

Abstract
Background
Smooth muscle cells (SMC) are remarkably plastic. Their reversible differentiation is required for growth and wound healing, but also contributes to pathologies including atherosclerosis and restenosis. While key regulators of the SMC phenotype including myocardin (MYOCD) and KLF4 have been identified, a unifying epigenetic mechanism that confers reversible SMC differentiation has not been reported.
Methods and Results
Using human SMC, human arterial tissue, and mouse models, we report that SMC plasticity is governed by the DNA modifying enzyme ten-eleven translocation-2 (TET2). TET2 and its 5-hydroxymethylcytosine (5-hmC) product are enriched in contractile SMC but reduced in dedifferentiated SMC. TET2 knockdown inhibits expression of key pro-contractile genes including MYOCD and SRF with concomitant transcriptional upregulation of KLF4. TET2 knockdown prevents rapamycin-induced SMC differentiation, while TET2 overexpression is sufficient to induce a contractile phenotype. TET2 overexpression also induces SMC gene expression in fibroblasts. Chromatin immunoprecipitation demonstrates that TET2 coordinately regulates phenotypic modulation through opposing effects on chromatin accessibility at the promoters of pro-contractile versus dedifferentiation-associated genes. Notably, we find that TET2 binds, and 5-hmC is enriched, in CArG-rich regions of active SMC contractile promoters (MYOCD, SRF, and MYH11). Loss of TET2 and 5-hmC positively correlates with the degree of injury in murine models of vascular injury and human atherosclerotic disease. Importantly, localized TET2 knockdown exacerbates injury response while local TET2 overexpression restores the 5-hmC epigenetic landscape, contractile gene expression, and greatly attenuates intimal hyperplasia in vivo.
Conclusions
We identify TET2 as a novel and necessary master epigenetic regulator of SMC differentiation.
Keywords: smooth muscle, differentiation, gene expression/regulation, epigenetics, hyperplasia
Introduction
Unlike most mature cells, smooth muscle cells (SMC) are remarkably plastic and can dedifferentiate in response to environmental cues,1, 2 adding a layer of complexity to the regulation of gene expression. While several transcription factors have been identified, a global mechanism that coordinately regulates SMC phenotype has yet to be uncovered. How SMC genes become silenced and then reactivated is unknown and is an area of intense investigation. Recent demonstration that the ten-eleven-translocation (TET) family of proteins is involved in DNA demethylation3–5 prompted us to evaluate the role of the TET proteins in the modulation of SMC phenotype.
The TET proteins (TET1-3) are a recently discovered family of DNA demethylases. TET proteins oxidize 5-methylcytosine (5-mC) to generate 5-hydroxymethylcytosine (5-hmC), frequently coined the “6th” DNA base, in mammalian cells.4, 5 Through the base excision repair pathway, 5-hmC is then converted to unmethylated cytosine, leading to DNA demethylation and gene activation.6–8 As such, the 5-hmC modification and the TET enzymes have emerged as key activators of gene expression. Studies on TET proteins and 5-hmC function in embryonic stem cells (ESC) demonstrate that they play a major role in maintaining cellular pluripotency through the regulation of lineage specific genes.4, 9–11 In contrast to this role in ESC pluripotency, the TET proteins (and their 5-hmC product) have an opposing role in adult stem cells and somatic tissues. TET2 mutations have been described in several types of hematopoietic disorders where the loss of TET2 has been shown to promote hematopoietic stem cell self-renewal.12 TET2 and 5-hmC levels are increased during neurogenesis,13 and more recently, loss of TET2 and 5-hmC was demonstrated to be a key epigenetic event associated with aggressive melanoma.14 Collectively, these studies suggest a cell- and/or tissue-specific effect of the TET proteins and the 5-hmC mark on gene activation. Herein we present data using both in vitro and in vivo knockdown and overexpression approaches to functionally characterize the role of TET2 and its 5-hmC product in regulating the SMC phenotype. We find that TET2 expression is necessary for SMC differentiation, and that TET2 binds to the SRF and MYOCD promoters under differentiation conditions. Additionally we show that TET2 regulates the cellular program by directly modulating chromatin accessibility of target genes. Furthermore, we report that TET2 activated gene promoters are enriched with the 5-hmC mark, resulting in strong gene activation. Overall, we demonstrate a novel epigenetic pathway by which contractile and synthetic genes are regulated simultaneously in SMC. In addition to demonstrating a critical role for TET2 in adult somatic cells in health and disease, our study suggests that targeting the TET2-5-hmC pathway may be a therapeutic strategy for treatment of diseases associated with aberrant SMC differentiation.
Methods
Please refer to the Online Data Supplement for an extended description of the Materials and Methods. Primer sequences for qPCR analysis of gene expression are provided in Supplemental Tables 1–2.
Human atherosclerotic tissues
Research protocols were approved by the institutional review boards of the West Haven VA Hospital, Yale University, and the New England Organ Bank. Informed consent was obtained and human coronary arteries were obtained as previously described.15, 16 The presence of atherosclerosis was documented at the time of cardiac procurement by an experienced cardiac surgeon. Pliable, translucent coronary arteries were deemed normal and designated as healthy controls. Opaque coronary arteries were classified as atherosclerotic to various degrees (mild, moderate, and severe). The macroscopic diagnosis was confirmed by histology.
Femoral artery wire injury
All experiments were approved by the Institutional Animal Care and Use Committees of Yale University and in adherence to the NIH Guide for the Care and Use of Laboratory Animals. Femoral arteries were injured as previously described.17, 18 For localized virus delivery, viruses (1 × 107 pfu) were delivered to the artery at the time of injury by painting the virus mixture (15 μl concentrated viral supernatant and 35 μl Pluronic-127 gel) circumferentially around the injured femoral artery. The injured and uninjured femoral arteries were collected for cryosectioning 3 weeks post-surgery.
Cell culture
hCASMC were purchased from Cascade Biologics (Portland, OR) and cultured as previously described.19–22 MRC5 cells were purchased from ATCC and cultured in 10% FBS. HUVEC were cultured in EBM-2 basal medium and associated supplements (Lonza Biologicals). Passage 4–7 cells were used for all experiments.
Chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR)
ChIP was performed as previously described with slight modifications.23 Briefly, 5 × 106 cells were cross-linked and used for each immunoprecipitation. DNA was sheared to 500–1000 bp by sonication. Protein G Dynabeads (Invitrogen) were used to pulldown the antibody-antigen complexes immunoprecipitated with antibodies against TET2 (Abcam), H3K4me3 and H3K27me3 (Cell Signaling). H3 (Cell Signaling) and IgG were also included as positive and negative controls, respectively. Immunoprecipitated DNA was extracted with phenol-chloroform, ethanol precipitated and eluted. Recovered DNA was analyzed by qPCR. Primers spanning the promoter regions (within 2500 bp of transcription start site) of MYH11, MYOCD, SRF, and KLF4 genes were used to amplify input and immunoprecipitated DNA. Primers were designed to span a CArG (CC(A/T)6GG) element and sequences are listed in Supplementary Table 3. All samples were performed in at least triplicate, from at least two independent experiments, and data were normalized to % input.
Methylated-DNA capture (MethylCap)
Enrichment of methylated DNA was performed using the MethylCap kit (Diagenode).24 Genomic DNA was diluted to reach 0.1 μg/μl and then sheared to 200 – 500 bp. Methylated DNA was captured by incubation with a His6-GST-MBD protein (the methyl-binding domain (MBD) of the human MeCP2 protein fused with a glutathione-S-transferase (GST) and containing an N-terminal His6-tag) coupled to magnetic beads. The beads were washed and DNA eluted. The MBD-bound (methylated) and input fractions were analyzed by qPCR to confirm enrichment of the methylated genes. Primer sequences can be found in Supplementary Table 3.
Hydroxymethylated DNA immunoprecipitation (hMeDIP)
Immunoprecipitation of 5-hmC was performed using the hMeDIP kit (Diagenode) per the manufacturer’s instructions. Samples were sonicated as for MethylCap and 1 μg of fragmented genomic DNA was immunoprecipitated with a 5-hmC antibody. The DNA-antibody mixture was incubated with magnetic beads, washed and DNA isolated. Endpoint analysis was performed using locus-specific hMeDIP-qPCR. Primer sequences are listed in Supplementary Table 3.
Glucosylation-coupled methylation-sensitivity qPCR (GlucMS-qPCR)
Sequence specific detection of 5-hmC and 5-mC as a percentage of total C was performed using the Quest 5-hmC Detection Kit (Zymo Research) based on published methods.11 Genomic DNA (1 μg) was treated with T4 Phage β-glucosyltransferase, a highly specific 5-hmC glucosyltransferase, that adds a glucose moiety to 5-hmC (generating glucosyl-5-hmC, 5-ghmC). Glucosylated genomic DNA was then digested with MspI which recognizes and cleaves mC, 5-mC and 5-hmC but not 5-ghmC. Locus specific detection of 5-hmC was determined by qPCR using primers designed to flank at least one MspI site. Primer sequences used are listed in Supplementary Table 4.
Dot blot
Cellular genomic DNA was isolated using the Qiagen kit while genomic DNA from tissue sections was isolated using the Pinpoint™ Slide DNA Isolation System (Zymo Research). DNA was blotted on to a nitrocellulose membrane and incubated with anti-5-hmC (1:10000, Active Motif) overnight at 4°C. After incubating with HRP-conjugated IgG secondary antibody, the signal was visualized with ECL. Methylene blue (MB) staining of the membranes was used to assess equal DNA loading.
Statistical analysis
Data are expressed as mean ± SD. Comparisons between two groups were calculated using the Mann-Whitney test. The Kruskal-Wallis test followed by the Dunn’s multiple comparison test was performed for data between more than two groups. P-values were two-tailed and values <0.05 were considered to indicate statistical significance. Analyses were performed using Prism 4.0 software (GraphPad). Intra-class correlation coefficients were calculated using an online tool (http://department.obg.cuhk.edu.hk/researchsupport/IntraClass_correlation.asp).
Results
TET2 and 5-hmC are up-regulated during SMC differentiation
The role of the TET family of enzymes in an adult somatic cell such as SMC is yet to be determined. We evaluated the expression of TET proteins in human coronary artery SMC (hCASMC) cultures and found TET2 to be the most highly expressed isoform in these cells (Figure 1A). Remarkably, TET2 transcript and protein levels were significantly higher in hCASMC when compared to human ESC (hESC). To explore a possible role for the TET family members in modulating the SMC phenotype, we induced SMC differentiation by treatment with the mTORC1 inhibitor rapamycin21, 22 or dedifferentiation with platelet-derived growth factor-BB (PDGF-BB).25 Both TET2 transcript and protein levels were elevated with differentiation, but repressed with PDGF-BB treatment (Figure 1B, Supplementary Figure 1). The changes in TET2 were of greater magnitude and of clear statistical significance compared to the modest changes observed with TET1 and TET3 (Figure 1B, Supplementary Figure 1A–D). Notably, TET2 expression closely correlated temporally with SMC differentiation markers such as smooth muscle-myosin heavy chain (MYH11) and smooth muscle actin (ACTA2) but inversely correlated with KLF4 expression (Figure 1B and Supplementary Figure 1E–G). Given its abundant expression and modulation during SMC differentiation, we focused our further studies on TET2. We determined that Tet2 is abundantly expressed in multiple SMC-rich murine tissues (Supplementary Figure 2). In the vasculature, Tet2 was highly expressed in SMC isolated from the medial layer of normal mouse aorta but dramatically decreased with culture in 10% serum, in parallel with Myh11 expression (Figure 1C). These levels were partially rescued with rapamycin (Figure 1C). As the enzymatic function of the TET proteins is to convert 5-mC to 5-hmC,4, 5, 9 we measured 5-hmC levels in SMC. Genomic dot blot analysis revealed a global increase in 5-hmC levels in differentiated SMC and reduced 5-hmC levels in dedifferentiated cultures (Figure 1D). These results demonstrate that both TET2 and its 5-hmC product correlate with the SMC differentiated phenotype.
Figure 1.
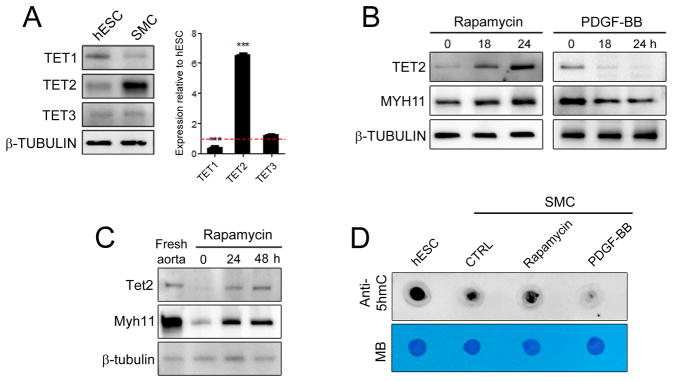
TET2 is associated with the differentiated SMC state. (A) Western blot showing protein levels of TET1, TET2 and TET3 in human coronary artery smooth muscle cells (SMC) compared to human embryonic stem cells (hESC). Corresponding relative mRNA levels are shown on the right (qPCR). Relative expression of hESC is set to 1.0 and denoted by the red line. Data are presented as mean ± SD for three independent experiments. ***P<0.001. (B) Western blot showing TET2 and MYH11 levels in hCASMC following 50 nM rapamycin or 5 ng/ml PDGF-BB treatment. (C) Western blot comparing Tet2 and Mhy11 levels from freshly isolated mouse aorta tissue and cultured cells. (D) Dot blots for global 5-hmC expression using genomic DNA isolated from hESC and hCASMC treated as in (B) for 48 hours. Methylene blue (MB) staining demonstrates equal loading.
TET2 and 5-hmC levels are high in mature SMC and are reduced following vascular injury and disease
To understand the physiological role of TET2 and 5-hmC in vivo, we examined TET2 and 5-hmC expression in murine and human models of vascular injury and disease. Intimal hyperplasia was induced using the mouse femoral artery wire injury model.18 In the uninjured vessels, Tet2-expressing cells comprised >95% of the total number of medial cells (n= 4). Tet2-expressing cells were reduced in the injured medial layer and only 16% of cells in the neointima were Tet2-positive at 3 weeks post-injury (n=5) (Figure 2A, quantified in Figure 2B). RNA analysis further confirmed that vessel injury reduced the levels of Tet2 and contractile genes in the media and neointima (Figure 2C). 5-hmC staining (Figure 2A, quantified in Figure 2D) and genomic levels (Figure 2E) closely paralleled Tet2 expression patterns. These results support our in vitro findings that high Tet2 and 5-hmC levels are associated with the mature, differentiated SMC phenotype, while significant loss of Tet2 and 5-hmC are characteristic of dedifferentiated SMC.
Figure 2.

TET2 is downregulated during vascular injury. (A) TET2 and 5-hmC immunostaining of cross-sections from uninjured contralateral control mouse femoral arteries (top row) or injured femoral arteries (bottom row) collected 3 weeks following wire injury. Nuclei are stained with DAPI. (B) Quantification of TET2 positive cells divided by total number of DAPI positive cells in the uninjured (n=4) compared to injured (n=5) mouse femoral arteries. **P<0.01 relative to uninjured media. (C) qPCR for Tet2, Myocd and Myh11 gene expression in uninjured compared to injured femoral arteries 3 weeks post-injury. RNA was isolated from media and neointima from sections on slides using the Pinpoint isolation system. The adventitia was removed to ensure that only the media and neointima were used for analysis. *P<0.05 relative to uninjured arteries. (D) Quantification of 5-hmC positive cells divided by total number of DAPI positive cells in the uninjured (n=4) versus injured (n=5) femoral arteries. **P<0.01 relative to uninjured media. (E) Dot blot for global 5-hmC levels of uninjured and injured samples (n=4). Genomic DNA was isolated from tissue sections using the Pinpoint isolation system as in (C). Fold induction levels were calculated based on pixel intensity using Image Lab Software (**P<0.01). M = media, N = neointima. Scale bar, 50 μm. Error bars represent mean ± SD. n represent the number of individual mice. Two or more slides (12–18 sections) per animal were used for analysis.
We next sought to assess TET2 and 5-hmC levels in human vascular disease. SMC dedifferentiation is a major contributor to atherosclerosis.26, 27 We analyzed human coronary artery biopsies from 16 individuals with normal arteries or demonstrating mild, moderate or severe atherosclerosis. MYH11 staining was used to identify differentiated SMC within the individual patient samples. MYH11 intensity inversely correlated with the severity of disease and TET2 expression correlated closely with the MYH11 staining (Figure 3A, quantified in Figure 3B). Consistently, global 5-hmC levels were also significantly reduced in atherosclerotic samples (Figure 3C). Taken together, these results implicate TET2 and its product, 5-hmC, as novel biomarkers for diseases associated with SMC dedifferentiation.
Figure 3.

TET2 and 5-hmC levels are high in mature human SMC and are lost in atherosclerotic lesions. (A) H&E staining (row 1), TET2 and MYH11 immunostaining (rows 2–5) of human coronary arteries with various degrees of atherosclerosis. The boxed areas in 4X sections are enlarged and shown in the lower panels. Scale bar, 50 μm. (B) Quantitation of the percent of TET2 expressing cells over total number of cells. n represent the number of individuals analyzed. Two sections from each patient (with five to eight random fields chosen) were captured and counted. Error bars represent mean ± SD. **P<0.01 relative to healthy subjects. (C) Dot blot of 5-hmC levels of the various human samples using the Pinpoint isolation system as in Figure 1E.
Disruption of TET2 results in impaired SMC differentiation
To assess a causal role for TET2 in SMC phenotypic modulation, we generated short hairpin RNA (shRNA) constructs targeting the human TET2 gene (shTET2) (Supplementary Figure 3A). TET2 knockdown was highly efficient and specific (Supplementary Figure 3B). Significant reductions in SMC differentiation gene expression and 5-hmC levels were observed with TET2 knockdown in hCASMC (Figure 4A–C). We found that TET2 is an obligate effector of rapamycin-induced differentiation, as TET2 knockdown prevented rapamycin-induced contractile gene expression (Figure 4D) and morphology (Supplementary Figure 3C). Additionally, we observed enhanced proliferation in shTET2 cultures as determined by propidium iodide staining and flow cytometry (Figure 4E), and mRNA levels for cyclins and cyclin-dependent kinase inhibitors supported these observations (data not shown). TET2 knockdown also significantly increased expression of markers associated with the synthetic SMC phenotype, such as KLF428–31, KLF532, osteopontin (OPN)33 and embryonic type nonmuscle myosin heavy chain-B (MYH10)34 compared to shCTRL cells (Figure 4F and Supplementary Figure 3D). On the other hand, TET2 over-expression promoted many aspects of differentiation in hCASMC, including increased contractile gene levels, elevated global 5-hmC expression (Figure 4G–H, Supplementary Figure 4A), a contractile morphology even in the absence of differentiation stimuli, decrease in KLF4 and other de-differentiation gene expression levels, and anti-proliferative gene expression (Supplementary Figure 4B–D). Together, these results suggest that TET2 is both necessary and sufficient for SMC differentiation.
Figure 4.

TET2 regulates SMC phenotype. (A) Western blot of contractile genes following TET2 knockdown (shTET2) in hCASMC. (B) Dot blot of 5-hmC in control (shCTRL) compared to shTET2 cells. (C–D) qPCR for contractile genes in shCTRL or shTET2 hCASMC treated with vehicle or 50 nM rapamycin for 24 hours. (E) Percentage of hCASMC in each cell cycle phase following TET2 knockdown compared to control. (F) Western blot for dedifferentiation genes in shTET2 cells compared to shCTRL cells. (G) Western blot of contractile markers in TET2 overexpressing hCASMC compared to control cells. (H) Dot blot of global 5-hmC in TET2 overexpressing hCASMC compared to control cells. (I) Western blot of SMC contractile markers in control and TET2 overexpressing MRC5 fibroblasts. (J) ACTA2 and MYH11 immunostaining of MRC5 fibroblasts infected with a control or TET2 overexpression virus. (K) Quantitation of ACTA2 and MYH11 positive cells in TET2 overexpressing MRC5 cells. Data are presented as mean ± SD for three independent experiments. *P<0.05, **P<0.01, ***P<0.001 compared control cells.
Ectopic expression of TET2 directs smooth muscle cell differentiation
As TET2 knockdown and overexpression had such profound effects on the SMC phenotype, we investigated whether ectopic expression of TET2 is sufficient to convert fibroblasts into mature SMC. The human fetal lung fibroblast line MRC5 expressed moderate levels of TET2 at baseline compared to hCASMC. Following five days of infection with lentivirus to overexpress TET2, TET2 levels were elevated almost 3-fold compared to control MRC5 cells. However, this was still less than the level of TET2 expression observed at baseline in hCASMC (Figure 4I). Moderate levels of ACTA2 and low levels of MYH11 were observed in MRC5 cells at baseline, as detected by western blot and immunostaining (Figure 4I and J, top panels). Introduction of TET2 into these fibroblasts was sufficient to upregulate numerous SMC markers such as ACTA2, MYH11, SRF and MYOCD as detected by western blot, immunostaining, and qPCR (Figure 4I–K, Supplementary Figure 5A). Significant increases in Myocd, Srf and Myh11 gene expression were also observed with TET2 overexpression in mouse NIH3T3 fibroblasts (Supplementary Figure 5B). The fibroblast marker S100A4 was also reduced with TET2 overexpression in MRC5 and NIH3T3 cells (Supplementary Figure 5A–B). These results demonstrate that activation of a SMC program in non-SMC can be achieved with by TET2. Interestingly, TET2 overexpression in human umbilical vein endothelial cells (HUVEC) upregulated only some SMC-specific genes such as MYH11 and ACTA2, while SRF and TAGLN remained unchanged. MYOCD mRNA was not detectable (data not shown), and levels of the endothelial-specific genes PECAM1 and CDH5 remained relatively unchanged (Supplementary Figure 5C–D).
Localized delivery of TET2 enhances SMC differentiation and can improve vascular repair
To assess the role of TET2 in SMC differentiation in vivo, we performed localized gene delivery of either control, TET2 knockdown, or TET2 overexpressing viruses to the site of femoral arterial wire injury. At 3 weeks post-injury, injured femoral arteries treated with a control virus showed moderate intimal hyperplasia, while local delivery of the TET2 knockdown virus markedly increased the neointimal area (Figure 5A). In stark contrast, arteries transduced with the TET2 overexpression virus showed significant suppression of neointimal hyperplasia (Figure 5A–B, and Supplementary Figure 6A). No difference in medial area was observed between the three groups (Supplementary Figure 6B). Consistent with the intimal hyperplasic phenotypes, TET2, MYOCD and MYH11 mRNA levels were reduced with TET2 knockdown but increased with TET2 overexpression relative to control virus (Figure 5C). MYH11, TET2 and 5-hmC levels correlated inversely with the extent of injury as demonstrated by immunostaining (Figure 5D, top panels). Expression of these genes was further reduced in the injured arteries transduced with the TET2 knockdown virus (Figure 5D, middle panels). In the injured femoral arteries transduced with TET2 overexpressing viruses, enhanced expression of Myh11 (Figure 5D) and Acta2 (Supplementary Figure 7A) were noted relative to the control injured samples. In addition, Tet2 and 5-hmC nuclear staining were also more evident throughout these injured samples (Figure 5D, bottom panels).
Figure 5.

TET2 modulation alters 5-hmC levels and the intimal hyperplastic response to arterial injury. (A) EVG staining 3 weeks post-injury of femoral arteries transduced with either a control, TET2 knockdown or TET2 overexpressing virus. (B) Quantitation of the neointima/media ratio in each group. (C) RNA was isolated from the neointima of the control, TET2 knockdown or TET2 overexpression injured mice using the Pinpoint Isolation System and gene expression for Tet2, Myocd and Myh11 in each group was determined by qPCR. (D) Representative images of Myh11, Tet2 and 5-hmC immunostaining from each group. Boxed areas are enlarged on the right. Scale bar, 50 μm. n represent the number of individual mice analyzed. Two or more slides (12–18 sections) per animal were used for analysis. Data are presented as mean ± SD. *P<0.05, **P<0.01, ***P<0.001 compared to the control group.
To further assess the effect of TET2 knockdown or overexpression on the injured vessels, we examined whether TET2 modulation affected endothelial recovery and/or changes in mononuclear cell infiltration. Complete endothelial recovery was observed in all treatment groups as detected by CD31 staining, and no differences in leukocyte (CD45) or macrophage (CD68) staining were noted (Supplementary Figure 7A). We observed significant upregulation of Col1a1, Col4a1, Col8a1 and Col8a2 in the control injured and TET2 knockdown injured samples compared to uninjured, while significant decreases in Col4a and Col8a2 expression were observed in the injured TET2 overexpression samples when compared to injured control (Supplementary Figure 7B,D). No significant differences in the cyclins or cyclin-dependent kinase inhibitors were observed between the groups at 3 weeks (Supplementary Figure 7C,E). Collectively, these data highlight that loss of TET2 diminishes the 5-hmC epigenetic landscape and plays a causative role in intimal hyperplasia post-injury, while reintroduction of TET2 potently antagonizes intimal hyperplasia.
TET2 binds to key SMC promoters and modifies histones at these promoters
The effects of TET2 on SMC phenotypic modulation suggested the possibility of a direct interaction between TET2 and master SMC genes that control SMC differentiation. To test this notion, we employed chromatin immunoprecipitation coupled with qPCR (ChIP-qPCR). We used an anti-TET2 antibody to immunoprecipitate protein/DNA complexes from hCASMC treated with vehicle, rapamycin or PDGF-BB for 24 hours and qPCR amplified CArG-containing regions of the human SRF, MYOCD and MYH11 promoters. Consistent with increased TET2 expression in SMC differentiation, TET2 binding to these promoters greatly increased during SMC differentiation induced by rapamycin (Figure 6A).
Figure 6.

TET2 binds to SMC promoters to regulate 5-hmC and modify histones at SMC loci. (A) ChIP assay demonstrating TET2 enrichment at SRF, MYOCD and MYH11 promoters during SMC differentiation. hCASMC were treated with rapamycin or PDGF-BB for 24 hours. Data are presented as mean relative enrichment over input ± SD of three biological repeats. *P<0.05, **P<0.01. (B–E) H3K4me3 and H3K27me3 ChIP-qPCR in shCTRL (gray bars) or shTET2 (white bars) hCASMC at various gene promoters. Primers were designed to encompass the CArG elements (denoted by the gray box). Primer locations are indicated in blue numbering, product is indicated as blue bar. Data are presented as mean relative enrichment over input ± SD of four biological replicates. *P<0.05, **P<0.01, ***P<0.001 compared to the shCTRL group. (F,H,J) DNA methylation as quantified by MethylCap. (G,I,K) 5-hMC levels as determined by GlucMS-qPCR or hMeDIP-qPCR. Data are presented as mean ± SD of four independent experiments. *P<0.05, **P<0.01, ***P<0.001 compared to the shCTRL group.
As TET2 is known to promote DNA demethylation,3, 5 we further sought to understand TET2-regulated epigenetic changes in SMC genes and evaluated the chromatin state of various SMC loci. We performed ChIP-qPCR, immunoprecipitating with antibodies recognizing the H3K4me3 (active) or H3K27me3 (inactive) marks and amplifying critical CArG-containing promoter regions of contractile SMC genes. ChIP analysis of control, untreated hCASMC cultured in 10% FBS revealed a euchromatic conformation at the MYOCD, SRF and MYH11 gene promoters (H3K4me3/H3K27me3 ratios of 2.5, 9, and 1.9, respectively) (Supplementary Figure 8A–C). No significant difference between the ratio of H3K4me3/H3K27me3 marks was observed at the KLF4 promoter in SMC at baseline (Supplementary Figure 8D). As expected, extensive H3K4me3 enrichment at contractile gene loci in SMC was seen following rapamycin-induced differentiation, whereas PDGF-BB promoted an open chromatin conformation in the KLF4 gene promoter (Supplementary Figure 8E). TET2 knockdown significantly diminished chromatin accessibility and increased the H3K27me3 mark at the MYOCD, SRF and MYH11gene loci in hCASMC, such that H3K4me3/H3K27me3 ratios were decreased by 3–8-fold (Figure 6B–D). In contrast, H3K4me3/H3K27me3 ratios were increased by more than 4-fold at the KLF4 loci following TET2 knockdown (Figure 6E). These results are consistent with the differentiation defects observed with TET2 knockdown in hCASMC (Figure 4) and indicate that TET2 differentially regulates the chromatin accessibility of contractile and dedifferentiation genes in an opposing manner.
Promoters of genes upregulated by TET2 are enriched for 5-hmC
5-hmC levels have been associated with gene activation, therefore we next used several methods to quantitate locus-specific 5-hmC expression at key contractile SMC promoters. We determined the effect of TET2 knockdown on locus-specific 5-hmC levels by glucosylation-coupled methylation-sensitivity qPCR (GlucMS-qPCR)11 and hydroxymethylated DNA immunoprecipitation-qPCR (hMeDIP-qPCR). The MYOCD, SRF and MYH11 promoters become hypermethylated in shTET2 cells compared to shCTRL cells (Figure 6F,H,J), accompanied by a significant and consistent decrease in 5-hmC levels relative to shCTRL cells at these loci (Figure 6G,I,K). TET2 overexpression increased 5-hmC levels at these loci (Supplementary Figure 9A). The effects of TET2 knockdown or overexpression on global nuclear 5-hmC were verified by immunostaining (Supplementary Figure 9B). Taken together, these data indicate that TET2 regulates SMC phenotype through the regulation of 5-hmC which, in turn, affects chromatin modification of key SMC genes. These data support a primary role for 5-hmC as a distinct epigenetic mark in SMC differentiation.
Discussion
In the present work, we identify TET2 as a novel epigenetic master regulator of SMC plasticity that acts upstream of both MYOCD/SRF and KLF4. Using both in vitro and in vivo approaches, we demonstrate that TET2 is necessary and sufficient for SMC differentiation, and that ectopic expression of TET2 is sufficient to direct SMC differentiation in fibroblasts. Endogenous TET2 is regulated by stimuli that modulate differentiation, including PDGF-BB and mTORC1 inhibition in vitro and following vascular injury in vivo. We report a striking correlation between diminished TET2 and 5-hmC expression and severity of atherosclerosis in human coronary arteries. Most notably, we demonstrate a causal role for TET2 in SMC phenotypic modulation in response to arterial injury, as TET2 loss of function exacerbates intimal hyperplasia post-injury, while TET2 overexpression greatly attenuates intimal hyperplasia.
The unique phenotypic plasticity of SMC is known to be transcriptionally regulated, with MYOCD/SRF35–39 and KLF428, 30 having been identified as key drivers of this plasticity, promoting the contractile and de-differentiated phenotypes, respectively. While these proteins interact with other factors that modulate chromatin,37, 40, 41 a unifying upstream regulator that promotes global changes in chromatin at both contractile and synthetic gene promoters has never before been identified. It is therefore particularly notable that we now report that TET2 functions as an upstream regulator of these genes. The epigenetic changes regulated by TET2 have broad effects on chromatin, resulting in induction of MYOCD, SRF, and contractile genes, with concomitant repression of KLF4 and other de-differentiation associated genes. TET2 broadly affects SMC phenotype in vitro, including gene expression, morphology, and proliferation, and this translates to corresponding effects on intimal hyperplasia in vivo, including changes in expression of Myocd, Myh11, and collagen gene expression. The TET2 knockout mouse is viable and fertile, but are susceptible to spontaneous myeloid leukemias.42 While dispensable during development, our data suggest that TET2 plays an important role during the response to vascular injury in the adult.
Mechanistically, we demonstrate that TET2 binding directly to the SRF, MYOCD and MYH11 promoters is strongly enriched in differentiated SMC and coordinately regulates phenotypic modulation through opposing effects on chromatin accessibility at contractile versus synthetic gene promoters. We propose that the potent effects of TET2 are due to its multiple modes of action, including binding directly to SM-specific promoters, regulating expression of the key transcription factors MYOCD and SRF, and inhibition of the repressor KLF4. TET2 alters DNA methylation, 5hmC modification, and histone methylation at these promoters.
Most studies of TET proteins to date have focused on embryonic and hematopoietic stem cells. Our study is the first to address the function of the TET proteins and the 5-hmC epigenetic mark in myocytes. We provide the first evidence that the 5-hmC modification correlates with the mature SMC phenotype and marks active contractile SMC gene promoters. Recent studies indicate that 5-hmC may represent more than just an intermediate base in the TET-mediated demethylation pathways but rather, have a larger role in regulating self-renewal and lineage commitment.14, 42, 43 Genome-wide analyses in mouse ESC have shown that altering DNA methylation via TET1 and 5-hmC also affects histone methylation,44, 45 adding to the complexity by which the TET proteins may regulate gene expression. We find that modulation of TET2, and subsequently 5-hmC, results in robust changes in histone methylation. A summary of our epigenetic data and schematic model is shown in Figure 7. The finding that 5-hmC modulates not only local DNA cytosine methylation but also histone methylation suggests that 5-hmC, generated by TET2, does not simply represent an intermediate in the 5-mC demethylation process, but functions as an epigenetic mark that modulates the SMC phenotype through recruitment of yet unidentified factors to promote chromatin remodeling.
Figure 7.
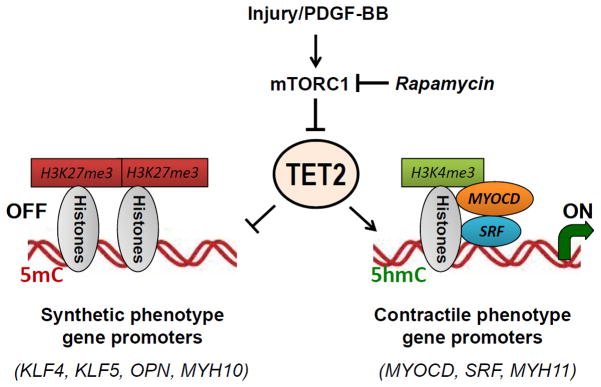
Proposed model of SMC phenotypic modulation control by TET2. Vascular injury releases a milieu of growth factors including PDGF-BB that can activate the mTORC1 pathway and promote SMC dedifferentiation to the synthetic phenotype. Rapamycin, an inhibitor of mTORC1, induces SMC differentiation. We now provide the first study to show that the mTORC1 pathway regulates SMC differentiation through regulation of TET2. In SMC, the balance between contractile and synthetic gene expression is governed by TET2, which results in changes in chromatin conformation and 5-hmC modifications at key promoters. An open chromatin conformation (as assessed by high levels of H3K4me3 and low expression of H3K27me3) as well as high levels of 5-hmC are seen at pro-contractile gene promoters in differentiated SMC, while TET2 promotes a closed chromatin formation at the KLF4 promoter.
High throughput screening in mouse ESC revealed that 5-hmC expressing regions are enriched for both the H3K4 and H3K27 trimethylation marks,45 where TET1 has been shown to maintain ESC identity by promoting the transcription of pluripotency-related factors while simultaneously participating in the silencing of developmental genes.44 We now identify a similar pattern of chromatin regulation in SMC and demonstrate that TET2 and 5-hmC have a similar dual function in adult SMC: TET2 promotes a euchromatic conformation at promoters of contractile-associated genes while concomitantly inducing a repressive chromatin state at promoters of dedifferentiation-associated genes. Furthermore, using the recently established GlucMS-qPCR technique and a modified hMeDIP protocol with the newly developed 5-hmC antibody, we were able to specifically and efficiently label 5-hmC at SMC gene promoter regions. This locus-specific analysis found that 5-hmC is abundant in active SMC promoters and low in promoters that are not transcribing, consistent with the notion that promoter hydroxymethylation leads to demethylation and gene activation.11, 44, 45 The mechanisms by which TET2 mediates opposing effects on differentiation and dedifferentiation genes require further investigation. By activating the promoters of key transcription factors, including MYOCD, SRF and MYH11, while repressing the KLF4 promoter, TET2 serves as an epigenetic master regulator of SMC differentiation. The concomitant activation of the promoters of MYOCD/SRF target genes (MYH11, ACTA2) may account for the potent pro-differentiation effects of TET2 (Figure 6). We noted TET2 binding to promoter regions containing CArG elements in contractile promoters, including a putative CArG element in the human MYOCD promoter, but important questions such as whether TET2 also interacts with non-CArG-rich regions of these promoters, and what directs TET2 repression of synthetic gene promoters will require further investigation.
Our data reveal that TET2 and 5-hmC levels are modulated by environmental signals in SMC. TET2 mRNA and protein and 5-hmC levels are repressed by PDGF-BB, the major stimulus of SMC de-differentiation in vivo and in vitro. Additionally, our data identify TET2 as a critical effector of the mTORC1 pathway in modulating SMC phenotype. We report that TET2 expression is in part regulated by the mTORC1 pathway, as rapamycin induces TET2 expression at both the mRNA and protein levels, and TET2 knockdown prevents rapamycin-induced differentiation. We have previously shown that mTORC1 inhibition promotes SMC differentiation at the level of transcription,21, 22 and rapamycin-eluting stents are highly effective against restenosis.46 Our data that local TET2 delivery to an injured artery prevents intimal hyperplasia suggests that manipulation of TET2 and 5-hmC expression may have important therapeutic implications for other diseases involving aberrant SMC plasticity, including cancer, aneurysm, transplant vasculopathy, and hypertension.
While TET2 mRNA has been reported to be expressed by many tissues, we noted that TET2 is very highly expressed in SMC relative to other TET family members. Our data suggest that despite persistent expression levels of TET1 and TET3, these enzymes do not compensate for TET2 knockdown or downregulation by PDGF-BB in SMC. We also observed robust TET2 expression in SMC-rich tissues and in cultured SMC relative to other cell types (fibroblasts and endothelial cells). TET2 overexpression was sufficient to induce a program of SMC-specific gene expression in human and mouse fibroblasts, similar to what has been reported for MYOCD overexpression in myoblasts,35 ESC,47 10T1/2 and 3T3 cells.38 We found very low level TET2 expression in HUVEC compared to SMC. We achieved a modest overexpression of TET2 in HUVEC that was still below that seen in SMC at baseline, and this induced only a subset of SMC genes. It is therefore possible that there is a threshold effect such that high levels of TET2 regulate SMC-specific genes. It is also feasible that as yet unidentified cell type-specific factors or other permissive chromatin modifications cooperate with TET2 in SMC and in fibroblasts.
In conclusion, this is the first report of TET protein function in myocytes, as well as the first demonstration of a TET protein regulated by the mTORC1 signaling pathway. Our data reveal that modulating TET2, and in turn, 5hmC levels, results in broad epigenetic changes in methylation and histones indicative of altered chromatin formations at both differentiation- and de-differentiation-specific SMC genes. Our findings support TET2 as a master epigenetic regulator of SMC phenotypic modulation. Altering key components of the TET2-5-hmC pathway may be a therapeutic target for treating diseases involving aberrant SMC plasticity that is involved in many cardiovascular associated disorders.
Supplementary Material
Clinical Summary - Original Research Put Into Perspective for the Practicing Clinician.
Vascular smooth muscle cells (SMC) retain the plasticity to dedifferentiate in response to extracellular cues, including inflammation and growth factors. Dedifferentiated SMC become proliferative, migratory, secrete copious extracellular matrix, and lose contractility. This contributes to many cardiovascular pathologies, including intimal hyperplasia/restenosis, atherosclerosis, aneurysm, transplant arteriosclerosis, and cancer. Genes associated with differentiated SMC, including contractile apparatus proteins, are known to be transcriptionally regulated. Our work now reveals the protein Ten-eleven translocation 2 (TET2) as a novel epigenetic master regulator of SMC plasticity. TET family proteins were recently found to regulate plasticity in embryonic and hematopoietic stem cells. We find that TET2, which demethylates DNA through production of 5-hydroxymethylcytosine (5-hmC), directly binds to chromatin and modifies both DNA and histone methylation at key SMC promoters. Notably, TET2 activates expression of pro-differentiation genes (e.g. myocardin, SRF, and MYH11) while concomitantly repressing the expression of dedifferentiation genes (e.g. KLF4). We find that expression of TET2 and its product 5-hmC correlates with SMC differentiation in atherosclerotic human coronary artery specimens. TET2 loss- and gain-of-function promote SMC dedifferentiation and differentiation, respectively, in vitro and in vivo. The drug-eluting stent agent rapamycin induces TET2 expression, and we find that TET2 is required for rapamycin-induced SMC differentiation. Most importantly, peri-adventitial viral delivery of TET2 prevented intimal hyperplasia in mouse femoral artery. Our study demonstrates that TET2 and 5-hmC are novel biomarkers of SMC contractile phenotype that actively promote differentiation, and that targeting TET2/5-hmC may have therapeutic potential for smooth muscle pathologies.
Acknowledgments
We thank Drs. Michael Simons and William Sessa for comments on the manuscript.
Funding Sources: This study was supported by grants from the NIH to KAM (HL091013), JH (HL074190, HL115247 and HL117798), an AHA SDG to JY, and the Brown-Coxe Fellowship to RL.
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40. doi: 10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- 2.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 3.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formlcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala HS, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, Lahesmaa R, Orkin SH, Rodig SJ, Daley GQ, Rao A. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–13. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J, Barbera AJ, Zheng L, Zhang H, Huang S, Min J, Nicholson T, Chen T, Xu G, Shi Y, Zhang K, Shi YG. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451–61. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Margues CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 12.Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A. 2011;108:14566–71. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orr BA, Haffner MC, Nelson WG, Yegnasubramanian S, Eberhart CG. Decreased 5-hydroxymethylcytosine is associated with neural progenitor phenotype in normal brain and shorter survival in malignant glioma. PLoS One. 2012;7:e41036. doi: 10.1371/journal.pone.0041036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, Xu W, Tan L, Hu Y, Zhan Q, Lee CW, Hu D, Lian BQ, Kleffel S, Yang Y, Neiswender J, Khorasani AJ, Fang R, Lezcano C, Duncan LM, Scolyer RA, Thompson JF, Kakavand H, Houvras Y, Zon LI, Mihm MC, Jr, Kaiser UB, Schatton T, Woda BA, Murphy GF, Shi YG. Loss of 5-hydroxmethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135–46. doi: 10.1016/j.cell.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, Sokol SI, Pfau S, Pober JS, Tellides G. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. 2009;119:1424–32. doi: 10.1161/CIRCULATIONAHA.108.827618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranjbaran H, Sokol SI, Gallo A, Eid RE, Iakimov AO, D’Alessio AC, Kapoor JR, Akhtar S, Howes CJ, Aslan M, Pfau S, Pober JS, Tellides G. An inflammatory pathway of IFN-gamma production in coronary atherosclerosis. J Immunol. 2007;178:592–604. doi: 10.4049/jimmunol.178.1.592. [DOI] [PubMed] [Google Scholar]
- 17.Blanc-Brude OP, Yu J, Simosa H, Conte MS, Sessa WC, Altieri DC. Inhibitor of apoptosis protein survivin regulates vascular injury. Nat Med. 2002;8:987–94. doi: 10.1038/nm750. [DOI] [PubMed] [Google Scholar]
- 18.Yu J, Rudic RD, Sessa WC. Nitric oxide-releasing aspirin decreases vascular injury by reducing inflammation and promoting apoptosis. Lab Invest. 2002;82:825–32. doi: 10.1097/01.lab.0000018828.61722.bd. [DOI] [PubMed] [Google Scholar]
- 19.Ding M, Carrao AC, Wagner RJ, Xie Y, Jin Y, Rzucidlo EM, Yu J, Li W, Tellides G, Hwa J, Aprahamian TR, Martin KA. Vascular smooth muscle cell-derived adiponectin: a paracrine regulator of contractile phenotype. J Mol Cell Cardiol. 2012;52:474–84. doi: 10.1016/j.yjmcc.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding M, Xie Y, Wagner RJ, Jin Y, Carrao AC, Liu LS, Guzman AK, Powell RJ, Hwa J, Rzucidlo EM, Martin KA. Adiponectin induces vascular smooth muscle cell differentiation via repression of mammalian target of rapamycin complex 1 and FoxO4. Arterioscler Thromb Vasc Biol. 2011;31:1403–10. doi: 10.1161/ATVBAHA.110.216804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin KA, Rzucidlo EM, Merenick BL, Fingar DC, Brown DJ, Wagner RJ, Powell RJ. The mTOR/p70 S6K1 pathway regulates vascular smooth muscle cell differentiation. Am J Physiol Cell Physiol. 2004;286:C507–C517. doi: 10.1152/ajpcell.00201.2003. [DOI] [PubMed] [Google Scholar]
- 22.Martin KA, Merenick BL, Ding M, Fetalvero KM, Rzucidlo EM, Kozul CD, Brown DJ, Chiu HY, Shyu M, Drapeau BL, Wagner RJ, Powell RJ. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem. 2007;282:36112–20. doi: 10.1074/jbc.M703914200. [DOI] [PubMed] [Google Scholar]
- 23.Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nature Protocols. 2006;1:729–48. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brinkman AB, Simmer F, Ma K, Kann A, Zhu J, Stunnenberg HG. Whole-genome DNA methylation profiling using MethylCap-seq. Methods. 2010;52:232–6. doi: 10.1016/j.ymeth.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 25.Kaplan-Albuquerque N, Garat C, Desseva C, Jones PL, Nemenoff RA. Platelet-derived growth factor-BB-mediated activation of Akt suppresses smooth muscle-specific gene expression through inhibition of mitogen-activated protein kinase and redistribution of serum response factor. J Biol Chem. 2003;278:39830–8. doi: 10.1074/jbc.M305991200. [DOI] [PubMed] [Google Scholar]
- 26.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–64. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–9. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adam PJ, Regan CP, Hautmann MB, Owens GK. Positive- and negative-acting Kruppel-like transcription factors bind a transforming growth factor beta control element required for expression of the smooth muscle cell differentiation marker SM22alpha in vivo. J Biol Chem. 2000;275:37798–806. doi: 10.1074/jbc.M006323200. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem. 2005;280:9719–27. doi: 10.1074/jbc.M412862200. [DOI] [PubMed] [Google Scholar]
- 30.Yoshida T, Kaestner KH, Owens GK. Conditional deletion of Krupple-like factor 4 delays down-regulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ Res. 2008;102:1548–57. doi: 10.1161/CIRCRESAHA.108.176974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshida T, Gan Q, Franke AS, Ho R, Zhang J, Chen YE, Hayashi M, Majesky MW, Somlyo AV, Owens GK. Smooth and cardiac muscle-selective knock-out of kruppel-like factor 4 causes postnatal death and growth retardation. J Biol Chem. 2010;285:21175–84. doi: 10.1074/jbc.M110.112482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105:158–66. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giachelli CM, Bae N, Almeida M, Denhardt DT, Alpers CE, Schwartz SM. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J Clin Invest. 1993;92:1686–96. doi: 10.1172/JCI116755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuro-o M, Nagai R, Nakahara K, Katoh H, Tsai RC, Tsuchimochi H, Yazaki Y, Ohkubo A, Takaku F. cDNA cloning of a myosin heavy chain isoform in embryonic smooth muscle and its expression during vascular development and in arteriosclerosis. J Biol Chem. 1991;266:3768–73. [PubMed] [Google Scholar]
- 35.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–56. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- 36.Wang DZ, Li S, Hockemeyer D, Sutherland L, Wang Z, Schratt G, Richardson JA, Nordheim A, Olson EN. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc Natl Acad Sci U S A. 2002;99:14855–60. doi: 10.1073/pnas.222561499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshida T, Sinha S, Dandre F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003;92:856–64. doi: 10.1161/01.RES.0000068405.49081.09. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci U S A. 2003;100:7129–34. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang DZ, Olson EN. Control of smooth muscle development by the myocardin family of transcriptional coactivators. Curr Opin Genet Dev. 2004;14:558–66. doi: 10.1016/j.gde.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Manabe I, Owens GK. Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differention of a novel P19-derived in vitro smooth muscle differentiation system. Cir Res. 2001;88:1127–34. doi: 10.1161/hh1101.091339. [DOI] [PubMed] [Google Scholar]
- 41.McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK. Control of SRF binding to CARG-box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. 2006;116:36–48. doi: 10.1172/JCI26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kallin EM, Rodriguez-Ubreva J, Christensen J, Cimmino L, Aifantis I, Helin K, Ballestar E, Graf T. Tet2 facilitates the derepression of myeloid target genes during CEBPa-induced transdifferentiation of pre-B cells. Mol Cell. 2012;48:266–76. doi: 10.1016/j.molcel.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doege CA, Inoue K, Yamashita T, Rhee DB, Travis S, Fujita R, Guarnieri P, Bhagat G, Vanti WB, Shih A, Levine RL, Nik S, Chen EI, Abeliovich A. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature. 2012;488:652–5. doi: 10.1038/nature11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–93. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, McLoughlin EM, Brudno Y, Mahapatra S, Kapranov P, Tahiliani M, Daley GQ, Liu XS, Ecker JR, Milos PM, Agarwal S, Rao A. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–7. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sousa JE, Costa MA, Abizaid A, Abizaid AS, Feres F, Pinto IM, Seixas AC, Staico R, Mattos LA, Sousa AG, Falotico R, Jaeger J, Popma JJ, Serruys PW. Lack of neointimal proliferation after implantation of sirolimus-coated stents in human coronary arteries: a quantitative coronary angiography and three-dimensional intravascular ultrasound study. Circulation. 2001;103:192–5. doi: 10.1161/01.cir.103.2.192. [DOI] [PubMed] [Google Scholar]
- 47.Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, Parmacek MS. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol. 2003;23:2425–37. doi: 10.1128/MCB.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.