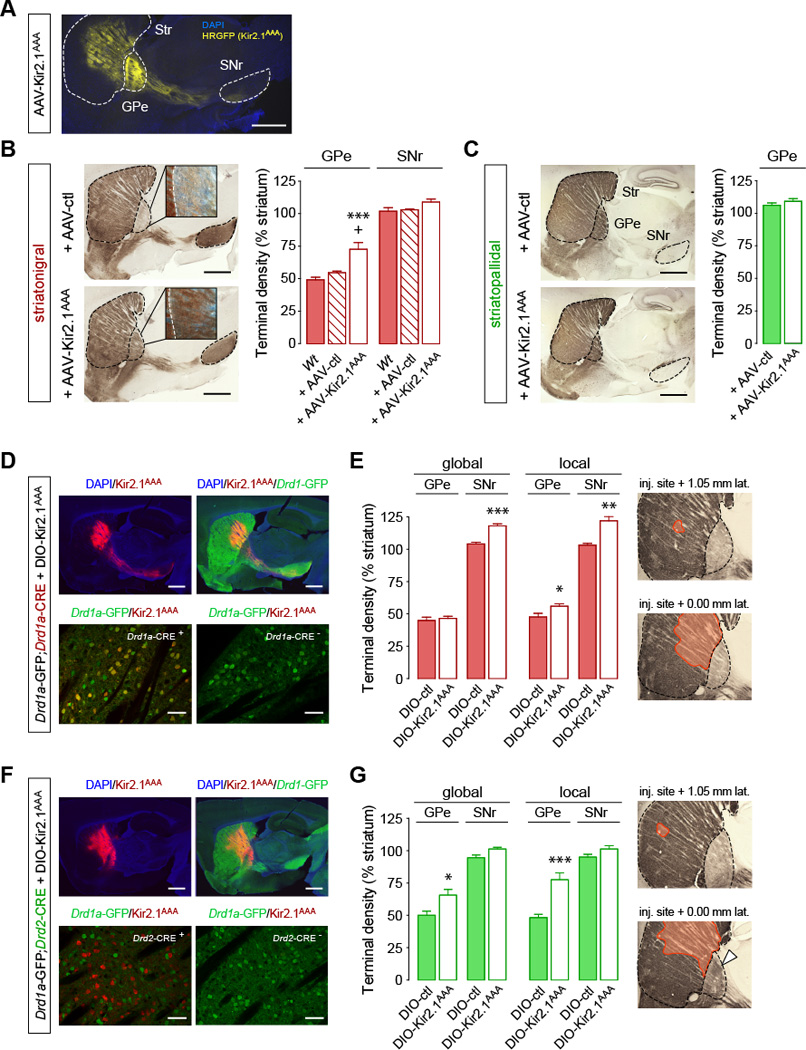
(A–C) Increasing MSN excitability promotes the growth of striatonigral terminal fields in the GPe of adult mice. (A) Sagittal sections showing Kir2.1AAA-ires-HRGFP expression in the striatum. Scale bar, 1 mm. (B,C) Quantification of striatonigral (B) and striatopallidal (C) terminal fields density in adult mice expressing the dominant negative Kir2.1AAA. ***p<0.001, compared to Wt; +p<0.05, compared to Wt + AAV-ctl; Data are presented as mean ± SEM (n=4–6 mice/group). Scale bar, 1 mm. Insets, 40x magnification (darkfield). (D–G) Excitability of striatopallidal pathway is sufficient to induce the growth of striatonigral bridging collaterals into the GPe. (D,F) Sagittal sections showing expression of DIO-Kir2.1AAA-ires-mCherry (red) and GFP (green) in Drd1a-GFP;Drd1a-CRE (D) and in Drd1a-GFP;Drd2-CRE (F) mice. Scale bar 1 mm (up), 50 µm (bottom). (E,G) Global and local quantification of striatonigral terminal density in Drd1a-GFP;Drd1a-CRE (E) and in Drd1a-GFP;Drd1a-CRE (G) + DIO-Kir2.1AAA mice. Global effect was assessed by quantifying striatonigral terminal density throughout the whole GPe. Local effect was assessed by quantifying terminal density in GPe regions containing mCherry-labeled terminal fields. *p<0.05, **p<0.01, ***p<0.005, compared to DIO-ctl; Data are presented as mean ± SEM (n=4–5 mice/group). Insets represent the striatal area expressing Kir2.1AAA-ires-mCherry (orange). Note that the increase in bridging collaterals is mainly restricted to the vicinity of Kir2.1AAA-infected striatopallidal terminals in line with the topographical organization of the striatopallidal connections (white arrows). See also Figure S1.
We then investigated whether the growth of bridging collaterals is regulated by neuronal excitability of the direct pathway (Drd1a-MSN) and/or by excitability of the indirect pathway (Drd2-MSN). AAVs expressing DIO-Kir2.1AAA (for which expression is dependent on CRE recombinase) were injected in the dorsal striatum of Drd1a-GFP;Drd2-CRE and Drd1a-GFP;Drd1a-CRE mice. This strategy allowed the quantification of Drd1a-GFP-labeled terminals after increasing MSN excitability selectively in the direct or indirect pathway (Figure 1D–G). Strikingly, expression of Kir2.1AAA in the direct pathway (Drd1a-CRE mice) had only a marginal effect on GPe collaterals, and instead led to an increase in terminal density in the SNr (Figure 1E). In contrast, expression of Kir2.1AAA in the indirect pathway (Drd2-CRE mice) increased terminal fields of the direct pathway in the GPe (Figure 1G). These findings show that neuronal excitability of the indirect pathway is sufficient to promote the growth of GPe direct pathway bridging collaterals. Interestingly, we observed that the increase in bridging collaterals was mainly restricted to the vicinity of Kir2.1AAA-infected striatopallidal terminals (see “global” vs. “local” effect in Figure 1G) in line with the topographical organization of striatopallidal connections (Alexander et al., 1986).
D2 receptors control the growth of bridging collaterals via their regulation of MSN excitability
What could be a physiological mechanism regulating excitability of the indirect pathway? Indirect pathway MSNs express D2Rs and are more excitable than direct pathway MSNs that express D1R (Gertler et al., 2008). Moreover, chronic upregulation of D2R in the striatum increases MSN excitability through the down-regulation of Kir channels (Cazorla et al., 2012), while chronic blockade of D2R using raclopride decreases excitability of the indirect pathway (Chan et al., 2012). Based on these findings we hypothesized that chronic changes in D2R function could regulate the extent of bridging collaterals via changes in excitability of the indirect pathway. To test this idea, mice overexpressing striatal D2Rs (D2R-OE mice (Kellendonk et al., 2006)) and Drd2 knock-out mice were crossed with BAC transgenic Drd1a-GFP and Drd2-GFP mice (Figure 2).
Figure 2. D2 receptors bi-directionally control the extent of bridging collaterals by regulating MSN excitability.

(A–D) GPe striatonigral bridging terminals are increased with D2R gain-of-function (D2R-OE mice) and decreased with D2R loss-of-function (Drd2−/− mice). (A,B) Sagittal sections and terminal density quantification of striatonigral (A) and striatopallidal (B) pathways in control and D2R-OE mice. (C,D) Sagittal sections and terminal density quantification of striatonigral (C) and striatopallidal (D) pathways in Drd2+/+ and Drd2−/− mice. *p<0.05, **p<0.01, ***p<0.005, compared to controls; #p<0.05, Drd2+/− vs. Drd2−/−; Data are presented as mean ± SEM (n=4–6 mice/group). (E) Increased bridging terminal density is restored in D2R-OE mice after striatal expression of AAV-Kir2.1Wt-ires-HRGFP. *p<0.05, ***p<0.005, compared to Ctl+AAV-ctl; ##p<0.01, ###p<0.005, compared to D2R-OE+AAV-ctl; Data are presented as mean ± SEM (n=4–6 mice/group). Scale bars, 1 mm. Insets, 40x magnification (A, darkfield; C, brightfield). See also Figure S2.
Similar to what we observed in Kir2.1AAA mice, D2R-OE;Drd1a-GFP mice showed a dramatic 1.75-fold increase in GFP-positive terminal density in the GPe without affecting the SNr (Figure 2A). The density of synaptophysin-positive terminals was increased in the GPe and co-localized with GFP-labeled terminals, indicating that the increase in GPe presynaptic terminals arises from MSNs of the direct pathway (Figure S2A,B). In addition, the distribution of terminals contacting PV-positive and PV-negative neurons was not changed in D2R-OE mice (Figure S2C). The increase in GPe terminal fields was specific for the direct pathway as GFP-labeled terminals emerging from the indirect pathway were not altered in D2R-OE;Drd2-GFP mice (Figure 2B). Analysis of the medial to lateral extent of bridging collaterals revealed that the highest effect size is observed in the medial part of the GPe despite an equal distribution of transgene expression in D2R-OE mice (Figure S2D). Associative cortico-striatal loops may therefore be more affected by the increase in bridging collaterals than sensorimotor loops. To visualize individual collaterals in the GPe further, low titer viruses (1.8 × 108 particles) were injected into the striatum to infect only a sparse number of neurons. We used AAV-DIO-Channelrhodopsin2 (ChR2) fused to eYFP as a tracer in combination with Drd1a-CRE mice to restrict the expression of ChR2-eYFP to the direct pathway. D2R-OE mice show an increase in the arborization of individual collaterals (Figure S2E). Quantification revealed that the number of higher order branches was increased in D2R-OE mice, whereas the number of primary branches emerging from passing axons was unaltered (Figure S2F). Remarkably, restoring Kir function in D2R-OE mice by injecting D2R-OE;Drd1a-GFP mice with an AAV expressing wild-type Kir2.1 (AAV-Kir2.1Wt) reversed the morphological phenotype (Figure 2E) confirming that D2Rs regulate the extent of bridging collaterals via its effect on neuronal excitability. Conversely, in D2R heterozygous (Drd2+/−;Drd1a-GFP) and homozygous (Drd2−/−;Drd1a-GFP) knock-out mice, we found a gene-dose-dependent decrease in GFP-positive terminal fields in the GPe (Figure 2C). GPe terminal fields from the indirect pathway were not altered in D2R-OE and knock-down mice (Figure 2D).
Bridging collaterals are dynamically regulated in the adult brain and are sensitive to chronic administration of haloperidol
Because this reversal demonstrates structural plasticity in the adult brain, we questioned whether dynamic changes in D2R expression could affect the bridging collaterals in adulthood. In D2R-OE mice, D2R overexpression can be switched off during adulthood with doxycycline (Kellendonk et al., 2006). Adult D2R-OE;Drd1a-GFP mice were fed doxycycline-supplemented food for 3, 7, 14 and 60 days to turn transgenic D2R expression off, and the density of GPe Drd1a-GFP-labeled terminal fields was measured (Figure 3A). We found that the increased GPe collateral terminal fields progressively reversed to almost control levels within 2 weeks of doxycycline treatment (Figures 3A,B and S3). Most importantly, switching-off expression in the adult for 2 weeks followed by re-initiation of D2R over-expression (2-month doxycycline withdrawal) led to re-growth of axonal terminal fields to nearly original levels, showing a remarkable degree of structural plasticity of bridging collaterals in adulthood that is sensitive to D2R levels.
Figure 3. Bridging collaterals are highly plastic in the adult animal and are sensitive to haloperidol.

(A) Transgenic D2R mRNA expression (upper row, in situ hybridization, scale bar 500 µm) is switched off in adult D2R-OE mice by doxycycline application (Dox on). Expression is reestablished after removal of doxycycline (Dox on then off). Increased striatonigral GPe collaterals (bottom row, Drd1a-GFP immunostaining, scale bar 100 µm) are reversed to normal after switching transgene expression off (Dox on). Collaterals grow back after re-expressing the transgene (Dox on then off). (B) Kinetics of collaterals retraction in the GPe of D2R-OE mice treated with doxycycline for increasing time periods and re-growth after re-expressing the transgene for 60 days (14 on + 60 off). Values are normalized to control mice and are presented as mean ± SEM (n=5–6 mice/group). (C) Reduction of striatonigral but not striatopallidal terminals in the GPe of control and D2R-OE mice treated for 14 days with haloperidol (1 mg/kg/day). *p<0.05, **p<0.01, ***p<0.005, compared to control; ##p<0.01, compared to D2R-OE; Data are presented as mean ± SEM (n=8–9 mice/group). See also Figure S3.
Because the density of bridging collaterals can be modulated in the adult by manipulating D2R expression, we asked whether the bridging collaterals are also sensitive to pharmacological manipulation of D2R function using chronic treatment of haloperidol, a D2R blocker that is commonly used as antipsychotic medication in humans. After 2 weeks of haloperidol treatment (1 mg/kg/day) bridging collaterals were decreased in both D2R-OE and wild-type mice without affecting the classical direct and indirect pathway terminals within the SNr and GPe, respectively (Figure 3C). The reduction in the density of collaterals was of similar magnitude in D2R-OE mice treated for 2 weeks with either haloperidol or doxycycline (haloperidol, −72.3%; doxycycline, −75.6%; Figure 3B,C). To use a different quantitative method independent of the GFP transgene we performed Western blots using the presynaptic markers synaptophysin and SNAP-25. Protein levels were increased in the GPe but not the striatum of D2R-OE mice (Figure S3C,D). These increases were reversed after switching off the transgene using doxycycline (Figure S3C,D).
Increased bridging collaterals are associated with increased inhibition of GPe cells after optogenetic stimulation of the direct pathway in vivo
Since MSNs are GABAergic, we investigated whether the direct pathway — like the indirect pathway — has the ability to inhibit GPe neurons via its bridging collaterals. Using optogenetics in anesthetized mice, we stimulated the direct and indirect pathways selectively while recording from GPe neurons. Drd1a-CRE and Drd2-CRE mice injected with AAV-DIO-ChR2 into the dorsal striatum (Figure 4A) were implanted into the injection site with an optic fiber connected to a laser source while a recording electrode was lowered into the GPe (Figure 4B). As expected, laser stimulation of indirect pathway MSNs (Drd2-CRE mice) led to massive inhibition of the majority of GPe cells, whereas control mice not injected with AAV-DIO-ChR2 (“No ChR2”) remained unaffected by laser stimulation (Figure 4C). Remarkably, laser stimulation of direct pathway striatal neurons (Drd1a-CRE mice) also led to an inhibition of basal firing rate of GPe cells, which was smaller than in Drd2-CRE mice but enlarged in Drd1a-CRE;D2R-OE mice (Figure 4C). Because the basal firing rate of GPe neurons is about 10 Hz, we used 100-msec bins to sample the responses. The latency of inhibition was comparable between Drd1a-CRE;control, Drd1a-CRE;D2R-OE and Drd2-CRE mice occurring for most activity recorded within the first 100-msec (Figure 4D). Because the indirect pathway (Drd2-CRE mice) is an established monosynaptic pathway, the data are consistent with a functional monosynaptic inhibition emerging also from the direct pathway. Figure 4E shows a comparison of relative spike numbers between the four groups.
Figure 4. Increased striatonigral bridging collaterals are associated with enhanced GPe inhibition.

(A,B) Set-up for the selective optogenetic activation of direct or indirect pathways and concurrent recording of GPe cells in vivo. (A) Example of CRE-positive Drd1a- and Drd2-MSNs (red) expressing ChR2-YFP (green) in the dorsomedial striatum. Scale bars, 20 µm. (B) Schematic depicting fiber-optic implantation in the dorsomedial striatum and positioning of the recording electrode in the GPe. (C–I) Optogenetic activation of the direct pathway reveals inhibition in the GPe that is enhanced in D2R-OE mice. (C) Peri-stimulus time histograms (PSTH) showing the mean spike frequency of all recorded GPe neurons before, during and after the 5-sec laser stimulation (100 ms-bins). Mice were injected with a control virus (No Chr2) or with a DIO-ChR2 virus for Drd1a-CRE;control, Drd1a-CRE;D2R-OE and Drd2-Cre mice. (D) Zoom-in of PSTH in (C) showing the latency of response 1000 ms before and after laser illumination. Arrows indicate the first 100 ms-bin recorded after laser stimulation. (E) Summary graphs of relative firing frequency showed in (C) for direct comparison of the four groups before, during and after the 5-sec laser illumination. (F) Change in firing rate during laser-induced stimulation of direct or indirect pathway expressed as a z-score of the pre-stimulation firing rate distribution. **p<0.01; n.s, not significant. All experimental groups differ from the control “No Chr2 group” (p<0.001). (G) Proportion of GPe cell units for which basal firing rate is significantly decreased after laser stimulation. **p<0.01; n.s, not significant. All experimental groups differ from the control group (p<0.001). (H) Distribution of GPe cells response after laser-induced activation of direct or indirect pathway (1-bin z-scores). A cut-off was determined at −2 (2 negative standard deviations) to define cells that are significantly inhibited after laser stimulation. (I) Degree of inhibition of GPe cells after laser-induced activation of direct and indirect pathways. **p<0.01. All experimental groups differ from the control group (p<0.001). Data are presented as mean ± SEM (No Chr2 injected: n=3 mice (33 neurons), Drd1a-CRE;control: n=5 (72), Drd1a-CRE;D2R-OE: n=5 (63), Drd2-CRE;control: n=6 (78).
To quantify the degree of inhibition we used z-score analysis (Figure 4F). The three experimental groups, Drd1a-CRE mice, Drd1a-CRE;D2R-OE and Drd2-CRE mice differed significantly from the non-ChR2 transfected control group (one-way ANOVA [F(3,242)=14.494; p<0.001], Scheffe corrected post-hoc comparisons, p<0.05) and from each other [F(2,212)=6.950, p<0.01] (Figure 4F). Pair-wise comparisons of the z-scores revealed that optical stimulation of striatopallidal neurons in Drd2-CRE mice was more effective than stimulation of striatonigral neurons at inhibiting GPe neurons in Drd1a-CRE mice. Similarly, Drd1a-CRE;D2R-OE mice (M=−3.7118, SEM=0.49125) showed significantly more inhibition than Drd1a-CRE mice (M=−1.9028, SEM=0.29643), while not differing from Drd2-CRE mice (M=−3.6796, SEM=0.39120).
Based on z-score analysis, we found the proportion of inhibited GPe neurons to be 68.4% in Drd2-CRE, 39.0% in Drd1a-CRE and 62.9% in Drd1a-CRE;D2R-OE mice (Figure 4G,H). A Pearson chi-square test revealed a higher proportion of inhibited cells in Drd1a-CRE;;D2R-OE mice than in Drd1a-CRE mice (p<0.01), with no significant differences compared to Drd2-CRE mice. Inhibited units showed 55%, 71%, and 90% reduction in activity compared to their pre-stimulation values in Drd1a-CRE, Drd1a-CRE;D2R-OE and Drd2-CRE mice, respectively (Figure 4I). The degree of inhibition differed between all three groups [F(2,116)=38.086, p<0.01]. Basal firing rates were not affected by the D2R transgene, but Drd2-CRE mice showed lower basal firing rates than control Drd1a-CRE “No ChR2” mice (Drd1a-CRE “No ChR2” mice: 13.3 ± 1.8 Hz, Drd1a-CRE + ChR2: 10.8 ± 1.0 Hz, Drd1a-CRE;D2R-OE + ChR2: 13.1 ± 1.1 Hz, Drd2-CRE + ChR2: 7.5 ± 0.6 Hz; one-way ANOVA [F(3,242)=7.560, p<0.01], post-hoc Scheffe corrected p<0.05 for comparison between Drd1a-CRE “No ChR2” and Drd2-CRE mice). This in vivo analysis of GPe firing is consistent with fast inhibition by direct pathway collaterals. It further suggests that increased density of bridging collaterals in D2R-OE mice strengthens inhibitory modulation of the GPe, a major node of the indirect pathway.
Changes in bridging collaterals are associated with disrupted motor behavior after stimulation of the direct pathway
Could increased inhibition in the GPe affect the behavioral balance of the direct and indirect pathways? Recently, Kravitz et al. have shown that in vivo stimulation of the direct pathway increases locomotion while stimulation of the indirect pathway inhibits locomotion (Kravitz et al., 2010). Nevertheless, under non-stimulated conditions initiation of movements requires the concurrent and coordinated activation of both pathways (Cui et al., 2013). Here, we hypothesize that the increased inhibition of the GPe associated with the increase in bridging collaterals should affect behavioral activation after direct pathway stimulation and thereby change the coordinated balance of the pathways.
We first analyzed locomotor activity in mice co-injected with AAV-DIO-ChR2 and the non-conditional AAV-Kir2.1AAA that leads to an increase in bridging collaterals without affecting the classical terminal fields (Figure 1B). Co-injection with a control virus led to increased locomotor activity after direct pathway stimulation (Figure 5A,B) and decreased locomotor activity after stimulation of the indirect pathway (Figure 5C,D) as previously described (Kravitz et al., 2010). This effect was specific to laser stimulation as the general motor activity of the mice was not affected during habituation (Figure S5A–F) or when the laser was switched off. Mice injected with DIO-ChR2 that do not express CRE recombinase (CRE-negative mice) remained unaffected by laser stimulation (Figure S5G,H). We then tested mice with increased collateral density in the GPe. Surprisingly, co-injection with AAV-Kir2.1AAA led to a decrease in locomotor activity after stimulating the direct pathway (p=0.005; laser/control vs. laser/Kir2.1AAA; Figure 5A,B), while inhibition of locomotor activity after stimulating the indirect pathway remained unchanged (Figure 5C,D).
Figure 5. Increased bridging collaterals are associated with disrupted behavioral activation of the direct pathway.

(A–D) Behavioral activation after direct pathway stimulation is disrupted in AAV-Kir2.1AAA-injected mice. (A,C) Traces show locomotor activity of Drd1a-CRE;DIO-ChR2 (A) or Drd2-CRE;DIO-ChR2 (C) + AAV-ctl or +AAV-Kir2.1AAA mice during open field performance. Note that AAV-ctl and AAV-Kir2.1AAA are CRE-independent and are expressed in both pathways. (B,D) Summary of locomotor activity during a 30-sec session measured in (A,C) respectively. #p<0.05, ###p<0.001, repeated two-way ANOVA. *p<0.05, ***p<0.001 compared to laser; Data are presented as mean ± SEM (n=5 mice/group). A significant interaction between Kir2.1AAA and laser illumination was observed in the direct pathway (B, p<0.001) but not in the indirect pathway (D). Data are mean only in A,C. Gray boxes represent s.e.m. of basal locomotion (pre+post). See also Figure S5.
Because D2 receptor up-regulation increases bridging collaterals, an effect that is rescued by haloperidol, we also assessed direct pathway-dependent locomotor activation in D2R-OE mice treated with chronic haloperidol. Drd1a-CRE mice crossed with D2R-OE mice were injected with AAV-DIO-ChR2 into the dorsal striatum and were then treated with either saline or haloperidol (1 mg/kg/day) for 2 weeks before analysis of motor behaviors (Figures 6 and S6). As observed in the Kir2.1AAA experiment, direct pathway stimulation increased locomotor activity in Drd1a-CRE;control mice but decreased locomotor activity in Drd1a-CRE;D2R-OE mice (p=0.001; laser/control vs. laser/D2R-OE). The effect size was larger in D2R-OE mice than in AAV-Kir2.1AAA-injected mice, probably due to the fact that the expression area of Kir2.1AAA is restricted to the injection sites whereas D2R are upregulated in the entire striatum of D2R-OE mice (Kellendonk et al., 2006). Basal (unstimulated) locomotor activity was not altered in D2R-OE mice (Figures 6 and S6).
Figure 6. Disrupted behavioral activation of the direct pathway in D2R-OE mice is rescued by haloperidol.

(A,B) Laser stimulation of the direct pathway induces motor inhibition in D2R-OE mice and is rescued after chronic treatment with haloperidol. (A) Traces show laser-induced locomotor activity of Drd1a-CRE;DIO-ChR2;control and Drd1a-CRE;DIO-ChR2;D2R-OE mice treated with either saline or haloperidol (1 mg/kg/day) for 2 weeks. (B) Summary of locomotor activity during a 30-sec session measured in (A).###p<0.0001, repeated two-way ANOVA. ***p<0.0001 compared to laser; Data are presented as mean ± SEM (n=6–8 mice/group). A significant interaction between D2R manipulation (D2R-OE and/or haloperidol) and laser illumination was observed (p<0.0001). Data are mean only in A. Gray boxes represent s.e.m. of basal locomotion (pre+post). Chronic haloperidol has no effect on behavioral inhibition after indirect pathway activation (see Figure S6).
We also tested the effects of chronic haloperidol treatment (2-weeks, 1 mg/kg/day) on direct pathway-dependent locomotion. Laser stimulation of the direct pathway in Drd1a-CRE;control mice treated with haloperidol efficiently induced locomotion to levels of saline-treated mice despite decreased baseline activity (p=0.001; pre/post control+saline vs. pre/post control+haloperidol; Figures 6 and S6). Remarkably, chronic treatment with haloperidol reversed the locomotor phenotype observed in D2R-OE mice to the level of control mice. This reversal was also observed using a same-subject longitudinal design in a subgroup of mice that was tested before and after chronic haloperidol treatment (Figure S6E). Chronic haloperidol treatment had no effect on motor activity after stimulation of the indirect pathway in Drd2-CRE mice (Figure S6) despite the fact that the indirect pathway predominantly expresses D2Rs.
Discussion
Neuronal activity has been found to be important during development for regulating axonal projections and establishing proper connectivity between different brain regions (Catalano and Shatz, 1998; De Marco Garcia et al., 2011; Hua et al., 2005). De Marco et al. have recently shown that during embryonic development decreasing excitability via overexpression of inward rectifying potassium (Kir) channels impairs proper axonal path finding of cortical interneurons (De Marco Garcia et al., 2011). In the adult animal, decreasing excitability of olfactory sensory neurons via Kir overexpression also disrupts the formation of a proper olfactory map (Yu et al., 2004). However, olfactory sensory neurons regenerate throughout adulthood and the olfactory system is known for its high degree of plasticity. Here, using Kir-expressing viruses we show that manipulating excitability in the adult brain alters axonal terminal fields in the basal ganglia, a structure that is a priori not known for its structural plasticity. In contrast to what has been observed during development and the olfactory system, structural plasticity of bridging collaterals is not cell-autonomous since collaterals of direct pathway grow after increasing excitability of indirect pathway neurons.
It is currently unclear why this anatomical re-organization is specific for the bridging collaterals of the direct pathway and does not affect the classical terminal fields of the direct and indirect pathways. We believe that this is related to the non-cell autonomous nature of the mechanism. Altered activity of the indirect pathway may lead to the release of growth factors within the GPe (by indirect pathway terminals or GPe target neurons) that are specifically recognized by direct pathway bridging collaterals. This is particularly exemplified by the locally-restricted growth of bridging collaterals that remain in the vicinity of AAV-DIO-Kir2.1AAA-infected striatopallidal projections.
What are the potential physiological mechanisms involved in this anatomical rearrangement? Since neuronal excitability of indirect pathway neurons seemed to be critical for the growth of direct pathway collaterals, we focused on regulatory mechanisms specific to this neuronal population. Striatopallidal neurons of the indirect pathway predominantly express D2 receptors. Chronic D2R overexpression increases excitability of the indirect pathway via down-regulation of Kir channels whereas chronic blockade of D2Rs with raclopride decreases striatopallidal excitability (Cazorla et al., 2012; Chan et al., 2012). Changes in excitability of indirect pathway neurons through regulation of Kir potassium conductances or by chronic blockade of DA D2 receptors revealed plasticity in bridging collaterals into the GPe. Surprisingly, this anatomical rearrangement is bi-directionally regulated by D2Rs and is extremely plastic in adult animals. For instance, increased collaterals in D2R-OE mice disappear two weeks after restoring D2R expression to normal levels and then reappear after reinstating D2R overexpression (by switching the transgene off and then on). Importantly, pharmacological blockade of D2R function in the adult animal with chronic administration of haloperidol also led to a decrease in direct pathway collaterals without affecting the classical terminal fields of the direct and indirect pathways. This demonstrates that chronic administration of antipsychotic medication in adulthood leads to specific changes in neuronal connectivity in the adult basal ganglia circuitry.
GABAergic neurons of the indirect pathway are known to inhibit the GPe. In line with this we found that optogenetic activation of the indirect pathway in vivo inhibited GPe activity. Surprisingly, we found that optogenetic activation of the direct pathway also inhibited GPe neurons in vivo with the same latency as activation of the indirect pathway. This inhibition was significantly increased in D2R-OE mice that possess excess bridging collaterals suggesting that increased bridging collaterals could be responsible for the increase in inhibition via monosynaptic inhibition. The direct and the indirect pathways have classically been thought as two functionally opposing pathways (Albin et al., 1989; Maia and Frank, 2011). This holds true for motor behaviors and is consistent with optogenetic studies showing direct pathway stimulation produces locomotor activation whereas indirect pathway stimulation inhibits locomotion (Kravitz et al., 2010). The opposing function of both pathways is also discussed in the context of motivation and cognition, with the direct pathway being involved in learning from positive outcomes and the indirect pathway in learning from negative outcomes (Durieux et al., 2009; Ferguson et al., 2010; Frank et al., 2004; Graybiel, 2000; Hikida et al., 2010; Kravitz et al., 2010; Kravitz et al., 2012; Tai et al., 2012). Other models propose coordinated activation of both pathways that could be simultaneous or sequential in nature (Hikosaka et al., 2000; Mink, 2003; Nambu, 2008) or could rather involve the direct and the hyper-direct pathway (Leblois et al., 2006). Coordinated activation of direct and indirect pathways in motor control is supported by a recent study using in vivo calcium imaging showing that both pathways are co-activated during locomotor initiation (Cui et al., 2013). The density of bridging collaterals could be the mean to regulate the coordinated actions of both pathways.
The fact that the observed anatomical changes are selective for the bridging collaterals of the direct pathway and that the behavioral activation of the direct – but not indirect – pathway is disrupted implies a functional role of these collaterals in regulating striatal-dependent behaviors. Our in vivo recordings suggest that bridging collaterals may form a functional bridge between the direct and the indirect pathways, hence acting as a concurrent “brake” when the “accelerator” (direct) pathway is activated (Graybiel, 2000). To test this idea, we used the open field experiment designed by Kravitz et al. (Kravitz et al., 2010) as a read-out for direct and indirect pathways strength. In both mouse models with increased bridging collaterals (AAV-Kir2.1AAA and D2R-OE mice) behavioral activation of the direct pathway was disrupted, in line with the observation of the selective anatomical changes in the direct pathway. In fact, direct pathway stimulation leads to decreased locomotor activity in Kir2.1AAA and D2R-OE mice, while wild-type mice show increased locomotor activity. Strikingly, this decrease is reversed by chronic pharmacological blockade of D2Rs emphasizing the importance of D2R function in this plasticity.
The decrease in locomotor activity after direct pathway activation in Kir2.1AAA and D2R-OE mice may be explained by the increased inhibition of GPe neurons, therefore reinforcing the “brake” action of the indirect pathway. In contrast, retracting collaterals should weaken the indirect pathway. Following this idea, we expected that haloperidol-treated mice that have less bridging collaterals would show facilitated locomotor activity after direct pathway stimulation due to decreased opposition by collateral input to the GPe. However, we did not observe this, possibly due to a ceiling effect of optogenetic stimulation.
Although the behavioral data are consistent with increased inhibition by GPe collaterals, it is possible that other mechanisms are contributing to the behavioral inhibition after direct pathway stimulation. For example, increased D2R expression and increased striatal excitability may have led to reversible changes within the striatum that do not allow optimal activation of the direct pathway thereby impairing the efficacy of the striato-nigral synapse. Moreover, functional connectivity within the basal can also be due to plasticity in striatal input. In line with this hypothesis, altered activity within the striatum during postnatal development has been shown to regulate the strength of excitatory input to the striatum (Kozorovitskiy et al., 2012).
The direct and indirect pathways are important not only for regulating motor activity but also for motivational and cognitive behaviors (Maia and Frank, 2011). Regulating the extent of bridging collaterals may therefore be important not only for refining locomotion but also for regulating cognitive and motivational behaviors. For instance, D2R-OE mice show deficits in motivation and cognition that resemble negative and cognitive symptoms observed in patients with schizophrenia (Bach et al., 2008; Drew et al., 2007; Kellendonk et al., 2006; Simpson et al., 2012; Ward et al., 2012). Both the deficits in motivation and the anatomical changes are reversed after reversing D2R levels to normality in adulthood. It is therefore possible that the changes in bridging collaterals are responsible for the motivational deficits. In contrast, the cognitive deficits persists after reversing D2R over-expression suggesting that the anatomical changes are not sufficient to explain the cognitive deficits (Bach et al., 2008; Kellendonk et al., 2006). To address causality between bridging collaterals and motivation, the collaterals will have to be manipulated directly without affecting neuronal excitability or D2R function. Anatomical changes in the bridging collaterals may also contribute to basal ganglia disorders such as Parkinson’s disease, in which an imbalance of the direct and indirect pathway has been postulated (Albin et al., 1989; Maia and Frank, 2011). In this context, it would be interesting to examine bridging collaterals in mouse models of Parkinson’s disease.
Finally, there is evidence for increased dopamine release and D2Rs stimulation in the striatum (associative caudate) of patients with schizophrenia (Howes et al., 2012; Kegeles et al., 2010; Simpson et al., 2010). D2R-OE mice were originally designed to model this aspect of the disorder (Kellendonk et al., 2006). We therefore hypothesize that increased bridging terminals and the resulting changes in the balance of the direct and indirect pathway may also exist in patients with schizophrenia. Advanced brain-imaging technologies capable of identifying changes at the level of terminal fields may be used in the future to test this hypothesis. In this context it is interesting that chronic treatment with haloperidol, a widely-used antipsychotic medication to treat patients with schizophrenia, reverses both the anatomical alteration and the disrupted behavioral consequence of direct pathway stimulation in D2R-OE mice. The longitudinal study following the same animals before and after treatment strongly suggests that haloperidol corrects the direct pathway-dependent behavioral deficits by reversing the increase in collaterals. Because all effective pharmacotherapies for positive symptoms of schizophrenia target D2Rs, we propose here that these medications possibly attenuate positive symptoms by correcting an anatomical imbalance between the direct and indirect pathways.
Experimental Procedures
Animals
All animal protocols used in the present study were approved by the New York State Psychiatric Institute and Columbia University Institutional Animal Care and Use Committees. The generation of the D2R-OE mice has been described previously(Kellendonk et al., 2006). TetO-D2R mice have been back-crossed for over 15 generations onto the C57BL/6J background and CaMKIIα-tTA mice back-crossed for over 20 generations onto 129SveVTac. To generate D2R-OE mice, tetO-D2R/C57BL6 mice were crossed with CaMKIIa-tTA/129SveV mice. Only double transgenic mice express transgenic D2Rs (D2R-OE mice). Littermates carrying a single transgene (tetO or tTA) or no transgene were used as controls. Drd2-GFP (S118Gsat/Mmnc), Drd1a-GFP (X60Gsat/Mmmh), Drd2-CRE (ER44Gsat/Mmcd) and Drd1a-CRE (FK150Gsat/Mmcd) mice were purchased from MMRRC, and Drd2−/−- (B6.129S2-Drd2tm1Low/J) from Jackson Laboratory. These strains were crossed to obtain D2R-OE;Drd1a-GFP, D2R-OE;Drd2-GFP, Drd2−/−;Drd1a-GFP, Drd2+/−;Drd1a-GFP, Drd2+/−;Drd2-GFP, Drd1a-GFP ;Drd1a-CRE and Drd1a-GFP;Drd2-CRE mice. All controls were littermates. Only adult mice were used in this study. For unknown reasons, no Drd2−/−;Drd2-GFP mice could be obtained. Mice were housed under a 12 h-light/dark cycle in a temperature-controlled environment with food and water available ad libitum.
Drug treatments
Expression of transgenic D2R in D2R-OE mice was switched off by feeding the mice with regular chows supplemented with doxycycline (40 mg/kg diet). Re-expression of transgenic D2R was allowed by replacing doxycycline-supplemented food with regular pellets for 60 days. Re-expression of transgenic D2R was assessed by in situ hybridization(Kellendonk et al., 2006).
Chronic treatment with haloperidol (1 mg/kg/day) was performed using mini-osmotic pumps (Alzet, model 2002) to provide a steady delivery rate of 0.5 µl/h for 14 days. Haloperidol was dissolved in 8.5% lactic acid (6 parts) then neutralized with 1N NaOH (4 parts). Minipumps filled with either lactic acid/NaOH or haloperidol were implanted subcutaneously in the interscapular region under isoflurane anaesthesia.
Histology and quantification of terminal field density
Terminals quantification was estimated by immunohistochemistry against aequorea GFP expressed in Drd1a-GFP and Drd2-GFP mice. Mice were anaesthetized with a mixture of ketamine and xylazine (100mg/kg and 10mg/kg) injected i.p. and transcardiacally perfused with first PBS and then 4% paraformaldehyde. Following perfusion, brains were post-fixed in 4% paraformaldehyde for 16 h, extensively washed and sliced into 50-µm sagittal sections on a vibratome (Leica Microsystems). Immunostaining was performed on free-floating sections from either right or left hemisphere. Every fourth section covering GPe and SNr in their entirety (~6 slices per mouse) was stained using a rabbit polyclonal antibody against aequorea GFP (1:2000, Molecular Probes). The signal was revealed using a biotinylated anti-rabbit (1:1000, Jackson ImmunoResearch), Vectastain ABC Elite kit (Vector Labs) and 3,3’ diaminobenzidine (Vector Labs). Contrast was intensified using 0.025% Nickel Cobalt and 0.02% Nickel Ammonium Sulphate. Sections were dehydrated, cleared and mounted on slides with Permount (Fisher Scientific). Brightfield and darkfield photomicrographs were taken using an AxioImager 2 microscope (Zeiss) connected to an AxioCam video camera and to Neurolucida v.10 (MBF Bioscience). Terminal density was evaluated using GFP staining. Quantification was performed using ImageJ v.1.46r by experimenters blind of genotypes/injections using two random counting frame per structure and per slice. Optical density in the striatum was not affected by genotype or Kir channel expression and used as an internal control so all values are in percentage of striatal optical density. All effects were cross-examined and validated using dark field microscopy (Zeiss) at a higher magnification (10x and 40x) and in some occurrences using synaptophysin quantification (mouse monoclonal anti-synaptophysin, 1:100, Abcam).
Note that antibodies directed against aequorea GFP expressed in Drd1a-GFP and Drd2-GFP mice do not recognize renilla GFP expressed by AAV-ires-HRGFP.
Immunofluorescence was performed on free-floating 50-µm thick sections using rabbit anti-aequorea GFP (1:2000, Molecular Probes), chicken anti-aequorea GFP (1:1000, Abcam), rabbit anti-DsRed (1:1000, Clontech), rabbit anti-CRE (1:1000(Kellendonk et al., 1999)), mouse anti-parvalbumin (1:2000, Sigma), and the appropriate fluorescent-labeled secondary antibodies. Sections were mounted on slides with VectaShield+DAPI (Vector Labs). Images were acquired using an AxioImager fluorescence microscope (Zeiss) or a LSM 510 META confocal system (Zeiss) mounted on an inverted Axiovert 200M microscope (Zeiss) at the HICCC confocal facility (Columbia University).
Virus constructs
AAV2/1-cmv-Kir2.1AAA -ires-HRGFP and AAV2/1-cmv-Kir2.1Wt -ires-HRGFP viruses were generated by inserting mouse Kir2.1AAA or Kir2.1Wt sequences, respectively, into the multiple cloning site of the pAAV-ires-HRGFP vector (Agilent Technologies) as described in (Cazorla et al., 2012). AAV2/1-syn-DIO-Kir2.1AAA-ires-mCherry was generated by replacing the ef1α promoter and ChR2-eYFP sequence with the synapsin promoter and Kir2.1AAA -ires-mCherry sequence into the pAAV-ef1α-DIO-hChR2(H134R)-eYFP vector (kind gift of Karl Deisseroth).
AAV2/1-cmv-Kir2.1AAA-ires-HRGFP (3.09 × 1012 particles/ml) and AAV2/1-cmv-Kir2.1Wt -ires-HRGFP (1 × 1011 particles/ml) were produced at U Penn Vector Core (University of Pennsylvania), AAV2/1-syn-DIO-Kir2.1AAA-ires-mCherry (1 × 1013 particles/ml) was produced at Vector Biolabs, AAV2/1-cmv-HRGFP (3.75 × 1012 particles/ml) was obtained from Vector Biolabs, and AAV2-ef1α-DIO-mCherry (1 × 1012 particles/ml) and AAV5-ef1α-DIO-ChR2(H134R)-eYFP (4 × 1012 particles/ml) were obtained from UNC Vector Core (University of North Carolina). DIO constructs allow gene expression only in cells that express CRE recombinase.
Stereotaxic injections and fiberoptic implantations
Mice were anaesthetized with a mixture of ketamine and xylazine (100mg/kg and 10mg/kg) injected i.p. and placed into a stereotaxic apparatus equipped with a temperature controller to regulate body temperature. Viruses were bilaterally delivered at an average rate of 100 nl/min using glass pipettes (10–15 µm). Two sites of injections were used into the mediodorsal striatum to allow for a broad expression (site A: AP +1.3, ML ± 1.4, DV −3.2; site B: AP +0.8, ML ± 2.0, DV −3.4). 0.4–0.5 µl of AAV2/1-cmv-Kir2.1AAA-ires-HRGFP, AAV2/1-cmv-HRGFP, AAV2/1-syn-DIO-Kir2.1AAA-ires-mCherry and AAV2-ef1α-DIO-mCherry, and 0.5–1.0 µl of AAV2/1-cmv-Kir2.1Wt-ires-HRGFP (lower titer) were injected per site. For double virus injections (AAV2/1-cmv-Kir2.1AAA-ires-HRGFP + AAV5-ef1α-DIO-ChR2(H134R)-eYFP and AAV2/1-cmv-HRGFP + AAV5-ef1α-DIO-ChR2(H134R)-eYFP), an equal volume of each virus was mixed and a final volume of 1.0–1.5 µl was injected per site. After each injection, the pipette remained in situ for 10 min to minimize leaking. For tracing experiments, sparse expression of ChR2 was obtained by injecting low titer of AAV5-ef1α-DIO-ChR2(H134R)-eYFP (4×1012 particles/ml diluted 1:10 in autoclaved, filtered PBS/5% glycerol; 0.44 µl) in the mediodorsal striatum (AP +1.0, ML ± 1.4, DV −3.1) of Drd1a-CRE;control and Drd1a-CRE;D2R-OE mice. Expression of the virus was allowed for a period of 4 weeks.
For optical fibers implantation, the injection glass pipette was removed and fiberoptic implants were placed in the dorsomedial striatum (AP +0.8, ML ± 1.8, DV −3.2). The implant was secured to the skull using dental cement (Dentsply). After wound closure, mice received analgesics and antibiotics and were returned to their home cage for > 4 weeks. Virus spreading and gene expression were assessed after each experiment. Variability was very low and no mice were removed from any experiment.
In vivo optical stimulation
Optical fibers were constructed in-house by interfacing a 200-µm, 0.37-numerical-aperture optical fiber (Fiber Instrument Sales) with a 1.25mm stick zirconium ferrule. The fiber extended 4 mm beyond the end of the ferrule. Fibers were attached with epoxy resin into the ferrules, then cut with a diamond pen and polished. All fibers were calibrated to have >80% efficiency of light transmission. 200-µm core diameter fiberoptic patch-cords were also constructed in house using an FC/PC connectorization kit (Thor Labs).
In vivo recording
Anesthetized mice (chloral hydrate) were implanted with an optic fiber for light stimulation into the site of ChR2 injection in the dorsomedial striatum (AP +1.5, ML ± 1.1, DV −3.3). A glass electrode (impedance 12–14 MΩ) filled with 2M NaCl was lowered into the GPe (AP −0.1, ML: ± 1.8, DV −2.5). The electrode was lowered using a hydraulic microdrive to detect spontaneously active pallidal neurons. From this starting point, the GPe was sampled in 4 locations 0.15 mm-apart and arranged in a 2×2 spaced grid moving in a clockwise direction. The starting locations were counterbalanced across animals and groups. Pallidal neurons were identified using a combination of sterotaxic position and narrow action potential width (< 1 ms). After 2–3 mins of stable recording, optical stimulation (473 nm; 2 mW) was applied for 5s as recording continued. Neuronal activity was amplified and filtered (1000x gain, 100–10K Hz band pass), and fed to an audio monitor and to a computer interface with custom-designed acquisition and analysis software (Neuroscope).
Traces from continuous recordings were analyzed offline, first by applying a window discriminator to identify spikes, then from the spike table to calculate firing rates. Peri-stimulus time histograms were obtained by sampling spike frequency with 100-msec bins 5 seconds before during and after laser illumination. Relative spike frequency is expressed as a function of baseline firing obtained during the 5 seconds preceding laser illumination. Average firing rate during the five seconds of laser stimulation was expressed as a percent change and a z-score of the pre-stimulation firing rate distribution which was determined from five 1-sec bins pseudorandomly sampled from the 60 sec prior to stimulation. Cells with z-scores more negative than −2 (cut-off for a one-tailed p<0.05) were defined as being significantly inhibited by striatal efferent stimulation. Proportion of cells with significant changes in spike firing following laser stimulation and amplitude of inhibition calculated as ([1-post/pre firing rate]*100) were also analyzed and compared.
Data analysis and statistics
Data were analyzed using Prism 5 (GraphPad) and Statview. Terminal fields density was analyzed using unpaired Student’s t test when only two conditions were compared within a group (GPe or SNr) or one-way ANOVA followed by a Bonferroni post-hoc test when more than two conditions were compared within a group. In vivo recordings were analyzed using a paired t test to compare pre to post firing rate distribution, a Pearson’s chi-squared test to compare the proportion of inhibited units across different groups and one-way ANOVA followed by post-hoc comparisons for the degree of inhibition (expressed as both z-score and % change from pre-stimulation spike firing). Motor activity was analyzed using two-way ANOVA with repeated measures. p values of repeated ANOVAs are given for each type of activity. Virus expression (AAV-ctl or AAV-Kir2.1AAA), genotypes (control or D2R-OE mice), treatment (saline or haloperidol) and laser illumination are independent variables. All statistical analyzes were conducted with an alpha level of 0.05. Data are presented as mean ± SEM unless otherwise stated. n values indicate the number of mice per group.
Supplementary Material
Acknowledgements
We would like to thank Bhavani Ramesh for excellent technical help and Pierre Trifilieff, Jonathan Javitch, Mark Ansorge and members of the Kellendonk lab for thoughtful discussions of the manuscript. We would like to thank Karl Deisseroth for the ChR2 construct. This work was supported by the National Institute of Mental Health, the Silvio O. Conte Center for Schizophrenia Research MH086404 (CK, HM, MOC, NC, SR), and R01MH093672 (CK, MC). Behavioral analyses were performed with support from the Rodent Neurobehavioral Analysis Core at the New York State Psychiatric Institute. MC, CK, HM, NC, SR designed experiments; MC, MS, FDC, MOC performed experiments; SEA supervised the ChR2 studies; MC, CK wrote the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annual Review of Neuroscience. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- Bach ME, Simpson EH, Kahn L, Marshall JJ, Kandel ER, Kellendonk C. Transient and selective overexpression of D2 receptors in the striatum causes persistent deficits in conditional associative learning. Proc Natl Acad Sci U S A. 2008;105:16027–16032. doi: 10.1073/pnas.0807746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano SM, Shatz CJ. Activity-dependent cortical target selection by thalamic axons. Science. 1998;281:559–562. doi: 10.1126/science.281.5376.559. [DOI] [PubMed] [Google Scholar]
- Cazorla M, Shegda M, Ramesh B, Harrison NL, Kellendonk C. Striatal D2 receptors regulate dendritic morphology of medium spiny neurons via Kir2 channels. J Neurosci. 2012;32:2398–2409. doi: 10.1523/JNEUROSCI.6056-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Peterson JD, Gertler TS, Glajch KE, Quintana RE, Cui Q, Sebel LE, Plotkin JL, Shen W, Heiman M, et al. Strain-specific regulation of striatal phenotype in Drd2-eGFP BAC transgenic mice. J Neurosci. 2012;32:9124–9132. doi: 10.1523/JNEUROSCI.0229-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, Costa RM. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 2013;494:238–242. doi: 10.1038/nature11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marco Garcia NV, Karayannis T, Fishell G. Neuronal activity is required for the development of specific cortical interneuron subtypes. Nature. 2011;472:351–355. doi: 10.1038/nature09865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew MR, Simpson EH, Kellendonk C, Herzberg WG, Lipatova O, Fairhurst S, Kandel ER, Malapani C, Balsam PD. Transient overexpression of striatal D2 receptors impairs operant motivation and interval timing. J Neurosci. 2007;27:7731–7739. doi: 10.1523/JNEUROSCI.1736-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux PF, Bearzatto B, Guiducci S, Buch T, Waisman A, Zoli M, Schiffmann SN, de Kerchove d’Exaerde A. D2R striatopallidal neurons inhibit both locomotor and drug reward processes. Nat Neurosci. 2009;12:393–395. doi: 10.1038/nn.2286. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Eskenazi D, Ishikawa M, Wanat MJ, Phillips PE, Dong Y, Roth BL, Neumaier JF. Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci. 2010;14:22–24. doi: 10.1038/nn.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MJ, Seeberger LC, O’Reilly R C. By carrot or by stick: cognitive reinforcement learning in parkinsonism. Science. 2004;306:1940–1943. doi: 10.1126/science.1102941. [DOI] [PubMed] [Google Scholar]
- Fujiyama F, Sohn J, Nakano T, Furuta T, Nakamura KC, Matsuda W, Kaneko T. Exclusive and common targets of neostriatofugal projections of rat striosome neurons: a single neurontracing study using a viral vector. Eur J Neurosci. 2011;33:668–677. doi: 10.1111/j.1460-9568.2010.07564.x. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, Surmeier DJ. Dichotomous anatomical properties of adult striatal medium spiny neurons. J Neurosci. 2008;28:10814–10824. doi: 10.1523/JNEUROSCI.2660-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Graybiel AM. The basal ganglia. Curr Biol. 2000;10:R509–R511. doi: 10.1016/s0960-9822(00)00593-5. [DOI] [PubMed] [Google Scholar]
- Hikida T, Kimura K, Wada N, Funabiki K, Nakanishi S. Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron. 2010;66:896–907. doi: 10.1016/j.neuron.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Takikawa Y, Kawagoe R. Role of the basal ganglia in the control of purposive saccadic eye movements. Physiol Rev. 2000;80:953–978. doi: 10.1152/physrev.2000.80.3.953. [DOI] [PubMed] [Google Scholar]
- Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, Kapur S. The Nature of Dopamine Dysfunction in Schizophrenia and What This Means for Treatment: Meta-analysis of Imaging Studies. Arch Gen Psychiatry. 2012 doi: 10.1001/archgenpsychiatry.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua JY, Smear MC, Baier H, Smith SJ. Regulation of axon growth in vivo by activity-based competition. Nature. 2005;434:1022–1026. doi: 10.1038/nature03409. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Emson PC. Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. J Neurosci. 1990;10:3421–3438. doi: 10.1523/JNEUROSCI.10-10-03421.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, Hwang DR, Huang Y, Haber SN, Laruelle M. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67:231–239. doi: 10.1001/archgenpsychiatry.2010.10. [DOI] [PubMed] [Google Scholar]
- Kellendonk C, Simpson EH, Polan HJ, Malleret G, Vronskaya S, Winiger V, Moore H, Kandel ER. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning.[see comment] Neuron. 2006;49:603–615. doi: 10.1016/j.neuron.2006.01.023. [DOI] [PubMed] [Google Scholar]
- Kellendonk C, Tronche F, Casanova E, Anlag K, Opherk C, Schutz G. Inducible site-specific recombination in the brain. J Mol Biol. 1999;285:175–182. doi: 10.1006/jmbi.1998.2307. [DOI] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, Sabatini BL. Recurrent network activity drives striatal synaptogenesis. Nature. 2012;485:646–650. doi: 10.1038/nature11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci. 2012;15:816–818. doi: 10.1038/nn.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblois A, Boraud T, Meissner W, Bergman H, Hansel D. Competition between feedback loops underlies normal and pathological dynamics in the basal ganglia. J Neurosci. 2006;26:3567–3583. doi: 10.1523/JNEUROSCI.5050-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque M, Parent A. The striatofugal fiber system in primates: a reevaluation of its organization based on single-axon tracing studies. Proc Natl Acad Sci U S A. 2005;102:11888–11893. doi: 10.1073/pnas.0502710102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia TV, Frank MJ. From reinforcement learning models to psychiatric and neurological disorders. Nat Neurosci. 2011;14:154–162. doi: 10.1038/nn.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Micklem BR, Henny P, Brown MT, Williams C, Bolam JP, Nakamura KC, Magill PJ. Dichotomous organization of the external globus pallidus. Neuron. 2012;74:1075–1086. doi: 10.1016/j.neuron.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink JW. The Basal Ganglia and involuntary movements: impaired inhibition of competing motor patterns. Arch Neurol. 2003;60:1365–1368. doi: 10.1001/archneur.60.10.1365. [DOI] [PubMed] [Google Scholar]
- Nambu A. Seven problems on the basal ganglia. Curr Opin Neurobiol. 2008;18:595–604. doi: 10.1016/j.conb.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Simpson EH, Kellendonk C, Kandel E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron. 2010;65:585–596. doi: 10.1016/j.neuron.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson EH, Waltz JA, Kellendonk C, Balsam PD. Schizophrenia in translation: dissecting motivation in schizophrenia and rodents. Schizophr Bull. 2012;38:1111–1117. doi: 10.1093/schbul/sbs114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LH, Lee AM, Benavidez N, Bonci A, Wilbrecht L. Transient stimulation of distinct subpopulations of striatal neurons mimics changes in action value. Nat Neurosci. 2012;15:1281–1289. doi: 10.1038/nn.3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Bertran-Gonzalez J, Herve D, Fisone G, Girault JA. Looking BAC at striatal signaling: cell-specific analysis in new transgenic mice. Trends Neurosci. 2009;32:538–547. doi: 10.1016/j.tins.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Ward RD, Simpson EH, Richards VL, Deo G, Taylor K, Glendinning JI, Kandel ER, Balsam PD. Dissociation of hedonic reaction to reward and incentive motivation in an animal model of the negative symptoms of schizophrenia. Neuropsychopharmacology. 2012;37:1699–1707. doi: 10.1038/npp.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Richard S, Parent A. The organization of the striatal output system: a single-cell juxtacellular labeling study in the rat. Neurosci Res. 2000;38:49–62. doi: 10.1016/s0168-0102(00)00140-1. [DOI] [PubMed] [Google Scholar]
- Yu CR, Power J, Barnea G, O’Donnell S, Brown HE, Osborne J, Axel R, Gogos JA. Spontaneous neural activity is required for the establishment and maintenance of the olfactory sensory map. Neuron. 2004;42:553–566. doi: 10.1016/s0896-6273(04)00224-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.