

Abstract
In the central nervous system (CNS), oligodendrocyte maturation and axonal myelination occur on a predictable schedule, but the underlying timing mechanisms are largely unknown. In the present study, we demonstrate that Nkx2.2 homeodomain transcription factor is a key regulator for the timing of oligodendrocyte differentiation during development. Whereas induced expression of Nkx2.2 in early oligodendrocyte precursor cells (OPCs) causes precocious differentiation of oligodendrocytes, conditional ablation of Nkx2.2 temporally delays oligodendrocyte maturation. Moreover, Nkx2.2 can directly bind to the promoter of platelet-derived growth factor receptor alpha (Pdgfra) and repress its gene expression. Genetic ablation of Pdgfra mimics the effect of Nkx2.2 overexpression in accelerating OPC differentiation in the developing spinal cord. Together, our findings strongly suggest that Nkx2.2 functions as a major ‘switch’ to turn off Pdgfra signaling in OPCs and initiate the intrinsic program for oligodendrocyte differentiation.
Keywords: Spinal cord, Tet-on, Transcription factor, Mouse
INTRODUCTION
A requisite component of nervous system development is the achievement of proper axonal myelination for rapid and accurate transmission of electric activities. In the central nervous system (CNS), myelin sheaths are elaborated by oligodendrocytes (OLs), and the myelination process is preceded by molecular and morphological differentiation of oligodendrocyte precursor cells (OPCs). It was observed that OPCs differentiate on a predictable schedule both in vivo and in vitro, but the molecular pathways that control the timing of OPC differentiation have not been clearly defined.
It has been recently shown that multiple classes of transcription factors are involved in the regulation of the OL differentiation process. They include the negative differentiation regulators Id2, Id4 and Hes5 (Kondo and Raff, 2000; Liu et al., 2006; Wang et al., 2001), and positive regulators such as Olig1 (Lu et al., 2002), Mrf (Myrf - Mouse Genome Informatics) (Emery, 2010), Mash-1 (Ascl1 - Mouse Genome Informatics) (Sugimori et al., 2008), Sip1 (Gemin2 - Mouse Genome Informatics) (Weng et al., 2012), Nkx2.2 (Qi et al., 2001) and Sox10 (Soula et al., 2001). Among these transcription factors, Nkx2.2 (Nkx2-2 - Mouse Genome Informatics) is uniquely positioned as a candidate regulator for the timing of OL differentiation. In the developing mouse spinal cord, Nkx2.2 expression is upregulated in OPCs immediately before their differentiation but rapidly downregulated after OPC differentiation (Fu et al., 2002; Soula et al., 2001; Xu et al., 2000; Zhou et al., 2001). Thus, Nkx2.2 expression in differentiating OPCs correlates seamlessly with the onset of OL differentiation. Functional analyses revealed that Nkx2.2 plays an essential role in the terminal differentiation of OLs (Qi et al., 2001; Zhou et al., 2001). However, because of the neonatal lethality of Nkx2.2 mutants, it has remained unknown whether Nkx2.2 is absolutely required for OPC maturation or simply controls the timing of OL differentiation. More importantly, the molecular pathways downstream of Nkx2.2 in the control of OL differentiation remain to be defined.
Previous studies demonstrated that OL differentiation and maturation in culture are also regulated by a variety of extracellular signaling molecules such as Pdgf, bFgf (Fgf2 - Mouse Genome Informatics), Cntf and thyroid hormone (Barres et al., 1994; Noble et al., 1988; Raff et al., 1985; Raff et al., 1988). In particular, Pdgf signaling has been implicated in the temporal control of OPC differentiation during development. In the developing CNS, Pdgf receptor alpha (Pdgfra) is specifically expressed in immature OPCs, but its expression is rapidly extinguished as OPCs undergo terminal differentiation (Pringle et al., 1989; Richardson et al., 1988). It is well known that Pdgf functions as a major mitogen for OPC division both in vitro and during development (van Heyningen et al., 2001). At the same time, Pdgf signaling has also been shown to inhibit OL differentiation, and withdrawal of Pdgf from culture medium is sufficient to trigger the onset of OPC differentiation (Raff et al., 1985; Raff et al., 1988; van Heyningen et al., 2001). Consistently, disruption of Pdgfra in OPCs also resulted in premature oligodendrocyte differentiation in culture (McKinnon et al., 2005). These in vitro studies suggest that there is an intrinsic default program for OPC differentiation, and this differentiation program is normally suppressed by Pdgfra signaling in immature progenitor cells. However, the involvement of Pdgfra signaling in the timing of OPC differentiation has so far not been verified by in vivo genetic study. More importantly, the molecular mechanisms that lead to the transcriptional repression of Pdgfra expression in OPCs and therefore trigger the differentiation process remain to be defined.
In this study, we report that Nkx2.2 controls the timing of OPC differentiation, possibly by directly repressing the expression of Pdgfra, which negatively regulates the differentiation of OLs during development. Alterations of the expression of Nkx2.2 or Pdgfra in OPCs in inducible transgenics or conditional mutants can change the timing of OL differentiation in the developing CNS.
RESULTS
Induced Nkx2.2 expression in early OPCs leads to precocious OL differentiation as well as inhibited OPC proliferation and migration
To determine the developmental role of Nkx2.2 in timing OPC differentiation, we first generated the TetO-Nkx2.2 transgenic line and then crossed it with the Sox10-rtTA knock-in mouse line (Ludwig et al., 2004). In the double transgenic (DTG) mice, Nkx2.2 protein could be induced by Dox treatment in cells of OL lineage at a specific time point. In addition, the Nkx2.2 transgene was tagged with human influenza hemagglutinin (HA) epitope to differentiate from the endogenous expression of Nkx2.2.
When Dox was administered from embryonic day (E) 12.5 to E15.5 before Nkx2.2 upregulation and OL differentiation, HA expression was specifically detected in Sox10-expressing OL cells in the spinal cords of DTG mice (Fig. 1A-C′), indicating the successful induction of exogenous Nkx2.2 protein in OPCs (>70% of the population). At E15.5, Mbp protein expression was not found in the control spinal cord (Fig. 1D). However, strong expression of Mbp was found in both the gray and white matters of DTG spinal cords (Fig. 1E). Moreover, all the induced Mbp+ cells co-expressed HA (Fig. 1E-E′, arrowheads), indicating that the precocious Mbp expression was induced by expression of exogenous Nkx2.2 protein. The premature OL differentiation was further confirmed by expression of other myelin markers such as Mag (Fig. 1F,G), and the display of multipolar processes instead of the typical bipolar processes for OPCs at this stage (Fig. 1E-E′, insets). Prolonged Dox treatment from E12.5 to E18.5 further promoted differentiation of OPCs and led to a significant reduction of the number of Olig2+ and Sox10+ cells in DTG spinal cords compared with the controls (supplementary material Fig. S1).
Fig. 1.

Precocious OL differentiation induced by overexpression of Nkx2.2 in early OPCs. (A) Time window of Dox treatment. (B-G) Immunostaining of transverse spinal cord sections from control and DTG embryos after Dox exposure (E12.5-E15.5) with antibodies against Sox10 and HA (B-C′), Mbp and HA (D-E′), Mag (F-G). Double-positive cells are represented by white arrowheads. (H) Quantification of Olig2+, Sox10+ or Mbp+ cells per section in control and DTG mouse spinal cords at E15.5 (mean ± s.e.m., n=3). **P<0.01. Scale bars: 50 μm.
Strikingly, Nkx2.2 induction in DTG spinal cord tissues also led to a drastic reduction in the total number of Sox10+ and Olig2+ cells (Fig. 1B,C,H; supplementary material Fig. S1B,C,F). This decrease could be caused by diminished OPC proliferation and/or increased cell death. To investigate these possibilities, we first carried out cell proliferation assays in E15.5 embryos after 2-hour short-term bromodeoxyuridine (BrdU) labeling, and found that the percentage of dividing OPCs, represented by Olig2+/BrdU+ cells in the Olig2+ population, in DTGs was about half of that in controls (supplementary material Fig. S2A-B′,E). Cell death assays revealed few phospho-Caspase-3 positive apoptotic cells in both control and DTG spinal cords at this stage (supplementary material Fig. S2C-D′). Therefore, overexpression of Nkx2.2 reduced early OPC proliferation and had little effect on the survival of OPCs. Noticeably, OPC distribution was significantly altered in the DTG spinal cord at this stage, with the majority of OPCs being restricted to the ventral half compared with the evenly dispersed OPCs in the control (supplementary material Fig. S2F,F′), suggesting that Nkx2.2 overexpression also repressed the migration of OPCs.
Inactivation of Nkx2.2 in OPCs caused a temporal delay in OPC differentiation
Previously, we observed that OPC maturation was significantly retarded in the conventional Nkx2.2 knockout mice (Qi et al., 2001). However, it was not clear whether OPC differentiation could recover at later stages, as the conventional Nkx2.2 mutants die soon after birth. To address if Nkx2.2 is absolutely essential for OPC differentiation, we generated the CNP+/cre;Nkx2.2fl/fl conditional mutant mice in which Nkx2.2 is specifically deleted in OPCs. Conditional ablation of Nkx2.2 did not affect the total size of the Olig2+ OL population at all stages examined (supplementary material Fig. S3). However, expression of the myelin genes Mbp, Plp (Plp1 - Mouse Genome Informatics) and Apc was dramatically reduced from postnatal day (P) 0 to 13, but was fully recovered at P21 (Fig. 2; supplementary material Fig. S4). Thus, Nkx2.2 mutation caused a temporal delay in OPC differentiation. Based on the results from studies of both transgenics and conditional mutants, we concluded that Nkx2.2 controls the timing of OL differentiation and is not absolutely required for OL differentiation.
Fig. 2.

Delayed OL differentiation and maturation following conditional ablation of Nkx2.2 in OPCs. Spinal cord sections from Cnp+/cre;Nkx2.2+/fl and Cnp+/cre;Nkx2.2fl/fl were subjected to Mbp (A-D′) and Plp (E-H′) in situ hybridization at P3, P7, P13 and P21.
Nkx2.2 negatively regulates the expression of Pdgfra in OPCs by directly binding to the Pdgfra promoter
In light of the previous finding that Pdgfa is a major mitogen for OPCs in the developing CNS, the reduced OPC proliferation in Nkx2.2 transgenics raises the possibility that Nkx2.2 induction may suppress Pdgfra expression or signaling in OPCs. Consistent with this idea, Pdgfra expression in transgenic spinal cords was completely inhibited after Dox induction (Fig. 3A-D). By contrast, Pdgfra expression was markedly enhanced in both the conventional (Fig. 3E-H) and conditional Nkx2.2 mutants (supplementary material Fig. S3E-H′). Together, these data indicated that Nkx2.2 repressed the expression of Pdgfra during the OPC differentiation process.
Fig. 3.
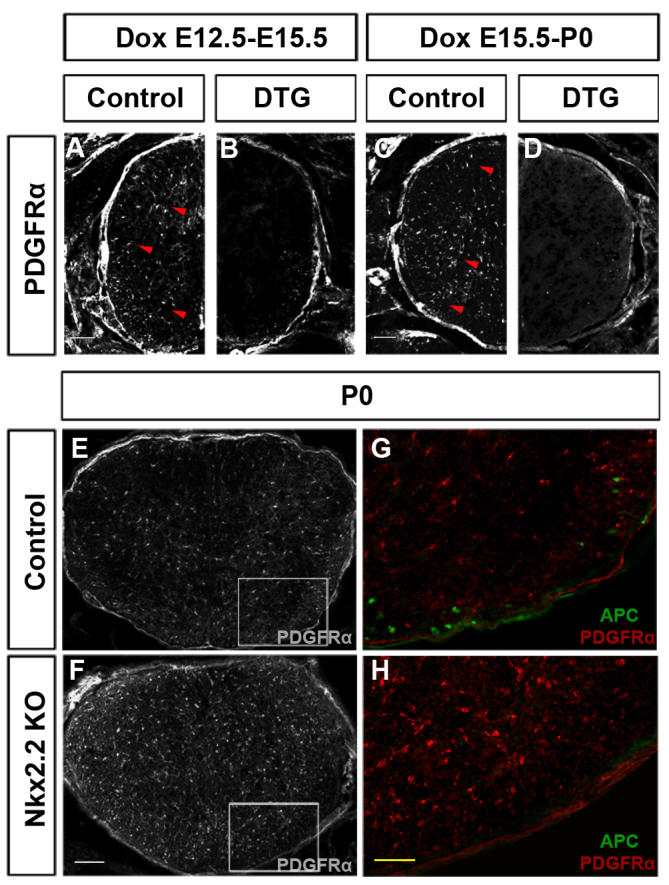
Negative regulation of Pdgfra expression by Nkx2.2. (A-D) Inhibition of Pdgfra expression in Nkx2.-overexpressing transgenics. Spinal cord sections from control and DTG mice after Dox treatment (E12.5-E15.5) or (E15.5-P0) were immunostained with anti-Pdgfra antibody. Pdgfra+ OPCs were represented by arrowheads. (E,F) Increased expression of Pdgfra in P0 Nkx2.2 knockout spinal cords. (G,H) Higher magnifications of the boxed areas in E,F with original anti-Pdgfra and anti-APC double immunostaining. KO, knockout. Scale bars: 50 μm (A-D,G,H); 100 μm (E,F).
To test whether Pdgfra is a direct downstream target gene of Nkx2.2, we analyzed the genomic sequence upstream of Pdgfra gene with a novel computational method (Hill et al., 2011) and predicted a potential Nkx2.2 binding site (highest score, Fig. 4A). Electrophoretic mobility shift assay (EMSA) demonstrated that Nkx2.2 protein formed complexes with the sequence corresponding to this putative binding site (Fig. 4B, arrow). These complexes were further shifted upward (supershift) with the addition of Nkx2.2 antibody (Fig. 4B, asterisk). Meanwhile, mutation of this core binding site suppressed the binding of Nkx2.2 to this labeled DNA sequence. Therefore, Nkx2.2 transcription factor can directly bind to the Pdgfra promoter sequence. We next cloned the potential promoter region of mouse Pdgfra (Bergeron et al., 2011) into the luciferase vector for gene expression assay (Fig. 4A,C). Transient transfection experiments in 293T cells showed a much higher luciferase activity from the Pdgfra promoter-luciferase construct than that from the control vector with the basic promoter (Fig. 4C). However, when the Nkx2.2-expressing vector was co-transfected, the luciferase measurement from the Pdgfra promoter construct was significantly reduced (Fig. 4C), indicating an inhibition of Pdgfra promoter activity by Nkx2.2 expression.
Fig. 4.

Binding and activity assays of Pdgfra promoter. (A) Schematic of the genomic sequence upstream Pdgfra, including the putative promoter cloned into the luciferase vector and selected sequence for EMSA probe (red represents core binding sequence). (B) EMSA analysis of Nkx2.2 binding. Probe (see A) spanning ∼40 bp surrounding the predicted binding sequence was incubated with nuclear extracts from mock-transfected cells (Ctrl NE) or nuclear extracts from transfected cells (Nkx2.2 NE). The core site ATAAGTGG was replaced with TTTTTTTT as mutated probe (Mut. Probe). Supershift experiments were performed using monoclonal anti-Nkx2.2 antibody (α-Nkx2.2). The arrow shows the Nkx2.2-specific-bound complexes with lower mobility. The asterisk marks α-Nkx2.2-Nkx2.2-probe complexes with the lowest mobility. (C) Co-transfection of pcDNA3.1-Nkx2.2 and pGL3-Pdgfra promoter significantly inhibits the luciferase activity (mean ± s.e.m., n=3). Data are normalized using Renilla luciferase.
Based on these observations, we propose that Nkx2.2 represses Pdgfra expression in OPCs by directly binding to its promoter sequence and interfering with its transcriptional activity.
Conditional ablation of Pdgfra gene in the OL lineage mimics the phenotypes of Nkx2.2 overexpression in OPCs
Our expression and gene regulation studies strongly suggested that Pdgfra functions downstream of Nkx2.2 in the control of OL differentiation. Despite the ample in vitro evidence that Pdgfra is a negative regulator of OL maturation, its in vivo developmental role in OL differentiation has not been demonstrated. Thus, we generated the Olig1cre/Pdgfraflox/flox conditional knockout animals to inactivate Pdgfra in early OPCs (Lu et al., 2002). As expected, in the conditional mutant spinal cords, Pdgfra expression was nearly completely absent throughout embryonic stages (Fig. 5A′-D′). Similar to the findings in Nkx2.2 transgenics, Sox10+ cells (Fig. 5E-H′) were drastically reduced in numbers and confined to the ventral spinal cord, probably due to the decreased cell proliferation and migration. Moreover, from E14.5 to E18.5, premature Mbp+ or Plp+ OLs appeared in the conditional mutants in the ventral region (Fig. 6A-C′,E-G′). Noticeably, the reduced number and distribution pattern of Mbp+ cells (Fig. 6A′-C′) in the mutants were identical to those of Sox10+ cells in the same tissues (Fig. 5F′-H′). Intriguingly, at P9, the number of Mbp+/Plp+ cells was drastically reduced compared with that in the control animals (Fig. 6D,D′,H,H′), as a result of the diminished OL population in the absence of Pdgfra signaling. The precocious differentiation and reduced number of OLs in Pdgfra conditional mutants mimic the phenotypes observed in Nkx2.2 overexpressing transgenics (Fig. 1; supplementary material Figs S2, S5, S6), in keeping with the notion that Pdgfra is a downstream target of Nkx2.2 transcription repressor.
Fig. 5.

Expression of Pdgfra and Sox10 in Pdgfra conditional mutants. (A-H′) Spinal cord sections from control (Olig1+/cre;Pdgfra+/+) and double mutant (Olig1+/cre;Pdgfrafl/fl) mice at various embryonic stages were subjected to in situ hybridization with Pdgfra (A-D′) or Sox10 (E-H′) riboprobes. Pdgfra was completely abolished, whereas Sox10 expression was reduced and restricted to the ventral half.
Fig. 6.

Accelerated OL differentiation and myelin gene expression in Pdgfra conditional mutants. (A-H′) Spinal cord sections from control (Olig1+/cre;Pdgfra+/+) and double mutant (Olig1+/cre;Pdgfrafl/fl) mice at various developmental stages were subjected to in situ hybridization with Mbp (A-D′) or Plp (E-H′) riboprobes. Premature Mbp+/Plp+ OLs were detected in E14.5-E18.5 mutant spinal cords.
DISCUSSION
Nkx2.2 is a crucial intracellular regulator for the timing of OL differentiation
One of the long-standing questions in OL development is: what are the molecular mechanisms that control the timing of OL differentiation? Recent studies have provided valuable insights into how this developmental process is tightly regulated by a number of extrinsic signaling mechanisms, including extracellular ligands, secreted molecules and axonal activity (Emery, 2010). Meanwhile, it has long been proposed an ‘internal’ clock measuring the time or the number of cell divisions in OPCs (Temple and Raff, 1986). To date, a number of extracellular and intrinsic factors have been suggested as part of these clock mechanisms (Emery, 2010). However, the in vivo developmental roles of many of these factors are yet to be confirmed by genetic studies, and their potential interactions have not been established at the molecular and genetic levels.
In this study, we provided genetic evidence that Nkx2.2 is a key intracellular factor that controls the timing of OL differentiation in the developing CNS. Studies in inducible transgenic mice demonstrated that elevated Nkx2.2 expression in early OPCs led to precocious differentiation and myelin protein production (Fig. 1; supplementary material Fig. S1). Conversely, conditional knockout of Nkx2.2 in OPCs resulted in a significant but transient delay of OPC maturation and myelin gene expression, and the delayed differentiation was completely overcome later in young adults (Fig. 2; supplementary material Fig. S4). Therefore, Nkx2.2 is not absolutely necessary for OL differentiation; instead it governs the timing of OL differentiation during development by switching on the intrinsic program for OL maturation. Given that Nkx2.2 is transiently upregulated in OPCs immediately before their differentiation in the white matter, where they make direct contacts with axons, it is conceivable that Nkx2.2 may be activated by axon-derived signals and functions as an intracellular relay mechanism between environmental signaling and the intrinsic OL differentiation program.
Nkx2.2 controls the timing of OL differentiation by directly suppressing Pdgfra expression
Several lines of evidence from this and other studies suggest that Nkx2.2 controls the timing of OPC differentiation through repressing Pdgfra gene expression in a direct manner. First, it was previously shown that Nkx2.2 primarily functions as a transcription repressor in the control of cell-fate specification in the developing CNS and pancreas by recruiting other transcriptional repressors such as Groucho4 (Muhr et al., 2001; Zhou et al., 2001), histone deacetylases (HDACs) and the DNA methyltransferase Dnmt3a (Papizan et al., 2011). Second, overexpression of Nkx2.2 in transgenics completely extinguished Pdgfra expression. Conversely, null mutations of Nkx2.2 led to a significant increase in Pdgfra expression (Fig. 3E-H; supplementary material Fig. S3E-H′). Third, a potential Nkx2.2 binding site is identified in the putative promoter of Pdgfra, and EMSA and antibody supershifting experiments confirmed that Nkx2.2 protein could directly bind to this consensus binding sequence (Fig. 4). More importantly, in vitro luciferase reporter assay showed that Nkx2.2 expression can inhibit gene transcription activity driven by the putative Pdgfra promoter harboring this binding sequence (Fig. 4C). Finally, Pdgfra conditional mutants and Nkx2.2 overexpressing transgenics exhibited nearly identical phenotypes in terms of the reduced number and ventral confinement of Sox10+ OLs (supplementary material Fig. S2F-F′; Figs 5, 6) owing to the impaired OPC proliferation/migration and premature OL differentiation in the absence of Pdgfra signaling. These findings lead us to propose that Pdgfra is a direct downstream target of Nkx2.2 transcription repressor during OL lineage progression. Through repressing the expression of the membrane receptor Pdgfra, OPCs no longer respond to external mitogen Pdgfa, cease proliferation and activate the intrinsic cell differentiation program. Consistent with this concept, inhibition of Pdgfra expression and enhancement of Mbp expression were also observed in primary OPCs when Nkx2.2 was overexpressed (supplementary material Fig. S5; Fig. 6). However, we cannot exclude the possibility that Nkx2.2 may also influence other factors at the same time to regulate OPC differentiation and maturation.
Pdgfra signaling controls OPC differentiation possibly by repressing positive differentiation factors
The role of Pdgf signaling in OL development has been extensively studied in the past three decades. It was reported that Pdgf controls OPC proliferation, migration and survival via Pdgfra, which is the only isoform of Pdgf receptors expressed by OPCs (McKinnon et al., 1990; Pringle et al., 1989). During OPC differentiation, the expression of Pdgfra in OPCs is progressively decreased and completely extinguished in mature OLs. Purified OPCs start to undergo terminal differentiation upon Pdgf withdrawal from culture medium. In this study, we provided the first in vivo evidence that Pdgfra signaling also controls OPC differentiation and maturation during development. Similar to the phenotypes described in Nkx2.2-overexpressing transgenics, conditional ablation of Pdgfra in OPCs caused precocious OPC differentiation in association with an inhibition of cell proliferation and migration (Figs 5, 6). Consistently, mutations of Pdgfra downstream signaling components Shp2 (Ptpn11 - Mouse Genome Informatics) or Erk2 (Mapk1 - Mouse Genome Informatics) also led to an altered timing of OL maturation (Fyffe-Maricich et al., 2011; Zhu et al., 2010). Together, these findings indicated that Pdgfra is a crucial negative regulator of OPC maturation in the developing CNS.
At this stage, it is not clear how extinction of Pdgfra expression or signaling in OPCs initiates the intrinsic default OL differentiation program. One possibility is that Pdgfra signaling may suppress the expression and/or function of the positive activators of OL maturation, including the OL-specific transcription factors Sox10, Sip1 or Mrf, expression of which is both necessary and sufficient for OL differentiation (Emery et al., 2009; Liu et al., 2007; Stolt et al., 2002; Weng et al., 2012). In support of this idea, in both Pdgfra conditional knockout mice and Nkx2.2 overexpressing transgenics, we observed a significant higher level of cellular Sox10 expression based on the intensity of immunostaining (supplementary material Fig. S2F,F′) or in situ hybridization (Fig. 5E-H′). If this hypothesis is correct, the Nkx2.2 transcriptional repressor may initiate the intrinsic differentiation program by derepressing these positive regulatory factors in response to axon-derived signals or other environmental cues.
MATERIALS AND METHODS
Generation of TetO-Nkx2.2 transgenic mouse line and induced expression of Nkx2.2 transgene in OLs
For generation of the TetO-Nkx2.2 mouse line, full-length Nkx2.2 cDNA tagged with HA epitope was cloned into the pTRE2 plasmid (Clonetech) for pronuclear microinjection followed by implantion into pseudopregnant host mice. The transgenic founder mice were identified through PCR and Southern blotting. The time- and cell-specific overexpression of Nkx2.2 was achieved by crossing the TetO-Nkx2.2 transgenic line with the Sox10-rtTA knock-in mouse line (Ludwig et al., 2004). To induce overexpression of Nkx2.2 in OPCs, doxycycline hydrochloride (Sigma) was administered at indicated times both in the drinking water (4 mg/ml) and food (Bio-serv). Single transgenic Sox10-rtTA control and Sox10-rtTA;TetO-Nkx2.2 DTG mice were used for further study. All experimental procedures conformed to National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee at the University of Louisville. Two founder lines were established and showed the same results in Nkx2.2 induction kinetics and OL differentiation.
Conditional ablation of Pdgfra or Nkx2.2 in OL lineage
Pdgfra+/fl mice obtained from Jackson Laboratories (Tallquist and Soriano, 2003) were mated to Olig1+/cre knock-in line with Neo (Lu et al., 2002) to obtain the Olig1+/cre;Pdgfra fl/+ double heterozygous mice, and conditional Pdgfra mutants were generated by interbreeding of double heterozygous. The generation of Nkx2.2 conditional mutants will be described elsewhere (Mastracci et al., 2013). Cnp+/cre;Nkx2.2fl/fl mice were obtained through intercross between Nkx2.2 floxed mouse lines and Cnpcre transgenic mice (Lappe-Siefke et al., 2003).
Immunofluorescent staining and in situ RNA hybridization
Postnatal mice were fixed by cardiac perfusion with 4% paraformaldehyde (PFA). Brain and spinal cord tissues were dissected out and post-fixed in 4% PFA at 4°C overnight. Following fixation, tissues were transferred to 20% sucrose in PBS overnight, embedded in optimal cutting temperature compound (OCT) media and then sectioned (16 μm thickness) on a cryostat. Procedures for immunofluorescence staining were described previously (Zhu et al., 2011). The dilution ratio of primary antibodies is as follows: anti-mouse Nkx2.2 (1:50, Developmental Studies Hybridoma Bank), anti-mouse Olig2 (1:3000, gift from Drs Stiles and Alberta, Harvard Medical School, MA, USA) (Lu et al., 2002), anti-mouse Sox10 (1:3000) (Stolt et al., 2002), anti-mouse Apc/Rb1cc1 (1:50, Oncogene), anti-mouse Mag (1:300, Chemicon), anti-mouse Mbp (1:5000, Chemicon), anti-mouse Gfap (1:300, Chemicon), anti-HA (1:300, Sigma), anti-phospho-Caspase-3 (1:200, BD), anti-BrdU (1:50, supernatant prepared in our lab), anti-Sox6 (1:200, Abcam) and anti-Pdgfra (1:100, Santa Cruz). The Alexa-488 or Alexa-594 conjugated secondary antibodies were obtained from Molecular Probes. The nucleic acid dye 4′,6-diamidino-2-phenylindole (DAPI) was obtained from Roche. In situ RNA hybridization was performed as described (Schaeren-Wiemers and Gerfin-Moser, 1993) with minor modifications.
Cell proliferation assay
BrdU (Sigma, 60 μg/g body weight) was administered to pregnant mice via intraperitoneal injection 2 hours before embryos were dissected. Immunofluorescent staining for BrdU was performed as described above, except that sections were incubated in 2 N HCl (30 minutes) and 0.1 M borate buffer (10 minutes) before blocking steps.
Electrophoretic mobility shift assay
EMSA was performed using 3′-end biotin-labeled 40 bp (ACAAAGGCAGGACCAGATAAGTGGCTCCGAAGGGATAAAG) probes for the putative Nkx2.2-binding site (positions -722 to -683) in the Pdgfra promoter region, as predicted by the method described previously (Hill et al., 2011). Briefly, the probe was incubated with 10 μg nuclear extracts from 293T cells transfected with pcDNA3.1-Nkx2.2 or pcDNA3.1 blank plasmids for 20 minutes at room temperature. Other steps were performed according to the LightShift Chemiluminescent EMSA Kit protocol (Pierce). For supershift experiments, Nkx2.2-specific antibody was added to the assay.
Luciferase assay
The upstream genomic sequence (potential promoter) which spans from -1271 to +68 bp relative to the transcription start site of Pdgfra gene was amplified by PCR and cloned into the pGL3 basic luciferase reporter vector (Promega). 293T cells plated in 24-well tissue culture plates were transfected with pGL3 basic, pGL3-Pdgfra promoter, pcDNA3.1 basic or pcDNA3.1-Nkx2.2 using Lipofectamine 2000 (Sigma). Transfection efficiency was controlled by co-transfection of cells with Renilla luciferase-encoding plasmid pRL-TK (Promega). Luciferase activity was determined by the Dual-Luciferase Reporter Assay System (Promega) and measured by a luminometer (Berthold Detection Systems). Promoter activity was calculated as a firefly/Renilla luciferase activity ratio.
Primary OPC culture and Nkx2.2 overexpression in vitro
Cerebral cortices from P2-4 s.d. rats were dissected out, minced and digested in 0.25% trypsin at 37°C. The digestion was stopped by the addition of Dulbecco’s modified Eagle’s medium (DMEM)/F12 containing 10% fetal bovine serum (FBS). The dissociated cells were plated in a 75 cm2 tissue culture flask coated with 100 μg/ml poly-L-lysine, and the whole medium was changed next day. After 10 days’ culture, the cells were rinsed three times with culture medium and pre-shaken for 1 hour at 200 rpm to remove microglia. Then flasks were sealed and shaken at 250 rpm at 37°C for 15-18 hours. The medium with the detached cells was collected and first plated on tissue culture dishes (non PDL coated) for 30-60 minutes at 37°C with a gentle swirling of the dishes to eliminate contaminating dead cells and residual microglia. The nonadherent cells were collected, centrifuged and replated in DMEM containing 10% FBS. On the following morning, the culture medium was changed to defined media consisting of B27/neurobasal medium and 10 ng/ml Pdgfaa.
For Nkx2.2 overexpression in vitro, rat Nkx2.2 cDNA was cloned into the pCDH-MCS-EF1-CopGFP vectors by primers as follows: forwards 5′-GGAATTCGCCACCATGTCGCTGACCAACACAAAGACG-3, reverse 5′-CGGGATCCtcaTGCATAATCAGGCACATCGTAAGGATACCAAGTCCACTGCTGGGCCTGGACC-3′. Lentiviruses were prepared by co-transfected the pCDH-MCS-EF1-CopGFP or pCDH-rNkx2.2-EF1-CopGFP with pMD2.G and psPAX2 packaging vectors (Addgene) into 293T cells. Harvested viral supernatants were used to infect OPCs. Six days after infection, OPCs were collected for the next experiments.
Statistical analyses
Statistical significance of the difference was evaluated by Student’s t-test. P<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We are grateful to Drs Charles Stiles, David Rowitch and Richard Lu for providing the Olig1-cre mouse line, and to Dr Klaus Nave for the CNP-Cre mouse line.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
Q.Z. and H.L. analyzed the phenotype of Nkx2.2 transgenic mice; Q.Z. performed bioinformatic analysis of the Nkx2.2 binding site, EMSA and luciferase assay; X.Z. and H.H. analyzed Nkx2.2 cko mice; K.Z. and Z.Z. characterized Pdgfra cko mice; M.W. generated the Sox10rtTA mice; Y.C. produced the TRE-Nkx2.2 transgenic mice; T.M. and L.S. produced the Nkx2.2flox mice; and Q.Z. and M.Q. wrote the manuscript.
Funding
This work is supported by the National Institutes of Health (NIH) [R01-NS37717]; the National Multiple Sclerosis Society [RG3276]; the National Natural Science Foundation of China [31071879, 31000488 and 31000945]; the Key Project of Zhejiang Provincial Natural Science Foundation of China [2100730]; the Zhejiang Provincial Science and Technology Key Project [2011C13030]; and the Zhejian Key Laboratory of Organ Development and Regeneration. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.095323/-/DC1
References
- Barres B. A., Lazar M. A., Raff M. C. (1994). A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development 120, 1097–1108 [DOI] [PubMed] [Google Scholar]
- Bergeron F., Bagu E. T., Tremblay J. J. (2011). Transcription of platelet-derived growth factor receptor α in Leydig cells involves specificity protein 1 and 3. J. Mol. Endocrinol. 46, 125–138 [DOI] [PubMed] [Google Scholar]
- Emery B. (2010). Regulation of oligodendrocyte differentiation and myelination. Science 330, 779–782 [DOI] [PubMed] [Google Scholar]
- Emery B., Agalliu D., Cahoy J. D., Watkins T. A., Dugas J. C., Mulinyawe S. B., Ibrahim A., Ligon K. L., Rowitch D. H., Barres B. A. (2009). Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138, 172–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H., Qi Y., Tan M., Cai J., Takebayashi H., Nakafuku M., Richardson W., Qiu M. (2002). Dual origin of spinal oligodendrocyte progenitors and evidence for the cooperative role of Olig2 and Nkx2.2 in the control of oligodendrocyte differentiation. Development 129, 681–693 [DOI] [PubMed] [Google Scholar]
- Fyffe-Maricich S. L., Karlo J. C., Landreth G. E., Miller R. H. (2011). The ERK2 mitogen-activated protein kinase regulates the timing of oligodendrocyte differentiation. J. Neurosci. 31, 843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J. T., Anderson K. R., Mastracci T. L., Kaestner K. H., Sussel L. (2011). Novel computational analysis of protein binding array data identifies direct targets of Nkx2.2 in the pancreas. BMC Bioinformatics 12, 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T., Raff M. (2000). The Id4 HLH protein and the timing of oligodendrocyte differentiation. EMBO J. 19, 1998–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C., Goebbels S., Gravel M., Nicksch E., Lee J., Braun P. E., Griffiths I. R., Nave K. A. (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 33, 366–374 [DOI] [PubMed] [Google Scholar]
- Liu A., Li J., Marin-Husstege M., Kageyama R., Fan Y., Gelinas C., Casaccia-Bonnefil P. (2006). A molecular insight of Hes5-dependent inhibition of myelin gene expression: old partners and new players. EMBO J. 25, 4833–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Hu X., Cai J., Liu B., Peng X., Wegner M., Qiu M. (2007). Induction of oligodendrocyte differentiation by Olig2 and Sox10: evidence for reciprocal interactions and dosage-dependent mechanisms. Dev. Biol. 302, 683–693 [DOI] [PubMed] [Google Scholar]
- Lu Q. R., Sun T., Zhu Z., Ma N., Garcia M., Stiles C. D., Rowitch D. H. (2002). Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109, 75–86 [DOI] [PubMed] [Google Scholar]
- Ludwig A., Schlierf B., Schardt A., Nave K. A., Wegner M. (2004). Sox10-rtTA mouse line for tetracycline-inducible expression of transgenes in neural crest cells and oligodendrocytes. Genesis 40, 171–175 [DOI] [PubMed] [Google Scholar]
- Mastracci T. L., Lin C. S., Sussel L. (2013). Generation of mice encoding a conditional allele of Nkx2.2. Transgenic Res. 22, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon R. D., Matsui T., Dubois-Dalcq M., Aaronson S. A. (1990). FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron 5, 603–614 [DOI] [PubMed] [Google Scholar]
- McKinnon R. D., Waldron S., Kiel M. E. (2005). PDGF alpha-receptor signal strength controls an RTK rheostat that integrates phosphoinositol 3′-kinase and phospholipase Cgamma pathways during oligodendrocyte maturation. J. Neurosci. 25, 3499–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhr J., Andersson E., Persson M., Jessell T. M., Ericson J. (2001). Groucho-mediated transcriptional repression establishes progenitor cell pattern and neuronal fate in the ventral neural tube. Cell 104, 861–873 [DOI] [PubMed] [Google Scholar]
- Noble M., Murray K., Stroobant P., Waterfield M. D., Riddle P. (1988). Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte/type-2 astrocyte progenitor cell. Nature 333, 560–562 [DOI] [PubMed] [Google Scholar]
- Papizan J. B., Singer R. A., Tschen S. I., Dhawan S., Friel J. M., Hipkens S. B., Magnuson M. A., Bhushan A., Sussel L. (2011). Nkx2.2 repressor complex regulates islet β-cell specification and prevents β-to-α-cell reprogramming. Genes Dev. 25, 2291–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle N., Collarini E. J., Mosley M. J., Heldin C. H., Westermark B., Richardson W. D. (1989). PDGF A chain homodimers drive proliferation of bipotential (O-2A) glial progenitor cells in the developing rat optic nerve. EMBO J. 8, 1049–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y., Cai J., Wu Y., Wu R., Lee J., Fu H., Rao M., Sussel L., Rubenstein J., Qiu M. (2001). Control of oligodendrocyte differentiation by the Nkx2.2 homeodomain transcription factor. Development 128, 2723–2733 [DOI] [PubMed] [Google Scholar]
- Raff M. C., Abney E. R., Fok-Seang J. (1985). Reconstitution of a developmental clock in vitro: a critical role for astrocytes in the timing of oligodendrocyte differentiation. Cell 42, 61–69 [DOI] [PubMed] [Google Scholar]
- Raff M. C., Lillien L. E., Richardson W. D., Burne J. F., Noble M. D. (1988). Platelet-derived growth factor from astrocytes drives the clock that times oligodendrocyte development in culture. Nature 333, 562–565 [DOI] [PubMed] [Google Scholar]
- Richardson W. D., Pringle N., Mosley M. J., Westermark B., Dubois-Dalcq M. (1988). A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell 53, 309–319 [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N., Gerfin-Moser A. (1993). A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100, 431–440 [DOI] [PubMed] [Google Scholar]
- Soula C., Danesin C., Kan P., Grob M., Poncet C., Cochard P. (2001). Distinct sites of origin of oligodendrocytes and somatic motoneurons in the chick spinal cord: oligodendrocytes arise from Nkx2.2-expressing progenitors by a Shh-dependent mechanism. Development 128, 1369–1379 [DOI] [PubMed] [Google Scholar]
- Stolt C. C., Rehberg S., Ader M., Lommes P., Riethmacher D., Schachner M., Bartsch U., Wegner M. (2002). Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 16, 165–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimori M., Nagao M., Parras C. M., Nakatani H., Lebel M., Guillemot F., Nakafuku M. (2008). Ascl1 is required for oligodendrocyte development in the spinal cord. Development 135, 1271–1281 [DOI] [PubMed] [Google Scholar]
- Tallquist M. D., Soriano P. (2003). Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development 130, 507–518 [DOI] [PubMed] [Google Scholar]
- Temple S., Raff M. C. (1986). Clonal analysis of oligodendrocyte development in culture: evidence for a developmental clock that counts cell divisions. Cell 44, 773–779 [DOI] [PubMed] [Google Scholar]
- van Heyningen P., Calver A. R., Richardson W. D. (2001). Control of progenitor cell number by mitogen supply and demand. Curr. Biol. 11, 232–241 [DOI] [PubMed] [Google Scholar]
- Wang S., Sdrulla A., Johnson J. E., Yokota Y., Barres B. A. (2001). A role for the helix-loop-helix protein Id2 in the control of oligodendrocyte development. Neuron 29, 603–614 [DOI] [PubMed] [Google Scholar]
- Weng Q., Chen Y., Wang H., Xu X., Yang B., He Q., Shou W., Chen Y., Higashi Y., van den Berghe V., et al. (2012). Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 73, 713–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Cai J., Fu H., Wu R., Qi Y., Modderman G., Liu R., Qiu M. (2000). Selective expression of Nkx-2.2 transcription factor in chicken oligodendrocyte progenitors and implications for the embryonic origin of oligodendrocytes. Mol. Cell. Neurosci. 16, 740–753 [DOI] [PubMed] [Google Scholar]
- Zhou Q., Choi G., Anderson D. J. (2001). The bHLH transcription factor Olig2 promotes oligodendrocyte differentiation in collaboration with Nkx2.2. Neuron 31, 791–807 [DOI] [PubMed] [Google Scholar]
- Zhu Y., Park J., Hu X., Zheng K., Li H., Cao Q., Feng G. S., Qiu M. (2010). Control of oligodendrocyte generation and proliferation by Shp2 protein tyrosine phosphatase. Glia 58, 1407–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q., Whittemore S., DeVries W., Zhao X., Kuypers N., Qiu M. (2011). Dorsally-derived oligodendrocytes in the spinal cord contribute to axonal myelination during development and remyelination following focal demyelination. Glia 59, 1612–1621 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.