

Abstract
The breast and ovarian cancer type 1 susceptibility protein (BRCA1) has pivotal roles in the maintenance of genome stability. Studies support that BRCA1 exerts its tumour suppression function primarily through its involvement in cell cycle checkpoint control and DNA damage repair. In addition, recent proteomic and genetic studies have revealed the presence of distinct BRCA1 complexes in vivo, each of which governs a specific cellular response to DNA damage. Thus, BRCA1 is emerging as the master regulator of the genome through its ability to execute and coordinate various aspects of the DNA damage response.
Our genetic material is continuously challenged by genotoxic stress. DNA damage can arise during normal cellular metabolic processes such as DNA replication, from endogenous sources such as free radicals, or from exogenous agents such as ultraviolet light and ionising radiation. To ensure genome stability, cells have evolved the ability to sense DNA damage, activate the cell cycle checkpoint and initiate DNA repair (BOX 1). Failure to properly repair damaged DNA contributes to tumori-genesis1. This is exemplified by numerous cancer-predisposing clinical syndromes that are attributed to mutations in components involved in cellular processes that counteract genotoxic stress. Indeed, it has been proposed that activation of the DNA damage response is a general protective mechanism during early tumori-genesis, and that inactivating mutations of this anti-cancer barrier correlates with genomic instability and tumour progression2,3.
Box 1. Mechanisms that maintain genome stability.
Genome instability is a hallmark of cancer cell development. Mounting evidence indicates that genome instability contributes to tumorigenesis121. Therefore, to suppress tumorigenesis, cells have evolved several mechanisms to protect genome integrity, including cell cycle checkpoint controls and DNA repair1. It is now established that there is crosstalk between cell cycle checkpoints and DNA repair to ensure that cell cycle progression is halted soon after DNA damage is detected. This allows the DNA repair machinery to repair the damage before cells continue with DNA replication and cell division.
Cell cycle checkpoints are control mechanisms that result in cell cycle arrest at the G1–S interphase, during S phase and at the G2–M interphase. This ensures faithful inheritance of the genetic material. In addition, other checkpoints, including the spindle assembly checkpoint, exist during mitosis to ensure proper segregation of sister chromatids122.
Two major repair pathways operate to repair DNA double-stranded breaks: homologous recombination (HR)-mediated repair and non-homologous end joining. The error-free HR-mediated repair uses a homologous template (BOX 2), and non-homologous end joining involves the direct ligation of DNA ends, which is sometimes accompanied by a loss of genetic information.
With the development of RNA interference technology and advanced proteomics, we have witnessed unprecedented expansion in our knowledge of the DNA damage response over the past few years. These technological advances have revealed a highly complex protein network that allows the timely, efficient and coordinated activation of cellular responses to DNA damage. Specifically, the tumour suppressor breast and ovarian cancer type 1 susceptibility protein (BRCA1) has taken centre stage, being intimately involved in diverse cellular processes that ensure genome stability and promote cell survival. Through its ability to form complexes with other proteins, BRCA1 has dedicated and complementary roles in the DNA damage response, including cell cycle checkpoint control and DNA repair. Thus, BRCA1 is emerging as a master regulator of genome integrity. In this Review we discuss recent progress in elucidating the roles of BRCA1 in DNA damage responses, with an emphasis on how the temporal and spatial regulation of BRCA1 enables it to act as the master regulator of genome integrity.
Domain organization of BRCA1
Germline mutations of the human BRCA1 gene account for most familial cases of breast and ovarian cancer. BRCA1, which is located on chromosome 17q21 (REF. 4), encodes a protein of 1,863 amino acids. BRCA1 harbours a highly conserved amino-terminal RING domain and tandem BRCT domains at the end of its carboxyl terminus. The RING domain is a motif found in many E3 ubiquitin ligases and is recognized as an important structure that mediates protein ubiquitylation. The BRCT domain is a motif that binds phosphorylated proteins5,6 (FIG. 1) and is present in many proteins that respond to DNA damage. The fact that these highly conserved domains are frequently targeted by many clinically important mutations indicates that they are integral for BRCA1 function. Although BRCA1 was cloned more than a decade ago4, it was not until recently that the roles of the RING and BRCT domains have begun to emerge. Studies have now revealed the mechanism by which these domains confer the tumour suppressor functions of BRCA1. Intriguingly, evolutionarily conserved stretches of amino acids can also be found on other parts of the BRCA1 coding region, suggesting that much about BRCA1 remains to be unravelled.
Figure 1. BRCA1 domain organization and interaction partners.
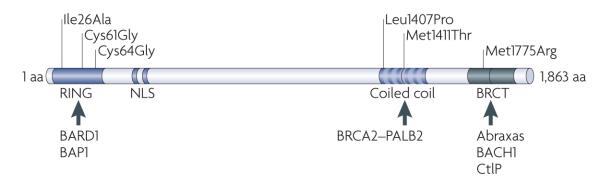
The human breast and ovarian cancer type 1 susceptibility gene (BRCA1) encodes a protein of 1,863 amino acids. BRCA1 has two highly conserved motifs at its termini: the RING domain and tandem BRCT domains. The RING domain sequence encodes a folded protein structure and confers the E3 ubiquitin ligase activity of BRCA1, and the BRCT domains bind phosphorylated proteins that are primarily involved in the DNA damage response. BRCA1 exists as a stable heterodimer through its RING-mediated interaction with BRCA1-associated RING domain protein 1 (BARD1), which is inhibited by BRCA1-associated protein 1 (BAP1). The sites at which other BRCA1-interacting proteins bind are also shown. Important clinical mutations routinely used in research settings are indicated, such as Cys61Gly and Cys64Gly, which target the RING domains, and Met1775Arg, which disrupts the BRCA1 BRCT domain interaction with phosphorylated proteins and the accumulation of BRCA1 at damage-induced foci. The IlE26Ala mutation abrogates the BRCA1 E3 ubiquitin ligase activity and is predicted to maintain the RING structure. In addition, mutations from patients with cancer at conserved residues Leu1407Pro and Met1411Thr on the BRCA1 coiled-coil domain were found to abrogate its interaction with partner and localizer of BRCA2 (PALB2; also known as FANCN) and to compromise homologous recombination-mediated DNA repair. BACH1, BRCA1-interacting protein carboxy-terminal helicase 1; CtIP, CtBP-interacting protein; NLS, nuclear localization sequence.
Sequences encompassing the RING domain of BRCA1 mediate its stable association with BRCA1-associated RING domain protein 1 (BARD1)7. Heteromeric formation of the BRCA1–BARD1 dimer has been implicated in the maintenance of genomic stability and tumour suppression through its involvement in DNA damage signalling, DNA repair and transcriptional regulation. Similar to BRCA1 mutations, BARD1 mutations have been identified in individuals with sporadic and hereditary tumours8,9. Moreover, a recent genome-wide association study revealed BARD1 single nucleotide polymorphisms that closely associated with familial neuroblastoma10. Establishing whether and how these BARD1 mutations contribute to tumorigenesis will require further work. BARD1 has a similar domain organization to BRCA1, comprising an N-terminal RING domain and tandem C-terminal BRCT domains. Although the physiological role of the BARD1 BRCT domains remains to be determined, overexpression of a BARD1 mutant that is devoid of its BRCT domains compromised homologous recombination (HR)-mediated DNA repair11. However, as this effect was observed in both BRCA1-sufficient and BRCA1-deficient cells, it remains unclear whether BARD1 contributes to DNA repair through its stable interaction with BRCA1.
The structure of the RING domains of BRCA1 and BARD1 in solution revealed that their respective zinc-binding residues are flanked by α-helices that combine to form a four-helix bundle with an extensive buried surface12. This dimeric configuration gives rise to the stability of the heterodimer. The BRCA1 RING domain, similarly to that of other RING-containing E3 ubiquitin ligases, has been shown to mediate the interaction with E2 ubiquitin-conjugating enzymes13,14, which allows the differential synthesis of monoubiquitin and polyubiquitin. Although it has yet to be determined how the BRCA1 RING domain selects these E2 enzymes in vivo, these studies imply that BRCA1 might regulate and conjugate different substrates with distinct ubiquitin species, which in turn can govern protein degradation, subcellular localization and/or activity15–18.
The BRCA1 BRCT domains are thought to be integral for the protein’s ability to regulate a diverse set of cellular processes that confer its tumour-suppressing activity. BRCA1 has been reported to interact with numerous cell cycle checkpoint and repair proteins through its BRCT domains, as well as chromatin remodelling factors and components of transcriptional machineries19–22. Recent evidence indicates that the BRCA1 BRCT domains are responsible for phosphorylation-dependent interactions with several of its bona fide associated proteins5,6, highlighting the potential for specific interactions and activation of certain BRCA1 functions during defined conditions.
BRCA1 localizes at DNA breaks
One of the first pieces of evidence that suggested a role of BRCA1 in the DNA damage response was the cytological observation that, following DNA damage, BRCA1 localizes at damage-induced foci (also known as ionising radiation-induced foci)23, which colocalize with foci where the DNA repair protein RAD51 accumulates. The DNA damage-induced foci, marked by the histone variant H2AX phosphorylated on Ser139 (known as γH2AX), represent sites of DNA breaks24,25. γH2AX is essential for the accumulation of numerous DNA damage repair factors, including BRCA1 (REF. 26), at sites of DNA breaks, suggesting that γH2AX is one of the initial recruiting factors for various checkpoint and DNA repair proteins to DNA breaks. The H2AX signalling cascade began to emerge with the discovery that mediator of DNA damage checkpoint 1 (MDC1) is the main downstream factor in the pathway and is required for the damage-induced focal accumulation of a growing list of DNA damage repair factors at DNA breaks. MDC1 contains tandem BRCT domains, which allow its direct interaction with γH2AX27 (because γH2AX is phosphorylated) and its accumulation at DNA breaks; this, in turn, promotes the formation of damage-induced foci where BRCA1 accumulates28–33.
Targeting BRCA1 to DNA breaks
Despite having BRCT domains, BRCA1 does not bind directly to γH2AX. The localization of BRCA1 to damage-induced foci occurs in a BRCT-dependent manner, but is independent of the two BRCT domain-binding proteins: CtBP-interacting protein (CtIP; also known as RBBP8)34 and BRCA1-interacting protein C-terminal helicase 1 (BACH1; also known as FANCJ and BRIP1)35. So how does BRCA1 localize to damage-induced foci? Phosphopeptide affinity proteomics analysis and affinity purification approaches identified the coiled-coil domain-containing protein abraxas (also known as CCDC98 and FAM175A) as a BRCA1 BRCT domain-binding protein36–38. Following its phosphorylation on Ser406, abraxas interacts specifically with the BRCA1 BRCT domain, suggesting that BRCA1 forms at least three distinct protein complexes in vivo (with abraxas, CtIP and BACH1). Interestingly, the ubiquitin-interacting motif (UIM)-containing receptor-associated protein 80 (RAP80; also known as UIMC1) was also identified as a component of the abraxas–BRCA1 complex36,39–41. Detailed analyses indicate that abraxas bridges the interaction between RAP80 and the BRCA1 BRCT domain and mediates the localization of BRCA1 to sites of DNA damage. The finding that RAP80 and its UIM are required for proper BRCA1 localization following DNA damage suggested the existence of a ubiquitin-dependent signalling pathway that controls the retention of the RAP80–abraxas–BRCA1 complex at DNA double-stranded breaks (DSBs). This was supported by the observation that ubiquitin conjugates reside at DSBs and that the RAP80 UIM specifically interacts with non-canonical ubiquitin chains, namely those linked by Lys63 and Lys6 (REF. 41) (FIG. 2).
Figure 2. BRCA1 participates in DNA damage signalling.

A model for targeting the breast and ovarian cancer type 1 susceptibility protein (BRCA1) to damage-induced foci at DNA double-stranded breaks that involves a ubiquitin-dependent signal transduction pathway. The current hypothesis is that DNA damage (1) first triggers the ataxia telangiectasia mutated (ATM)–ataxia telangiectasia and RAD3-related protein (ATR)-dependent phosphorylation of histone variant H2AX (2). Subsequently, phosphorylated H2AX directly recruits mediator of DNA damage checkpoint 1 (MDC1), which in turn promotes the accumulation of the E3 ubiquitin ligase complex RING finger protein 8 (RNF8)–ubiquitin-conjugating enzyme 13 (UBC13; also known as UBE2N) to mediate ubiquitylation of histones and other yet-to-be-identified substrates at or near the sites of DNA breaks (3). The newly identified E3 ubiquitin ligase RNF168 recognizes ubiquitylated histones and, in concert with UBC13, amplifies the local ubiquitylation events involving Lys63-linked ubiquitin chains (4). These ubiquitin chains are recognized by the ubiquitin-interacting motif (UIM)-containing receptor-associated protein 80 (RAP80; also known as UIMC1), which recruits the BRCA1A complex (including breast cancer type 1 susceptibility protein (BRCA1)) to DNA double-stranded breaks (5). Chromatin retention of BRCA1 promotes checkpoint kinase 1 (CHK1; also known as CHEK1) phosphorylation and G2–M checkpoint activation. Ub, ubiquitin.
All this circumstantial evidence supporting a regulatory role of ubiquitylation in the DNA damage signalling cascade finally came together with the identification of two E3 ubiquitin ligases — RING finger protein 8 (RNF8) and RNF168 — as crucial components in the transduction of DNA damage signals42–47. RNF8 is thought to be the initiating E3 ubiquitin ligase, which is brought to DNA damage sites by directly interacting with MDC1 (which binds γH2AX). Together with the E2 ubiquitin-conjugating enzyme 13 (UBC13; also known as UBE2N), which catalyses exclusively Lys63-linked ubiquitin chains48, RNF8 and RNF168 mediate histone and probably other substrate ubiquitylation reactions close to DSBs and promote the formation of ubiquitin conjugates at damage-induced foci. Likewise, both RNF8 and RNF168 are required for the proper localization of the RAP80–abraxas–BRCA1 complex following DNA damage, therefore supporting the earlier hypothesis that RAP80 is tethered to DNA breaks through its ability to directly interact with ubiquitin chains at damage-induced foci. Although the identity of the ubiquitylated substrates, besides histones, remains unclear, these studies have shed considerable light on the mechanistic basis underlying the spatial regulation of BRCA1 in response to DNA damage.
In addition, despite the well-known requirement of the RAP80 UIM for RAP80 localization to damage-induced foci, RAP80 UIM-deletion mutants still localized to damage-induced foci, albeit at a lower rate than wild-type RAP80 (REFS 36,42). Thus, it will be important to clarify whether an alternative route contributes to RAP80 targeting to the foci.
The functions of BRCA1 at damage-induced foci
Although recent studies have revealed that numerous levels of regulation ensure a specific BRCA1 concentration close to DSBs, exactly how BRCA1 enforces the DNA damage response at DSBs remains largely unknown. As BRCA1 has E3 ubiquitin ligase activity49, perhaps it is not surprising that BRCA1 might mediate protein ubiquitylation at DSBs, which in turn could facilitate the DNA damage response. Consistent with this possibility, early studies suggested that BRCA1 is required for the accumulation of ubiquitin conjugates at damage-induced foci50,51, and that the H2A type histones (that is, H2A and H2AX) might be targets of BRCA1-mediated ubiquitylation52,53. Similarly, the phosphorylated form of CtIP interacts with the BRCA1 BRCT domain54 and is modified with non-canonical ubiquitin chains in a BRCA1-dependent manner55. Although these data indicate that BRCA1 might regulate the ubiquitylation status of a range of proteins at DSBs, the biological importance of these ubiquitylation events awaits future investigation. Given that the BRCA1–BARD1 heterodimer catalyses the formation of different ubiquitin chains, it is tempting to speculate that each of these ubiquitin configurations might confer a specific function in the BRCA1 repertoire, including checkpoint control, DNA resection and DNA repair. Moreover, because Lys6- and Lys63-linked ubiquitin conjugates have been implicated at DSBs41,50, it will be interesting to test whether these putative BRCA1 substrates at DSBs are modified exclusively with these non-canonical ubiquitin chains56,57.
BRCA1 might also act as a scaffold protein at damage-induced foci to facilitate ataxia telangiectasia mutated (ATM)–ataxia telangiectasia and RAD3-related protein (ATR) signalling. Indeed, early studies revealed that BRCA1 regulates the phosphorylation status of many proteins involved in ATM–ATR signalling, including p53, Nijmegen breakage syndrome protein 1 (NBS1; also known as nibrin), checkpoint kinase 1 (CHK1; also known as CHEK1) and CHK2 (also known as CHEK2)58,59. Thus, it seems possible that BRCA1, perhaps through its association with these signalling proteins, might facilitate their activation by bringing them close to ATM and ATR at DNA breaks.
BRCA1–BARD1: one in many
BRCA1 exists as a stable heterodimer with BARD1 (REF. 60), which ensures genome stability through its role in protein ubiquitylation15–17,61. An early report suggested that the BRCA1–BARD1 heterodimer controls centrosome duplication by regulating the ubiquitylation of γ-tubulin62. In addition, evidence also indicates that this E3 ubiquitin ligase complex modulates cell cycle control, partly by inhibiting RNA processing in response to DNA damage. The clinical importance of the BRCA1–BARD1 ubiquitin ligase activity is shown by the presence of mutations on the RING domain of BRCA1 in tumour samples, which inactivate its enzymatic activity63. Biochemical studies revealed that BARD1 potentiates the ubiquitin ligase activity of BRCA1 (REFS 57,63), which can be reversed through the inhibition of BRCA1–BARD1 association by BRCA1-associated protein 1 (BAP1)64. The notion that the tumour-suppressing activity of the BRCA1–BARD1 heterodimer is interdependent is best supported by mouse models with mammary epithelial cell-targeted ablation of BRCA1, BARD1 or both, which revealed striking similarities in the frequency, latency, histopathology and cytogenetic features of the observed breast carcinomas65. Moreover, formation of the heterodimer is important for the maintenance of protein stability66, further suggesting that BRCA1 and BARD1 require each other for most, if not all, of their cellular functions.
To accommodate the diverse nature of BRCA1 functions in the DNA damage response, a multifactorial model was proposed that requires BRCA1 to exist as part of distinct macromolecular protein complexes67. This originated from earlier studies indicating that BRCA1 interacts with CtIP and BACH1, and that their respective association with BRCA1 dictated distinct damage-induced cell cycle checkpoints6,54. To date, at least three distinct BRCA1-containing protein complexes have emerged36,41. Although they all contain BARD1, each protein complex has distinct subunits, as BRCA1 is thought to interact with phosphorylated CtIP, BACH1 or abraxas through its BRCT domain in a mutually exclusive manner. Below, we summarise the distinct and overlapping functions of each of these BRCA1 macrocomplexes. For simplicity, we have adopted the existing nomenclature and refer to the three BRCA1 complexes as BRCA1A, BRCA1B and BRCA1C36 (TABLE 1).
Table 1.
BRCA1 macrocomplexes and their functions in DNA damage repair
| BRCA1 complex |
Function | Components* | Refs |
|---|---|---|---|
| Core complex |
Promotes E3 ubiquitin ligase activity |
BRCA1 and BARD1 (constitutive heterodimer) |
60 |
| BRCA1A‡ | Control of the G2–M checkpoint and BRCA1 accumulation at damage-induced foci |
BRCA1, abraxas (CCDC98), RAP80 (UIMC1), BRCC36, BRCC45 (BRE) and MERIT40 (NBA1) |
36–41, 72–74 |
| BRCA1B‡ | DNA replication and S phase progression |
BRCA1, BACH1 (FANCJ and BRIP1) and TOPBP1 |
35,67 |
| BRCA1C‡ | DNA resection and G2–M checkpoint control |
BRCA1, CtIP (RBBP8) and MRE11–RAD50–NBS1 (nibrin) (the MRN complex) |
34,87,89 |
| BRCC§ | Homologous recombination- mediated DNA repair |
BRCA1, BRCA2, PALB2 (FANCN), RAD51, BRCC36 and BRCC45 |
70, 106–107 |
BACH1, BRCA1-interacting protein carboxy-terminal helicase 1; BARD1, BRCA1-associated RING domain protein 1; BRCA, breast cancer susceptibility protein; CtIP, CtBP-interacting protein; MERIT40, mediator of RAP80 interactions and targeting subunit of 40 kDa; MRE11, meiotic recombination 11; NBS1, Nijmegen breakage syndrome protein 1; PALB2, partner and localizer of BRCA2; RAP80, receptor-associated protein 80; TOPBP1, topoisomerase II-binding protein 1.
Alternative names are indicated in brackets.
Macrocomplex formation is mediated through the BRCA1 BRCT domain.
Complex formation is mediated through the BRCA1 coiled-coil domain. As the BRCC complex contains BRCC36 and BRCC45, it is likely that PALB2 and BRCA2 are present in the other BRCA1 BRCT domain-dependent complexes.
BRCA1A is involved in G2–M checkpoint control
The BRCA1A complex is made up of RAP80, abraxas, BRCC36, BRCC45 (also known as BRE) and mediator of RAP80 interactions and targeting subunit of 40 kDa (MERIT40; also known as NBA1), which enable the targeting of BRCA1 to DSBs (FIG. 1). BRCA1A is involved in G2–M checkpoint control and ensures that entry into mitosis is transiently inhibited to avoid aberrant chromosome segregation36–38,40,41. It is notable that, although cells depleted of RAP80 and abraxas exhibit impaired accumulation of BRCA1 at damage-induced foci and display defects in BRCA1-dependent G2–M checkpoint activation, the phenotypes and magnitude of defects in these cells are mild compared with BRCA1-deficient cells. These observations suggest that BRCA1 potentially also regulates the G2–M checkpoint through different mechanisms (see below).
BRCA1 participates in G2–M checkpoint control partly by regulating the phosphorylation status of CHK1 (REF. 59). Intriguingly, the ability of BRCA1A to accumulate at the damage-induced foci correlates with proper G2–M arrest on DNA damage. As a result, deficiencies in the signalling proteins that are required for sustained accumulation of BRCA1 at DSB sites result in compromised G2–M checkpoint activation29,30,68. Although it remains unclear how the targeting of BRCA1 to DSBs promotes CHK1 phosphorylation, one possibility is that BRCA1 may modify the local chromatin structure through substrate ubiquitylation at DSBs to enhance ATM signalling. Alternatively, accumulation of BRCA1 at damage-induced foci may also enhance CHK1 phosphorylation indirectly by acting as a scaffold protein at DSBs59.
In addition to RAP80 and abraxas, BRCA1A contains BRCC36 and BRCC45 (REFS 69,70), both of which were identified in an early study as components of the BRCC complex, which also contains BRCA1, BRCA2 (also known as FANCD1), partner and localizer of BRCA2 (PAlB2; also known as FANCN) and RAD51 (REF. 70). Although there is no experimental evidence to indicate that the BRCA1A and BRCC complexes interact and assemble in vivo, the similar requirement for these complexes in cell survival and checkpoint control following DNA damage implies that this is a possibility. BRCC36 and BRCC45 were found to promote the E3 ubiquitin ligase activity of the BRCA1–BARD1 heterodimer70. Consistent with roles of BRCC36 and BRCC45 in the DNA damage response pathway, depletion of either of these factors abrogated the accumulation of BRCA1 at damage-induced foci and rendered cells hypersensitive to DNA damaging agents. Interestingly, part of the BRCC36 coding sequence shares homology with the JAB1/MPN/MOV34 metalloenzyme catalytic domain (JAMM; also known as MPN+) of JAMM family deubiquitinases71 and can hydrolyse Lys63-linked ubiquitin chains41. Although the exact role of the deubiquitinase activity of BRCC36 remains unknown, the fact that this BRCA1 complex has opposing activities that potentially regulate the ubiquitylation status of its bona fide substrates underlines the complex and dynamic regulation of BRCA1 functions at DSBs.
More recently, MERIT40 was identified as the fifth element of the BRCA1A complex72–74. MERIT40 stabilizes various components of the BRCA1A complex, which in turn allows optimal targeting of BRCA1 to DSBs. Interestingly, detailed bioinformatics analyses72 have shown that the structure of the BRCA1A complex has striking similarities to the domain organization of the 19S complex of the proteasome, which reveals that many BRCA1A components bind ubiquitin. With the exception of RAP80, which binds specifically to Lys63-linked ubiquitin chains, BRCC36, BRCC45, abraxas and MERIT40 were all shown to associate with both Lys48- and Lys63-linked ubiquitin polypeptides72. Whether this is achieved through the various putative ubiquitin-binding domains, namely the ubiquitin E2 variant (UEV) domain of BRCC45, the MPN+ domain of BRCC36, the MPN− domain of abraxas and the von Willebrand A (VWA) domain of MERIT40, remains to be determined. However, the association with ubiquitin chains is particularly interesting as it highlights an integral role for BRCA1A in ubiquitylation and suggests that assembly of BRCA1A might control the dynamics of local ubiquitylation events72. It is also noteworthy that, apart from UIM domain-containing RAP80, these putative ubiquitin domain-containing proteins seem to show a preference for ubiquitin chains of eight or more subunits. It is thus tempting to speculate that these ubiquitin binding events stabilize RAP80 UIM-mediated docking at ubiquitin-modified chromatin structures irrespective of chain or substrate specificities. This possibility and the identities of the proteins that are modified by Lys63-linked ubiquitin chains at the DNA damage sites warrant further investigation.
BRCA1B is involved in DNA replication
The BRCA1B complex is made up of BACH1 and topoisomerase II-binding protein 1 (TOPBP1) (FIG. 1). Replication stress and DNA synthesis across DSBs or DNA lesions often result in stalling or collapse of replication forks. To circumvent the deleterious nature of these DSBs, the cell has evolved strategies to transiently inhibit DNA synthesis, thereby allowing repair to occur before DNA synthesis resumes. These are collectively known as replication checkpoints and include the G1–S checkpoint and the intra-S phase checkpoint. BRCA1 has been ascribed roles in both of these checkpoint controls75,76, thereby ensuring genome stability during S phase of the cell cycle.
BACH1 was first described as a member of the DEAH helicase family that binds to the BRCT domain repeats of BRCA1 (REF. 35). Subsequent work indicated that BACH1 is phosphorylated on Ser990 in a cell cycle-dependent manner, and this phosphorylation event is required for its association with BRCA1 (REF. 6.) Interestingly, the BRCA1–BACH1 interaction can be readily detected during S phase6 and BRCA1 is required for progression through S phase77, which suggests that these two proteins probably work together during DNA replication. Moreover, the BRCA1–BACH1 complex is required for the ill-defined G2 accumulation checkpoint (which is distinct from the G2–M checkpoint) after DNA damage6,54. Because BACH1 is not involved in damage-induced CHK1 phosphorylation and the G2–M transition, the intrinsic differences between these checkpoint control mechanisms remain to be determined.
TOPBP1, which has been implicated in DNA replication and replication checkpoint control78,79, was only recently described as a component of the BRCA1–BACH1 complex67. It is detected after ionising radiation-induced DSBs or thymidine treatment, which depletes the cellular pool of dCTP and results in stalling and collapse of replication forks.
Several lines of evidence support a role for the BRCA1B complex in S phase to ensure genome integrity. First, all three proteins (BRCA1, BACH1 and TOPBP1) are loaded onto replication origins and facilitate DNA replication initiation by mediating the loading of the replication licensing factor CDC45L (REFS 67,80,81). Second, the three proteins are required for timely S phase progression77–79. Third, BACH1 directly interacts with TOPBP1 during S phase, and their molecular interaction is essential for optimal loading of replication protein A (RPA; which binds single-stranded DNA (ssDNA)) onto the chromatin, suggesting that the BACH1–TOPBP1 association monitors normal DNA replication and facilitates replication stress-induced checkpoint activation (Z. Gong and J.C., unpublished observations). Taken together, these results suggest that the BRCA1B complex probably has a role in DNA replication to ensure the proper activation of the replication checkpoint in response to stalled replication forks.
BRCA1C is involved in DNA end resection
The BRCA1C complex is composed of CtIP and the meiotic recombination 11 (MRE11)–RAD50–NBS1 complex (MRN complex), which has an essential role in damage detection and ATM signalling. Both CtIP and the MRN complex are thought to function in HR-mediated DNA repair. HR-mediated repair operates during the S and G2 phases of the cell cycle, when sister chromatids are available. To facilitate this DNA repair, DSBs need to be processed to generate ssDNA regions, which allow assimilation of the DNA recombination repair protein RAD51. Recent work by several groups suggested that optimal resection of DNA ends requires the concerted actions of the MRN complex, CtIP, the 5′–3′ double-stranded DNA exonuclease Exo I and the helicase Bloom syndrome protein (BLM) 82–86. Of these, CtIP is of particular interest because it lacks any known functional domains aside from a poorly understood SUMO-activating enzyme subunit 2-like motif at its C terminus. CtIP promotes DNA end resection by interacting and stimulating the nuclease activity of the MRN complex83. Phenotypical analyses revealed that depletion of CtIP markedly reduced ssDNA generation and damage-induced RPA focus formation. This, in turn, impaired the loading of the ATR–ATR-interacting protein (ATRIP) complex, which is important for G2–M checkpoint activation. Interestingly, CtIP is phosphorylated at Ser327 and interacts with BRCA1 during the S–G2 phase of the cell cycle, when HR-mediated repair is more prominent54, suggesting that BRCA1 might also participate in DNA end resection. In line with this possibility, the interaction of BRCA1 with components of the MRN complex is mediated by cyclin-dependent kinases and is dependent on phosphorylation, which can be further stimulated by genotoxic stress87. Moreover, BRCA1 is required for efficient ssDNA generation at DSBs87,88. This is supported by the observation that the accumulation of RPA (which indicates DNA ‘single-strandedness’) at damage-induced foci is reduced in BRCA1-deficient cells at early time points, which suggests defects in DNA resection. DNA resection is an early event in DNA repair and checkpoint activation, indicating that the BRCA1C complex is involved early in both HR-mediated repair and ATR–ATRIP-dependent G2–M checkpoint activation.
Because BRCA1 is not known to have any nuclease or helicase domains, it is tempting to speculate that BRCA1 might act as a scaffold to stabilize the MRN–CtIP complex formation, which is important for DNA end resection. Interestingly, a recent study in chicken DT40 cells suggested that BRCA1 might act as a switch to activate CtIP-dependent DNA end resection specifically during the S–G2 phase progression89. The authors found that a phosphorylation-defective CtIP mutant, which does not associate with BRCA1, displayed reduced accumulation in damage-induced ssDNA and was defective in HR-mediated repair. Whether BRCA1 serves a similar purpose in mammalian cells remains to be confirmed. Nevertheless, these studies highlight the intriguing possibility that BRCA1 might modulate HR-mediated repair partly through coupling DNA end resection and G2–M checkpoint control, thereby ensuring that appropriately processed substrates for HR-based DNA repair are ready when homologous templates are available.
Functional redundancy among BRCA1 complexes
In addition to the BRCA1A complex, CtIP is required for damage-induced phosphorylation of CHK1 and G2–M checkpoint activation54. Because deficiency in components involved in the pathways that ensure the accumulation of BRCA1 at damage-induced foci do not fully recapitulate the magnitude of defects in G2–M checkpoint activation, it is likely that the BRCA1C complex also contributes to part of the BRCA1-dependent G2–M checkpoint control. Consistent with this possibility, cells expressing a phosphorylation-defective CtIP mutant that cannot bind BRCA1 exhibit defects in damage-induced CHK1 phosphorylation and G2–M checkpoint control55. The BRCA1C complex might enforce the G2–M checkpoint control through its role in DNA end resection. Recent studies suggested that RPA-coated ssDNA is the recruiting factor for ATR–ATRIP loading onto the damaged chromatin, which in turn facilitates the activation of the G2–M checkpoint90. In addition, BRCA1 was also reported to interact with phosphorylated ATRIP91. Thus, it is likely that BRCA1 and its distinct macrocomplexes participate in many aspects of the cellular response to DNA damage to ensure efficient cell cycle arrest.
The apparent complexity of BRCA1 roles in the DNA damage response also stems from our limited understanding of the many pathways that govern BRCA1 tethering to chromatin. Although sustained localization of BRCA1 at DSBs requires the canonical H2AX pathway, the mechanistic basis underlying its accumulation onto ssDNA regions remains unknown92. In addition, because H2AX is dispensable for the initial recruitment of many DNA damage response elements, including the accumulation of BRCA1 onto DSBs93, it is possible that the transient localization of BRCA1 at DNA lesions might occur independently of H2AX and that this localization might suffice for at least some of its functions. Indeed, mice with deficiencies in various components of the DNA damage signalling pathway that are important for the accumulation of BRCA1 at damage-induced foci, including H2AX and MDC1, have a mild phenotype compared with wild-type mice, which indicates that these components are not essential for cell survival26,94,95.
Given the putative role of the MRN complex in DNA lesion detection, it is likely that some of these BRCA1 functions at damage-modified chromatin might be achieved through its direct interaction with the MRN complex96 without the requirement for its sustained retention at chromatin. Future studies will be needed to address these possibilities.
BRCA1 and DNA repair
Although recent studies have revealed new ways through which BRCA1 facilitates DNA repair, a role for BRCA1 in HR-mediated repair97 was first described a decade ago. This was partly on the basis of its stable complex formation with BRCA2 (REF. 98) (BOX 2), which has a well-defined role in HR-mediated DNA repair through its direct interaction with RAD51 (REFS 23,99,100). BRCA2 initiates HR-mediated DNA repair by recruiting RAD51 to sites of DNA damage and facilitating its assimilation onto ssDNA. Recent work identified a new component, PALB2, as an upstream factor that is required for chromatin loading of BRCA2 and RAD51 (REF. 101). Similar to BRCA2 deficiency, deficiency in PALB2 severely impairs HR-mediated repair. Interestingly, in addition to patients with breast and ovarian cancer, both PALB2 and BRCA2 were found to be mutated in individuals with the cancer-predisposing disease Fanconi anaemia (see below). Cells derived from patients with Fanconi anaemia are especially sensitive to DNA cross-linking agents and often display radial chromosomes, a hallmark of Fanconi anaemia cells102–105. Given that HR-mediated repair is one of the main pathways involved in the repair of DNA cross links, these observations indicate that both PALB2–BRCA2 and the BRCA1 pathway are involved in this process.
Box 2. Homologous recombination-mediated DNA repair involving BRCA2.
Homologous recombination (HR)-mediated DNA repair operates during S and G2 phases of the cell cycle, when sister chromatids are available. The use of identical sister chromatids as templates during HR-mediated repair minimizes errors, so this repair pathway is generally thought to be error free123.
HR-mediated DNA repair requires the recombinase RAD51, an evolutionarily conserved single-stranded DNA (ssDNA)-binding protein. RAD51 is loaded onto ssDNA, forming a nucleoprotein filament. Through the concerted action of numerous proteins (such as RAD54 and RAD51-associated protein 1 (RAD51AP1)), the RAD51-coated ssDNA catalyses strand invasion and results in the formation of a displacement loop (D-loop). Branch migration and resolution of the recombination intermediate (also known as the Holliday junction) by helicases and resolvases dictate the formation of recombinant products.
Studies have identified the breast and ovarian cancer type 2 susceptibility protein (BRCA2; also known as FANCD1) as an upstream factor required for RAD51 accumulation at DNA damage sites99. BRCA2 directly interacts with RAD51, is required for the loading of RAD51 onto ssDNA and is essential for HR-mediated repair. Partner and localizer of BRCA2 (PALB2; also known as FANCN) was recently identified as the recruiting factor for BRCA2 to DNA breaks101. PALB2- and BRCA2-deficiencies abrogated the formation of damage-induced foci at which RAD51 accumulates103, suggesting an epistatic relationship between the two repair factors.
How does BRCA1 regulate DNA repair?
Besides its possible roles in DNA end resection (discussed above), direct evidence for BRCA1 in DNA repair was also revealed by recent studies that identified PALB2 as the bridging factor required for the BRCA1–BRCA2 association106,107. The BRCA1–PALB2 interaction, mediated by their respective coiled-coil domains, was found to promote HR-mediated repair. Importantly, missense mutations identified in the PALB2-binding region on BRCA1 disrupted the specific interaction of BRCA1 with PALB2 (REF. 106) and compromised DNA repair in a gene conversion assay. This indicates that impaired HR-mediated repair is one of the fundamental causes of the genomic instability and tumorigenesis that is observed in a subset of patients with BRCA1 mutations. Although these studies revealed a molecular link between BRCA1 function and HR-mediated repair, the mechanism by which BRCA1 promotes HR through the PALB2–BRCA2–RAD51 axis remains unclear.
BRCA1 has been reported to facilitate the accumulation of BRCA2 and RAD51 in damage-induced foci67,92. Intriguingly, mutations that result in a truncated BRCA1 that lacks its BRCT domains, and thus cannot accumulate at damage-induced foci, still supported the partial RAD51 accumulation at DNA damage sites106,108. This suggests that BRCA1, in a manner that does not require its BRCT domains, might play an accessory part in HR through PALB2. Because cells with mutations in PALB2 that prevent binding to BRCA1 still retain their ability to form damage-induced foci at sites flanking a DSB106, it is possible that BRCA1 tethering at DSBs is normally an additional anchor point to facilitate PALB2 accumulation, which may be regulated by another factor that has yet to be identified. A non-mutually exclusive means by which BRCA1 might coordinate repair is supported by the fact that BRCA1 is also involved in DNA end resection, which is a prerequisite for RAD51 nucleoprotein filament formation. Given that other BRCA1 BRCT domain-binding proteins, including CtIP and BACH1, are found in PALB2 immunoprecipitates106, it remains to be tested whether BRCA1 has evolved to coordinate different steps in the HR reaction.
Despite the suggested requirement for the BRCA1 E3 ubiquitin ligase activity and its HR repair function in tumour suppression, a recent study challenged this notion and indicated that the ubiquitin ligase activity of BRCA1 may be dispensable for at least a subset of its functions109. Using isogenic embryonic stem cells that express an enzymatically inactive BRCA1 mutant (IlE26Ala mutant), it was found that cells lacking ubiquitin ligase activity and wild-type cells showed comparable levels of BRCA1- and HR-mediated DNA repair. Thus, it seems that the ubiquitin ligase activity of BRCA1 is not absolutely required for all of its functions in vivo. However, cells expressing the catalytically inactive IlE26Ala mutant showed increased rates of damage-induced chromosomal rearrangements109, which suggests that BRCA1-mediated protein ubiquitylation may be involved in a different aspect of the BRCA1 tumour suppression function. In this regard, BRCA1 is also known to participate in DNA decatenation by regulating the ubiquitylation status of topoisomerase IIA110. Furthermore, BRCA1 has also been ascribed a role in ensuring proper centrosome duplication during cell division through the ubiquitylation of γ-tubulin62. It will also be important to see whether the BRCA1 IlE26Ala mutation supports development and has a tumour suppressor role in mice, which would reveal the importance of the BRCA1 ubiquitin ligase activity in BRCA1 function.
How does BRCA1 relate to the Fanconi anaemia pathway?
Fanconi anaemia is a rare human genetic disorder that is characterized by various developmental defects and a high incidence of malignancies111. To date, at least 13 complementation groups have been assigned to this heterogeneous disease. Consistent with the observed increase in tumorigenesis among patients with Fanconi anaemia, Fanconi anaemia cells are highly sensitive to DNA cross-linking agents and show increased rates of chromosome breakage and sister chromatid interchanges. This phenotype is indicative of defective DNA repair and, in particular, of repair and removal of cross-linked DNA, which is a common cause of the clinical features of the affected individuals111,112. Interestingly, the successful identification of the proteins mutated in the 13 complementation groups revealed that three of them, namely BACH1, BRCA2 and PALB2, have also been found mutated in heritable breast and ovarian cancers, with BRCA2 and PALB2 being intimately involved in HR-mediated DNA repair (discussed above). These results indicate a functional relationship between the proteins involved in the Fanconi anaemia pathway and the proteins that interact with BRCA1.
The mechanism by which BRCA1 is involved in the Fanconi anaemia pathway remains controversial. Early reports suggested that BRCA1 interacts with Fanconi anaemia group D2 protein (FANCD2) and modulates its monoubiquitylation and the formation of damage-induced foci113, and that BRCA1 deficiency results in phenotypes that resemble Fanconi anaemia. However, despite the known role of BRCA1–BACH1 in DNA damage repair, recent studies indicated that BACH1-mediated resistance to the cross-linking agent mitomycin C (to which Fanconi anaemia cells are highly sensitive) is independent of its interaction with BRCA1 (REFS 114,115). This suggests that BRCA1 may participate in the Fanconi anaemia pathway independently of its binding to BACH1.
Interestingly, clinical observations in which patients with mutations in Fanconi anaemia genes other than those encoding BRCA1, BRCA2, PALB2 and BACH1 are not susceptible to breast and ovarian cancers suggest that the BRCA-related Fanconi anaemia components may participate in Fanconi anaemia-like functions that do not entirely overlap with other Fanconi anaemia proteins. It is conceivable that the repair of cross-linked DNA occurs in multiple steps that involve different repair factors or repair machineries (FIG. 3). As such, the Fanconi anaemia core (FANCA–FANCC, FANCe–FANCG, FANCL and FANCM) and FANCD2 in complex with FANCI might have an early role in the processing of cross-linked DNA, which may subsequently require the participation of HR repair factors (for example, BRCA1, BRCA2, PALB2 and BACH1) to repair the break. In line with this idea, DNA cross-linking agents trigger replication-dependent DSBs, which are commonly repaired through sister chromatid exchange116. Notably, in the absence of efficient or functional HR-mediated repair, cross link-induced DSBs give rise to radial chromosomes. Given that BRCA1 is intimately involved in HR-mediated repair, it is tempting to speculate that BRCA1 might mediate the repair of processed cross-linked DNA, failure of which contributes to the Fanconi anaemia-like phenotype observed in BRCA1 mutant cells. In support of this model, a recent study revealed the coupling of DNA replication and concentration of the BRCA-related Fanconi anaemia proteins (PALB2, BACH1 and FANCD1) to DNA cross links in vivo117. Although it remains to be seen whether BRCA1 recruitment to DNA cross links resembles that of BRCA-related Fanconi anaemia proteins, these results suggest that cross link repair involves at least two pathways and recognition signals.
Figure 3. The connection between the Fanconi anaemia pathway and the BRCA1-linked homologous recombination repair pathway.

Repair of inter-strand DNA cross links can involve both Fanconi anaemia (FANC) and breast cancer susceptibility (BRCA) proteins. When the replication machinery encounters cross-linked DNA, replication fork stalling and/or collapsing occurs (1). This type of replication stress might signal for the monoubiquitylation of FANCD2–FANCI and its consequent association with chromatin, which is mediated by the Fanconi anaemia core complex (FANCA–FANCC, FANCE–FANCG, FANCL and FANCM) (2). It is thought that the Fanconi anaemia core and FANCD2–FANCI may be involved in the repair or removal of DNA cross links. Subsequently, the BRCA1 core complex (BRCA1, BRCA2 (also known as FANCD1), partner and localizer of BRCA2 (PALB2; also known as FANCN) and BRCA1-interacting protein carboxy-terminal helicase 1 (BACH1; also known as FANCJ and BRIP1)) would be loaded onto sites of DNA damage (3). This would facilitate the accumulation and nucleation of RAD51 filaments (4), ultimately leading to the repair of inter-strand DNA cross links and the subsequent reinitiation of DNA replication (5).
Concluding remarks and perspectives
The multifunctional nature of BRCA1 in maintaining genome integrity can be appreciated by its ability to form various distinct protein complexes, each of which is dedicated to a specific cellular function in the DNA damage response pathway. Notably, these BRCA1-containing macrocomplexes have ascribed roles in cell cycle checkpoint control and DNA repair, and at times participate in different steps of a common reaction to complement each other and ensure a full response to DNA damage. This is not surprising considering that checkpoint control is tightly coupled with DNA repair. Thus, BRCA1 is emerging as the central component involved in multiple aspects of DNA damage responses, which together aim to promote cell survival and genomic stability following genotoxic stress. Interestingly, inactivation of the DNA damage mediator tumour suppressor p53-binding protein 1 (TP53BP1) was recently shown to alleviate senescence and cell death associated with BRCA1 deficiency118, suggesting that cell cycle checkpoint and repair defects in the absence of BRCA1 may be exploited by TP53BP1-dependent pathways and converted to deleterious signals. Given the prominent role of TP53BP1 and BRCA1 in non-homologous end joining and HR pathways, respectively, it will be interesting to see whether DNA lesions in the absence of BRCA1 may be processed by alternative repair pathways involving TP53BP1.
Although studies from recent years have greatly broadened our current views of the BRCA1-dependent DNA damage responses, it remains to be seen whether additional players might be involved in this intertwined BRCA1 network. As such, it would be interesting to scrutinize coding regions on BRCA1 other than the RING domain and tandem BRCT repeats, which are targeted by clinical mutations. The recent identification of PALB2 as a new BRCA1-binding factor illustrates the existence of many possible routes by which BRCA1 functions in the maintenance of genome stability and tumour suppression. For example, a recent study indicated that the central region of BRCA1 enables its interaction with DNA119. Whether and how these findings eventually translate into functional attributes of BRCA1 awaits further research.
Another major challenge in the field of BRCA1 research is to elucidate the roles of each protein in the BRCA1 complex. The presence of the deubiquitylase BRCC36 in the BRCA1A complex is especially intriguing and warrants further study. A recent report revealed that inhibition of BRCC36 partially restored the DSB-associated ubiquitin conjugates and damage-induced foci, where TP53BP1 accumulates, in cells depleted of the E3 ubiquitin ligase RNF8 (REF. 120). This suggests that BRCC36 normally deubiquitylates the substrate of RNF8 and UBC13 at DSBs. It remains to be determined whether BRCC36-dependent deubiquitylation is required for checkpoint recovery or whether it functions to supply a local pool of free ubiquitin by enzymatically deconjugating Lys63-linked polyubiquitin chains, which in turn allows BRCA1 to efficiently catalyse ubiquitylation of its substrates.
Studies from the past few years have provided the structural framework for our current understanding of the function of BRCA1 and its network of proteins that are involved in the DNA damage response pathway. To fully appreciate the role of BRCA1 in the maintenance of genome stability, more in-depth analyses will be needed to fill in the gaps in the numerous working models involving BRCA1. Although the link between BRCA1 and breast cancer is uniquely related to humans, the biological roles of BRCA1 homologues in other organisms await to be elucidated. Understanding the conserved and fundamental cellular functions of this tumour suppressor may reveal unprecedented insights into how and why functions of human BRCA1 have diverged during the course of evolution. This will probably require a combination of genetic, biochemical and structural approaches to investigate the biology of BRCA1 and its binding partners. Until then, the question of how BRCA1 exerts its tumour suppressor activity will linger for just a little longer.
Acknowledgements
This work was supported in part by grants from the National Institutes of Health (CA089239, CA092312 and CA100109 to J.C.), the Startup Fund (Department of Anatomy, The University of Hong Kong to M.S.Y.H.) and Seed Funding for Basic Research (The University of Hong Kong to M.S.Y.H.). J.C is a recipient of an Era of Hope Scholar award from the Department of Defence and a member of the Mayo Clinic Breast SPORE program (P50 CA116201).
Glossary
- E3 ubiquitin ligase
A protein or protein complex that covalently attaches ubiquitin moieties to its target protein by an isopeptide bond. E3 ubiquitin ligases usually provide the substrate specificity for a ubiquitylation reaction that involves an E1 ubiquitin-activating enzyme and an E2 ubiquitin-conjugating enzyme. Two major classes of E3 ubiquitin ligases have been defined based on their conserved HECT and RING domains.
- Homologous recombination
A DNA recombination pathway, which includes the repair of dsDNA breaks, that uses a homologous dsDNA molecule as a template for the repair of the broken DNA.
- E2 ubiquitin-conjugating enzyme
A protein that transfers the activated ubiquitin from the E1 ubiquitin-activating enzyme to an E3 ubiquitin ligase. E2 ubiquitin-conjugating enzymes determine ubiquitin chain specificities, and each E2 enzyme associates with several E3 ligases.
- Coiled-coil domain
A structural element that is important for mediating protein–protein interactions. The sequence of coiled-coil domains contains repetitive elements of seven apolar residues that form a heptad.
- Non-canonical ubiquitin chain
A diubiquitin or polyubiquitin chain comprising ubiquitin molecules that are conjugated by their Lys residues, other than Lys48. Some of these chains do not target proteins for proteosomal degradation.
- G2–M checkpoint
A DNA damage-induced transient cell cycle arrest at the G2–M border, usually associated with CHK1 phosphorylation and activation.
- Proteasome
A large protein complex that is responsible for breaking down polyubiquitylated proteins.
- G1–S checkpoint
A checkpoint that ensures growing conditions are optimal before cells are committed to one round of DNA replication and cell division.
- Intra-S phase checkpoint
ATM- and ATR-dependent transient inhibition of DNA replication in response to DNA damage. Defects in an ionising radiation-induced intra-S phase checkpoint cause radioresistant DNA synthesis.
- DEAH helicase family
A family of proteins that use ATP and unwind nucleic acids, which have a conserved DEAH box.
- Radial chromosome
An abnormal chromosome structure that results from pairing of homologous or non-homologous metaphase chromosomes. These structures are observed in chromosome spreads prepared from cells with an underlying chromosome instability, such as cells from patients with fanconi anaemia, Bloom syndrome and ataxia telangiectasia.
- Gene conversion
A non-reciprocal recombination process that results in an alteration of the sequence of a gene to that of its homologue.
- Isogenic
Here, refers to a genotype that has been engineered to be identical to another genotype, with the exception of one or more mutations of interest.
- DNA decatenation
An ATP-dependent process for the resolution of replicated sister chromatids that requires topoisomerase II activity.
- Chromatid exchange
The physical exchange of genetic material between identical sister chromatids. This process can be enhanced by treatment with DNA damaging agents, such as the cross-linking agent mitomycin C.
Footnotes
Competing interests statement The authors declare no competing financial interests.
DATABASES OMIM: http://www.ncbi.nlm.nih.gov/omim Fanconi anaemia
UniProtKB: http://www.uniprot.org abraxas ∣ ATM ∣ ATR ∣ ATRIP ∣ BACH1 ∣ BAP1 ∣ BARD1 ∣ BLM ∣ BRCA1 ∣ BRCA2 ∣ BRCC36 ∣ BRCC45 ∣ CDC45L ∣ CHK1 ∣ CHK2 ∣ CtIP ∣ FANCI ∣ FANCD2 ∣ MDC1 ∣ MERIT40 ∣ NBS1 ∣ PALB2 ∣ p53 ∣ RAD51 ∣ RAP80 ∣ RNF8 ∣ RNF168 ∣ TOPBP1 ∣ TP53BP1 ∣ UBC13
FURTHER INFORMATION Junjie Chen’s homepage: http://faculty.mdanderson.org/Junjie_Chen
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 3.Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 4.Miki Y, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 5.Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. doi: 10.1126/science.1088877. Together with reference 6, these studies laid the groundwork for how the BRCA1 BRCT domains interact with various binding partners.
- 6.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 7.Meza JE, Brzovic PS, King MC, Klevit RE. Mapping the functional domains of BRCA1. Interaction of the ring finger domains of BRCA1 and BARD1. J. Biol. Chem. 1999;274:5659–5665. doi: 10.1074/jbc.274.9.5659. [DOI] [PubMed] [Google Scholar]
- 8.Ghimenti C, et al. Germline mutations of the BRCA1-associated ring domain (BARD1) gene in breast and breast/ovarian families negative for BRCA1 and BRCA2 alterations. Genes Chromosomes Cancer. 2002;33:235–242. doi: 10.1002/gcc.1223. [DOI] [PubMed] [Google Scholar]
- 9.Thai TH, et al. Mutations in the BRCA1-associated RING domain (BARD1) gene in primary breast, ovarian and uterine cancers. Hum. Mol. Genet. 1998;7:195–202. doi: 10.1093/hmg/7.2.195. [DOI] [PubMed] [Google Scholar]
- 10.Capasso M, et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nature Genet. 2009;41:718–723. doi: 10.1038/ng.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westermark UK, et al. BARD1 participates with BRCA1 in homology-directed repair of chromosome breaks. Mol. Cell. Biol. 2003;23:7926–7936. doi: 10.1128/MCB.23.21.7926-7936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nature Struct. Biol. 2001;8:833–837. doi: 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 13.Christensen DE, Brzovic PS, Klevit RE. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nature Struct. Mol. Biol. 2007;14:941–948. doi: 10.1038/nsmb1295. Suggests that BRCA1 may promote the formation of distinct ubiquitin chains through selective interaction with E2 enzymes.
- 14.Brzovic PS, Lissounov A, Christensen DE, Hoyt DW, Klevit RE. A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1-directed ubiquitination. Mol. Cell. 2006;21:873–880. doi: 10.1016/j.molcel.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Parvin JD. The BRCA1-dependent ubiquitin ligase, γ-tubulin, and centrosomes. Environ. Mol. Mutagen. 2009;50:649–653. doi: 10.1002/em.20475. [DOI] [PubMed] [Google Scholar]
- 16.Heine GF, Parvin JD. BRCA1 control of steroid receptor ubiquitination. Sci. STKE. 2007;2007:pe34. doi: 10.1126/stke.3912007pe34. [DOI] [PubMed] [Google Scholar]
- 17.Starita LM, Parvin JD. Substrates of the BRCA1-dependent ubiquitin ligase. Cancer Biol. Ther. 2006;5:137–141. doi: 10.4161/cbt.5.2.2479. [DOI] [PubMed] [Google Scholar]
- 18.Eakin CM, Maccoss MJ, Finney GL, Klevit RE. Estrogen receptor α is a putative substrate for the BRCA1 ubiquitin ligase. Proc. Natl Acad. Sci. USA. 2007;104:5794–5799. doi: 10.1073/pnas.0610887104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nature Genet. 1998;19:254–256. doi: 10.1038/930. [DOI] [PubMed] [Google Scholar]
- 20.Scully R, et al. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl Acad. Sci. USA. 1997;94:5605–5610. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bochar DA, et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000;102:257–265. doi: 10.1016/s0092-8674(00)00030-1. [DOI] [PubMed] [Google Scholar]
- 22.Yarden RI, Brody LC. BRCA1 interacts with components of the histone deacetylase complex. Proc. Natl Acad. Sci. USA. 1999;96:4983–4988. doi: 10.1073/pnas.96.9.4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scully R, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. First evidence for a role of BRCA1 in HR-based DNA repair
- 24.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 25.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Celeste A, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stucki M, et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 28.Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- 29.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 30.Lou Z, Chini CC, Minter-Dykhouse K, Chen J. Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J. Biol. Chem. 2003;278:13599–13602. doi: 10.1074/jbc.C300060200. [DOI] [PubMed] [Google Scholar]
- 31.Goldberg M, et al. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- 32.Xu X, Stern DF. NFBD1/MDC1 regulates ionizing radiation-induced focus formation by DNA checkpoint signaling and repair factors. Faseb J. 2003;17:1842–1848. doi: 10.1096/fj.03-0310com. [DOI] [PubMed] [Google Scholar]
- 33.Xu X, Stern DF. NFBD1/KIAA0170 is a chromatin-associated protein involved in DNA damage signaling pathways. J. Biol. Chem. 2003;278:8795–8803. doi: 10.1074/jbc.M211392200. [DOI] [PubMed] [Google Scholar]
- 34.Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998;273:25388–25392. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 35.Cantor SB, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 36.Wang B, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim H, Huang J, Chen J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nature Struct. Mol. Biol. 2007;14:710–715. doi: 10.1038/nsmb1277. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z, Wu J, Yu X. CCDC98 targets BRCA1 to DNA damage sites. Nature Struct. Mol. Biol. 2007;14:716–720. doi: 10.1038/nsmb1279. [DOI] [PubMed] [Google Scholar]
- 39.Yan J, et al. The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 2007;67:6647–6656. doi: 10.1158/0008-5472.CAN-07-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- 41.Sobhian B, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. References 36–41 reveal mechanistically how BRCA1 is targeted to sites of DNA breaks.
- 42.Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl Acad. Sci. USA. 2007;104:20759–20763. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolas NK, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huen MS, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mailand N, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 46.Doil C, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 47.Stewart GS, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–434. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 48.Hofmann RM, Pickart CM. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 1999;96:645–653. doi: 10.1016/s0092-8674(00)80575-9. [DOI] [PubMed] [Google Scholar]
- 49.Lorick KL, et al. RING fingers mediate ubiquitinconjugating enzyme (E2)-dependent ubiquitination. Proc. Natl Acad. Sci. USA. 1999;96:11364–11369. doi: 10.1073/pnas.96.20.11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morris JR, Solomon E. BRCA1: BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum. Mol. Genet. 2004;13:807–817. doi: 10.1093/hmg/ddh095. Together with reference 56, these studies reveal that BRCA1-dependent non-canonical types of ubiquitin linkages are required for DNA repair.
- 51.Polanowska J, Martin JS, Garcia-Muse T, Petalcorin MI, Boulton SJ. A conserved pathway to activate BRCA1-dependent ubiquitylation at DNA damage sites. EMBO J. 2006;25:2178–2188. doi: 10.1038/sj.emboj.7601102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen A, Kleiman FE, Manley JL, Ouchi T, Pan ZQ. Autoubiquitination of the BRCA1•BARD1 RING ubiquitin ligase. J. Biol. Chem. 2002;277:22085–22092. doi: 10.1074/jbc.M201252200. [DOI] [PubMed] [Google Scholar]
- 53.Zhao GY, et al. A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol. Cell. 2007;25:663–675. doi: 10.1016/j.molcel.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 54.Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell. Biol. 2004;24:9478–9486. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006;20:1721–1726. doi: 10.1101/gad.1431006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishikawa H, et al. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J. Biol. Chem. 2004;279:3916–3924. doi: 10.1074/jbc.M308540200. [DOI] [PubMed] [Google Scholar]
- 57.Xia Y, Pao GM, Chen HW, Verma IM, Hunter T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem. 2003;278:5255–5263. doi: 10.1074/jbc.M204591200. [DOI] [PubMed] [Google Scholar]
- 58.Foray N, et al. A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J. 2003;22:2860–2871. doi: 10.1093/emboj/cdg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating CHK1 kinase upon DNA damage. Nature Genet. 2002;30:285–289. doi: 10.1038/ng837. Identifies BRCA1 function in the G2–M checkpoint control through regulation of CHK1 phosphorylation
- 60.Wu LC, et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nature Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 61.Baer R, Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr. Opin. Genet. Dev. 2002;12:86–91. doi: 10.1016/s0959-437x(01)00269-6. [DOI] [PubMed] [Google Scholar]
- 62.Starita LM, et al. BRCA1-dependent ubiquitination of γ-tubulin regulates centrosome number. Mol. Cell. Biol. 2004;24:8457–8466. doi: 10.1128/MCB.24.19.8457-8466.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hashizume R, et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001;276:14537–14540. doi: 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- 64.Nishikawa H, et al. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res. 2009;69:111–119. doi: 10.1158/0008-5472.CAN-08-3355. [DOI] [PubMed] [Google Scholar]
- 65.Shakya R, et al. The basal-like mammary carcinomas induced by BRCA1 or BARD1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc. Natl Acad. Sci. USA. 2008;105:7040–7045. doi: 10.1073/pnas.0711032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joukov V, Chen J, Fox EA, Green JB, Livingston DM. Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc. Natl Acad. Sci. USA. 2001;98:12078–12083. doi: 10.1073/pnas.211427098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Greenberg RA, et al. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. A proposed model of how BRCA1 regulates diverse cellular processes in the DNA damage response.
- 68.Fernandez-Capetillo O, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nature Cell Biol. 2002;4:993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 69.Chen X, Arciero CA, Wang C, Broccoli D, Godwin AK. BRCC36 is essential for ionizing radiation-induced BRCA1 phosphorylation and nuclear foci formation. Cancer Res. 2006;66:5039–5046. doi: 10.1158/0008-5472.CAN-05-4194. [DOI] [PubMed] [Google Scholar]
- 70.Dong Y, et al. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol. Cell. 2003;12:1087–1099. doi: 10.1016/s1097-2765(03)00424-6. [DOI] [PubMed] [Google Scholar]
- 71.Ambroggio XI, Rees DC, Deshaies RJ. JAMM: a metalloprotease-like zinc site in the proteasome and signalosome. PLoS Biol. 2004;2:E2. doi: 10.1371/journal.pbio.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang B, Hurov K, Hofmann K, Elledge SJ. NBA1, a new player in the BRCA1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009;23:729–739. doi: 10.1101/gad.1770309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feng L, Huang J, Chen J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009;23:719–728. doi: 10.1101/gad.1770609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shao G, et al. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009;23:740–754. doi: 10.1101/gad.1739609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu B, O’Donnell AH, Kim ST, Kastan MB. Phosphorylation of serine 1387 in BRCA1 is specifically required for the Atm-mediated S-phase checkpoint after ionizing irradiation. Cancer Res. 2002;62:4588–4591. [PubMed] [Google Scholar]
- 76.Xu B, Kim S, Kastan MB. Involvement of BRCA1 in S-phase and G2-phase checkpoints after ionizing irradiation. Mol. Cell. Biol. 2001;21:3445–3450. doi: 10.1128/MCB.21.10.3445-3450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kumaraswamy E, Shiekhattar R. Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol. Cell. Biol. 2007;27:6733–6741. doi: 10.1128/MCB.00961-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Makiniemi M, et al. BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J. Biol. Chem. 2001;276:30399–30406. doi: 10.1074/jbc.M102245200. [DOI] [PubMed] [Google Scholar]
- 79.Kim JE, McAvoy SA, Smith DI, Chen J. Human TopBP1 ensures genome integrity during normal S phase. Mol. Cell. Biol. 2005;25:10907–10915. doi: 10.1128/MCB.25.24.10907-10915.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Hatten RA, et al. The Xenopus Xmus101 protein is required for the recruitment of Cdc45 to origins of DNA replication. J. Cell Biol. 2002;159:541–547. doi: 10.1083/jcb.200207090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hashimoto Y, Takisawa H. Xenopus Cut5 is essential for a CDK-dependent process in the initiation of DNA replication. EMBO J. 2003;22:2526–2535. doi: 10.1093/emboj/cdg238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mimitou EP, Symington LS. SaE2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl Acad. Sci. USA. 2008;105:16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1•CtIP•MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 88.Schlegel BP, Jodelka FM, Nunez R. BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res. 2006;66:5181–5189. doi: 10.1158/0008-5472.CAN-05-3209. [DOI] [PubMed] [Google Scholar]
- 89.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 91.Venere M, Snyder A, Zgheib O, Halazonetis TD. Phosphorylation of ATR-interacting protein on Ser239 mediates an interaction with breast-ovarian cancer susceptibility 1 and checkpoint function. Cancer Res. 2007;67:6100–6105. doi: 10.1158/0008-5472.CAN-07-0369. The first documented BRCA1 accumulation at two distinct micronuclear domains, which correspond to single-stranded and double-stranded DNA breaks.
- 92.Bekker-Jensen S, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Celeste A, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nature Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 94.Celeste A, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lou Z, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 96.Zhong Q, et al. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science. 1999;285:747–750. doi: 10.1126/science.285.5428.747. [DOI] [PubMed] [Google Scholar]
- 97.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol. Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. The first functional evidence for a role of BRCA1 in HR repair.
- 98.Chen J, et al. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol. Cell. 1998;2:317–328. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- 99.Thorslund T, West SC. BRCA2: a universal recombinase regulator. Oncogene. 2007;26:7720–7730. doi: 10.1038/sj.onc.1210870. [DOI] [PubMed] [Google Scholar]
- 100.Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem. 2000;275:23899–23903. doi: 10.1074/jbc.C000276200. [DOI] [PubMed] [Google Scholar]
- 101.Xia B, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 102.Erkko H, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–319. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 103.Xia B, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nature Genet. 2007;39:159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 104.Reid S, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nature Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 105.Rahman N, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nature Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl Acad. Sci. USA. 2009;106:7155–7160. doi: 10.1073/pnas.0811159106. Together with reference 107, these studies identify PALB2 as the link between BRCA1 and proteins involved in HR-mediated repair.
- 107.Zhang F, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009;19:524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang J, et al. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol. Cell Biol. 2004;24:708–718. doi: 10.1128/MCB.24.2.708-718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reid LJ, et al. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc. Natl Acad. Sci. USA. 2008;105:20876–20881. doi: 10.1073/pnas.0811203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lou Z, Minter-Dykhouse K, Chen J. BRCA1 participates in DNA decatenation. Nature Struct. Mol. Biol. 2005;12:589–593. doi: 10.1038/nsmb953. [DOI] [PubMed] [Google Scholar]
- 111.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nature Rev. Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 112.Kennedy RD, D’Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19:2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 113.Garcia-Higuera I, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 114.Peng M, et al. The FANCJ/MutLα interaction is required for correction of the cross-link response in FA-J. cells. EMBO J. 2007;26:3238–3249. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nature Genet. 2005;37:953–957. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- 116.Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123:1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 117.Shen X, et al. Recruitment of Fanconi anemia and breast cancer proteins to DNA damage sites is differentially governed by replication. Mol. Cell. 2009;35:716–723. doi: 10.1016/j.molcel.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cao L, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by BRCA1 deficiency. Mol. Cell. 2009;35:534–541. doi: 10.1016/j.molcel.2009.06.037. In vivo evidence of an important link between BRCA1- and TP53BP1-dependent pathways.
- 119.Brazda V, Jagelska EB, Liao JC, Arrowsmith CH. The central region of BRCA1 binds preferentially to supercoiled DNA. J. Biomol. Struct. Dyn. 2009;27:97–104. doi: 10.1080/07391102.2009.10507299. [DOI] [PubMed] [Google Scholar]
- 120.Shao G, et al. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc. Natl Acad. Sci. USA. 2009;106:3166–3171. doi: 10.1073/pnas.0807485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bartek J, Lukas J, Bartkova J. DNA damage response as an anti-cancer barrier: damage threshold and the concept of ‘conditional haploinsufficiency’. Cell Cycle. 2007;6:2344–2347. doi: 10.4161/cc.6.19.4754. [DOI] [PubMed] [Google Scholar]
- 122.Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007;19:238–245. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 123.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]