

Abstract
Synthetic organic chemists have the power to replicate some of the most intriguing molecules of living nature in the laboratory and apply their developed synthetic strategies and technologies to construct variations of them. Such molecules facilitate biology and medicine, as they often find uses as biological tools and drug candidates for clinical development. In addition, by employing sophisticated catalytic reactions and appropriately designed synthetic processes, they can synthesize not only the molecules of nature and their analogues, but also myriad other organic molecules for potential applications in many areas of science, technology and everyday life. After a short historical introduction, this article focuses on recent advances in the field of organic synthesis with demonstrative examples of total synthesis of complex bioactive molecules, natural or designed, from the author’s laboratories, and their impact on chemistry, biology and medicine.
Keywords: chemistry, biology, medicine, natural products, anti-cancer agents, neurotoxins
1. Introduction
Among what matters the most is matter itself. It is, therefore, not a surprise that chemistry, the science of matter, is considered by many as the central science lying between physics and biology. Its power derives from its ability to analyse and synthesize molecules from atoms and other, more or less complex, molecules. The latter practice, synthesis, is of paramount importance to our well-being, for through it we create new chemical entities (i.e. molecules) from which we derive our most precious material items. A subdiscipline of synthesis is organic synthesis, the art and science of constructing substances, natural or designed, whose primary element is carbon. The flagship of organic synthesis is total synthesis, the endeavour of synthesizing the molecules of living nature in the laboratory. The ability of man to replicate the molecules of living creatures, and create other molecules like them, is a remarkable development in human history. Its birth goes back to 1828, when German chemist Friedrich Wöhler, a Foreign Member of the Royal Society (ForMemRS), synthesized urea, an example of a naturally occurring substance from the living world [1]. Such molecules are commonly known as natural products, a term usually referring to secondary metabolites. The creative nature of total synthesis earned this discipline the privilege of being called a fine art and a precise science. Technologies derived from it, and organic synthesis in general, have led to an impressive host of benefits to society, including useful products ranging from pharmaceuticals, dyes, cosmetics and agricultural chemicals to diagnostics and high-technology materials used in computers, mobile phones and spaceships [2].
2. Organic synthesis in perspective
The world has changed dramatically in the last two centuries as a result of scientific discoveries and their applications. One of the most profound of these discoveries is the advent of organic synthesis as marked by Wöhler’s synthesis of urea. And although its foundations go back before that era, this initial event, together with developments in structural theory and analytical techniques, gave momentum to its advancement and application in several fields. But what were the conditions and foundations that allowed this science to emerge? And from where did they come? To answer these questions, we must go back to ancient times, when humans were practising transformations of matter as a means to prepare food, medicines, dyes, tools and weapons. The artefacts left behind from ancient civilizations like those of the Egyptians, Babylonians, Greeks, Romans and Chinese provide evidence for such endeavours, although there was no significant understanding of the nature of these transformations. The curiosity about nature, however, drove the Ancient Greeks to think and speculate about matter, a practice that led to Democritus’ atomic theory.
The latter served as the basis from which the more precise atomic theory of the English chemist and physicist John Dalton, a Fellow of the Royal Society (FRS), emerged at the dawn of the nineteenth century. Dalton’s theory was one of the most influential theoretical developments in science of all time and gave enormous momentum to further the advancement of chemistry [3]. But before we move forward in time, we must mention the alchemists and their practices that can be traced back to thousands of years ago in the Middle East and the Orient, and prevailed later on during the Middle Ages in Europe. From these endeavours, modern chemistry emerged slowly in the eighteenth century. Among the main protagonists responsible for the transition to modern chemistry from alchemy was Irish-born Robert Boyle (FRS), who was both an alchemist and a modern chemist. He exposed his philosophies in his book The Sceptical Chymist, which was published in 1661, one year after the Royal Society was founded. Boyle promoted experimentation based on purity, precision and data.
Experimentation and quantitative analysis were moved to a higher level by French chemist Antoine-Laurent de Lavoisier (ForMemRS), who many consider as the father of modern chemistry, with Boyle viewed as the grandfather. Lavoisier described his chemical philosophy and methods in his Traité Élémentaire de Chimie that provided the foundation for the emergence of modern chemistry. His chemistry was primarily inorganic and was based on combustion and elemental analysis. Lavoisier published a list of chemical elements but had no means to distinguish between them and atoms; the latter had to await Dalton’s atomic theory and subsequent developments that took hold in the nineteenth century. Among these developments was the emergence of organic chemistry, the branch of chemistry dealing with organic compounds, those made of carbon and a few other elements, most commonly hydrogen, oxygen, nitrogen, sulfur, phosphorus and halogens.
The chemistry of natural products was born in the eighteenth century, primarily from the work of the apothecaries, the pharmacists of the time, among whom Swedish Carl Wilhelm Scheele was the most prominent. He, besides being credited with the identification of oxygen, discovered several naturally occurring organic acids, including citric, gallic, malic, lactic, oxalic and uric acids. Scheele also developed important practical laboratory techniques such as distillation and crystallization.
By the dawn of the nineteenth century, the stage was set for the arrival of organic chemistry in general and organic synthesis in particular. Thus, in addition to the advancement of Dalton’s atomic theory, a number of other important discoveries and ideas emerged and eventually gave rise to the understanding of the structure of the molecule and the art of its synthesis. Included among the initial prominent contributions to the establishment of the foundations of modern chemistry are those of English chemist Humphry Davy (FRS and President of the Royal Society), Swedish chemist Jöns Jakob Berzelius (ForMemRS), English chemists Alexander Williamson (FRS) and William Odling (FRS), and French chemist and physicist Joseph Gay-Lussac (ForMemRS). Their theories and discoveries served as the foundation from which further advancements occurred, including the distinction between atomic and equivalent weights, the structural theory and the tetrahedral nature of carbon. Among the protagonists of these developments were French chemists Jean-Baptiste André Dumas (ForMemRS), Auguste Laurent, Charles Gerhardt, Joseph Le Bel and C. Adolphe Wurtz (ForMemRS), German chemist Friedrich August Kekulé (ForMemRS), Italian chemists Amedeo Avogadro and Stanislao Cannizzaro (ForMemRS), Russian chemist Dmitri I. Mendeléev (ForMemRS), French physicist, chemist and mathematician Jean-Baptiste Biot (ForMemRS), French chemist and microbiologist Louis Pasteur (ForMemRS) and Dutch chemist Jacobus van’t Hoff (ForMemRS) [1,3].
3. Emergence and evolution of organic synthesis and total synthesis
The development of experimental methods for practical chemistry and the discoveries of naturally occurring substances such as urea, quinine, morphine and strychnine in the late eighteenth and early nineteenth centuries laid the foundations and provided the impetus for the emergence of organic synthesis [1].
As mentioned above, the first natural product to be synthesized in the laboratory was urea (for the molecular structure of urea and of the other milestone molecules mentioned in this article, see figure 1). This momentous event, albeit a serendipitous discovery, meant that man could construct organic compounds, the molecules of living nature, in the laboratory and without the aid of living creatures or their organs. This important singularity led to the downfall of vitalism, the understanding of the phenomenon of isomerism, and to a revolution in science that came to be known as organic synthesis. As urea was a naturally occurring organic compound, the milestone of its synthesis also marks the birth of total synthesis, the subdiscipline of organic synthesis dealing with the construction of nature’s organic molecules. The achievement of the synthesis of urea by Wöhler was followed by the total synthesis of acetic acid, a natural product containing two carbon atoms (as opposed to urea’s one), by German chemist Hermann Kolbe (ForMemRS) in 1845.
Figure 1.

Select historical milestone accomplishments in total synthesis (* formal total synthesis).
Soon after its occurrence, the advent of organic synthesis gave birth first to the dye industry and then to the pharmaceutical industry with the synthesis and commercialization of mauve (or mauveine) and acetylsalicylic acid (aspirin), respectively, triggering these industrial revolutions. The first discovery was made, also serendipitously, by English chemist William Henry Perkin (FRS) during his attempts to synthesize quinine (the miracle natural product used as medication to treat malaria), employing an erroneous recipe. At the time, Perkin was a student of German chemist August Wilhelm von Hofmann (FRS), who founded and directed the Royal College of Chemistry in London upon invitation by Queen Victoria. The second discovery was made by German chemist Felix Hoffmann at the Bayer company and was based on the isolation and structural elucidation of salicin, the active pain-relieving ingredient of the willow bark, whose medicinal properties were known from ancient times [2].
Indeed, natural products played a crucial role in the emergence and advancement of organic synthesis from its birth to the present day. Thus, from the early days of elemental analysis of natural products, these substances fascinated and challenged organic chemists, first with their structural elucidation and then with their total synthesis. By the dawn of the twentieth century, chemists had synthesized, besides urea and acetic acid, numerous natural and designed molecules, including indigo, alizarin, glucose, coniine and salicylic acid, the precursor of acetylsalicylic acid. They had also discovered several new reactions and applied them to the synthesis of a wide range of organic compounds, including many derivatives of benzene, collectively known as aromatic compounds [1–3].
The major achievements in organic synthesis and total synthesis of the last decades of the nineteenth century were widely recognized and appropriately hailed, as acknowledged by two Nobel Prizes in Chemistry awarded during the first 5 years of the Prize’s existence [4]. The first went to German chemist Emil Fischer (ForMemRS) in 1902 ‘in recognition of the extraordinary services he has rendered by his work on sugar and purine syntheses’, and the second to German chemist Adolf von Baeyer (ForMemRS) in 1905 ‘in recognition of his services in the advancement of organic chemistry and the chemical industry, through his work on organic dyes and hydroaromatic compounds’. Many more Nobel Prizes would follow with notable frequency and regularity, reflecting the impressive advances made continuously in these fields throughout the twentieth century, underscoring their importance to science and society. These advances were made possible not only by discoveries and inventions within the field of organic synthesis in terms of new synthetic reactions, methods and strategies, but also by the improvement of analytical techniques and instrumentation, as well as theories that led to better understanding of the nature of the chemical bond [5] and chemical reactivity. The isolation and structural elucidation of novel molecular architectures from natural sources provided fuel and inspiration to the practitioners of total synthesis. Among the most important new reactions to be discovered in the first part of the twentieth century were the catalytic hydrogenation reaction of unsaturated carbon–carbon bonds by French chemist Paul Sabatier (ForMemRS) and the Grignard reaction for the formation of carbon–carbon bonds by French chemist Victor Grignard. Sabatier and Grignard shared the 1912 Nobel Prize in Chemistry for their pioneering and influential discoveries. Another highly influential discovery of that era was the Diels–Alder reaction (4+2 cycloaddition for constructing six-membered ring compounds) made by German chemists Otto Diels and Kurt Alder in 1928. Their work was recognized in 1950 with the Nobel Prize in Chemistry. A number of relatively complex alkaloid natural products were synthesized, including tropinone, quinine, morphine and strychnine. The total synthesis of strychnine was accomplished by American chemist Robert Burns Woodward (ForMemRS), a major figure who led a revolutionary movement in the field in the 1950s and 1960s that culminated in his recognition by the Royal Swedish Academy of Sciences with the 1965 Nobel Prize in Chemistry ‘for his achievements in the art of organic synthesis’ [6]. By then, in addition to strychnine, he had synthesized quinine (formal total synthesis), reserpine, chlorophyll and cephalosporine, and then went on to complete the total synthesis of vitamin B12, the most complex natural product to be replicated in the laboratory at the time, in collaboration with Swiss chemist Albert Eschenmoser (ForMemRS) [7]. Woodward’s contributions also included the adoption of modern instrumentation for structural purification and elucidation purposes, as well as theoretical aspects of organic chemistry, for example the Woodward–Hoffmann rules.
In the meantime, the spectacular success of penicillin as a life-saving antibiotic generated impetus for the discovery of a wide range of new biologically active natural products from microorganisms, a surge at the helm of which were initially the pharmaceutical companies, soon to be joined by academic institutions. Many of these compounds became clinical agents to treat disease and some are in use even today. Their allure attracted the attention of synthetic organic chemists of the second half of the twentieth century and resulted in major achievements in the field of total synthesis. Human hormones such as the steroids and the eicosanoids (e.g. prostaglandins, thromboxanes and leukotrienes) played similar roles to those natural products derived from plants and microbes in challenging and inspiring young practitioners entering the field. One of these practitioners was American chemist Elias J. Corey (ForMemRS), whose legendary contributions helped shape organic synthesis in decisive ways during the second half of the twentieth century. His achievements included the introduction of the theory of retrosynthetic analysis, the development of several new synthetic methods, reagents and catalysts and the total synthesis of numerous bioactive naturally occurring substances, including several members of the prostaglandins, leukotriene and macrolide classes, ginkgolide B, maytansine and ecteinascidin 743. Corey was awarded the Nobel Prize in Chemistry in 1990 ‘for his development of the theory and methodology of organic synthesis’ [8–10].
The latter part of the twentieth century witnessed impressive advances in the area of new synthetic methodology, which propelled the art of organic synthesis to higher levels of elegance, practicality and efficiency. These new methods facilitated discovery research, product development and manufacturing of pharmaceuticals and other fine chemicals that benefited society. Among the most powerful of these useful reactions are the Wittig reaction for constructing carbon–carbon double bonds, developed by German chemist Georg Wittig, and the hydroboration reaction, developed by American chemist Herbert C. Brown. Brown and Wittig shared the 1979 Nobel Prize in Chemistry ‘for their development of the use of boron- and phosphorus-containing compounds, respectively, into important reagents in organic synthesis’. The contributions of English chemist Sir Derek H. R. Barton (FRS) and Norwegian chemist Odd Hassel to conformational analysis played a major role in shaping our understanding of molecular structure that facilitated chemical reactivity and selectivity. Barton’s discoveries extended well beyond stereochemistry and into other domains of organic synthesis such as biomimetic oxidative coupling reactions and radical chemistry. His pioneering studies in the latter field included deoxygenation and oxygenation methods (C−H activation/functionalization) that proved highly useful and inspiring to synthetic organic chemists of his and later generations. Barton and Hassel shared the 1969 Nobel Prize in Chemistry ‘for their contributions to the development of the concept of conformation and its application in chemistry’. American chemist Gilbert Stork (ForMemRS) and Albert Eschenmoser made pioneering contributions to organic synthesis of theoretical and practical importance. Thus in 1955, they independently proposed the so-called Stork–Eschenmoser hypothesis stating that polyunsaturated molecules possessing all trans olefinic bonds (e.g. squalene oxide, the biosynthetic precursor of steroid hormones) should undergo stereospecific cyclization to furnish a polycyclic system with all trans ring fusion stereochemistry (e.g. trans, trans, trans for dammaratienol, the product of squalene cyclization). This hypothesis was later verified experimentally by W. S. Johnson, who achieved the first biomimetic total synthesis of progesterone in 1971. Stork made several other seminal contributions to organic synthesis, including stereocontrol, cascade radical reactions and total synthesis. Eschenmoser’s contributions to organic synthesis are equally impressive and include regio- and stereocontrol reactions, method development, corrin chemistry and the aforementioned landmark total synthesis of vitamin B12. Other important reactions include phosphate and amide bond forming processes, discovered by Indian-born American biochemist H. Gobind Khorana (ForMemRS; 1968 Nobel Prize in Physiology or Medicine, shared with American biochemists Robert W. Holley and Marshall W. Nirenberg) and American biochemist R. Bruce Merrifield (1984 Nobel Prize in Chemistry), for the synthesis of oligonucleotides and peptides, respectively. In the meantime, catalytic asymmetric reactions for oxidation, reduction and a variety of other important processes (2001 Nobel Prize in Chemistry awarded to American chemist K. Barry Sharpless, Japanese chemist Ryoji Noyori (ForMemRS) and American chemist William S. Knowles), metathesis reactions (2005 Nobel Prize in Chemistry awarded to American chemists Robert H. Grubbs and Richard R. Schrock (ForMemRS) and French chemist Yves Chauvin) for the construction of olefinic bonds and cyclic structural motifs and polymers, and palladium-catalysed cross-coupling carbon–carbon bond-forming reactions (2010 Nobel Prize in Chemistry awarded to American chemist Richard F. Heck and Japanese chemists Ei-ichi Negishi and Akira Suzuki) changed the way synthetic chemists were thinking about and practising their science.
The impact of organic synthesis on science and technology does not stop with biology and medicine. It encompasses many other scientific and technological endeavours and facilitates their improvement, scope and reach. Among the most prominent fields that benefited enormously from applications of organic synthesis are those of molecular recognition and supramolecular chemistry, materials science and nanotechnology and chemical biology. Indeed, the universe of compounds synthesized by organic synthesis, natural and designed, is very large and could be almost infinite. Reflective of the progress made in organic synthesis in recent years are the numerous elegant total syntheses of biologically and medically important molecules achieved in laboratories around the world [11–15].
4. Endeavours in total synthesis
The selection of the target molecule from the myriad natural products for total synthesis by the practitioner of the art depends on the novelty of its molecular structure, biological activity and natural scarcity, among other criteria. Thus, some synthetic chemists may wish to use the structure of the molecule as an opportunity to discover and develop new reactions for unmet needs in organic synthesis in order to construct its unusual or sensitive structural motifs. Others may be interested to investigate and develop a scarce biologically active natural product, or a variation of it, as a biological tool or a pharmaceutical drug candidate for development as a clinical agent to use against disease. And yet others may wish to undertake a total synthesis campaign for the intellectual challenge and sheer excitement that it provides. To these reasons must be added the education and training of young students and the problem-solving skills they acquire during such endeavours, as well as the value of the fundamental discoveries that are often made whether through logical reasoning or serendipity.
Endeavours in total synthesis can be more or less challenging depending on the complexity of the molecular structure targeted. Simple and chemically stable molecules yield to synthesis more easily than those possessing complex and labile architectures. However, complexity does not always equate with size when it comes to molecules and their construction. Thus, a smaller molecule with unusual atom connectivities and structural motifs is always more challenging to synthetic organic chemists than one possessing a larger, but repetitive structure such as a polymer, a polypeptide or a polynucleotide.
The more challenging the total synthesis appears to be, the more chances it has to offer opportunities to discover and invent new synthetic strategies and technologies. And the higher the importance of the biology and medicine of the target molecule, the richer the harvest of the benefits and rewards of the endeavour is likely to be. Such campaigns often turn into interesting chemical biology studies and drug discovery programmes through molecular design and synthesis of analogues of the natural product. The select target molecules shown in figure 2 are but a few of those completed in our laboratories over the years [16]. The cases of calicheamicin 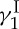 (1), Taxol (2) and brevetoxin B (3) are exemplary of the total synthesis endeavours we have been conducting, and will be highlighted below.
(1), Taxol (2) and brevetoxin B (3) are exemplary of the total synthesis endeavours we have been conducting, and will be highlighted below.
Figure 2.

Select molecules synthesized in the author’s laboratories.
5. The total synthesis of calicheamicin 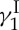
Calicheamicin 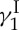 (1, figure 3) is a fascinating molecule whose intrigue stems from not only its phenomenal cytotoxic properties and potential as an anti-cancer agent but also its stunning molecular architecture and fascinating mechanism of action. At the time of its isolation from Micromonospora echinospora ssp. calichensis in the 1980s, neither its structure nor its mechanism of action was precedented. Particularly striking were the 10-membered enediyne, oligosaccharide and trisulfide structural motifs of the molecule of calicheamicin
(1, figure 3) is a fascinating molecule whose intrigue stems from not only its phenomenal cytotoxic properties and potential as an anti-cancer agent but also its stunning molecular architecture and fascinating mechanism of action. At the time of its isolation from Micromonospora echinospora ssp. calichensis in the 1980s, neither its structure nor its mechanism of action was precedented. Particularly striking were the 10-membered enediyne, oligosaccharide and trisulfide structural motifs of the molecule of calicheamicin 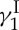 , all three of which are involved in its mode of action that leads to lethal double-strand cuts of the genetic material (double-helix DNA). This mechanism can be compared to that of a guided missile in which the enediyne moiety acts as the explosive payload (generating reactive benzenoid diradicals through Bergman cycloaromatization), the oligosaccharide domain as the delivery system (binding to the minor groove of DNA) and the trisulfide unit as the triggering device (initiating, upon activation, the Bergman cycloaromatization reaction). With all these exquisite features in place, the stage was set for what we expected to be an exciting adventure ahead as we embarked on the journey to the total synthesis of calicheamicin
, all three of which are involved in its mode of action that leads to lethal double-strand cuts of the genetic material (double-helix DNA). This mechanism can be compared to that of a guided missile in which the enediyne moiety acts as the explosive payload (generating reactive benzenoid diradicals through Bergman cycloaromatization), the oligosaccharide domain as the delivery system (binding to the minor groove of DNA) and the trisulfide unit as the triggering device (initiating, upon activation, the Bergman cycloaromatization reaction). With all these exquisite features in place, the stage was set for what we expected to be an exciting adventure ahead as we embarked on the journey to the total synthesis of calicheamicin 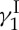 in the late 1980s. Indeed, we had no idea at the outset whether we could ever arrive at our destination, for the challenges facing us were formidable and unpredictable, owing to the demonic complexity of the molecule and its potential chemical instability.
in the late 1980s. Indeed, we had no idea at the outset whether we could ever arrive at our destination, for the challenges facing us were formidable and unpredictable, owing to the demonic complexity of the molecule and its potential chemical instability.
Figure 3.

Highlights of the total synthesis of calicheamicin 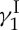 : (a) in retrosynthetic format and (b) in forward synthetic format.
: (a) in retrosynthetic format and (b) in forward synthetic format.
Arduous and difficult as the sail was, it led us, 5 years later, triumphantly to calicheamicin 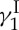 , our molecular ‘Ithaca’. Most importantly, we arrived there much wiser and quite content with the bounty of discoveries and inventions we collected on the way. These rewards came in the form of new synthetic methods and strategies, designed analogues of calicheamicin
, our molecular ‘Ithaca’. Most importantly, we arrived there much wiser and quite content with the bounty of discoveries and inventions we collected on the way. These rewards came in the form of new synthetic methods and strategies, designed analogues of calicheamicin 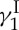 that exhibit similar biological properties despite their simpler structures, and a final confirmation of the originally assigned structure of the natural product. The details of our total synthesis of calicheamicin
that exhibit similar biological properties despite their simpler structures, and a final confirmation of the originally assigned structure of the natural product. The details of our total synthesis of calicheamicin 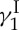 have been published and reviewed in other forums [17–19] and, therefore, will not be dealt with here beyond the highlights depicted in figure 3. As shown in figure 3a in retrosynthetic format, a number of strategic bond disconnections allowed the definition of a set of building blocks (i.e. 4, 11–16, figure 3b), which were constructed, coupled and elaborated appropriately to two larger intermediates, enediyne fragment 10 and oligosaccharide fragment 17. These two domains were then coupled through a glycosidation reaction to afford the entire framework of the molecule in the required atomic spatial arrangements. This advanced intermediate was then transformed to synthetic calicheamicin
have been published and reviewed in other forums [17–19] and, therefore, will not be dealt with here beyond the highlights depicted in figure 3. As shown in figure 3a in retrosynthetic format, a number of strategic bond disconnections allowed the definition of a set of building blocks (i.e. 4, 11–16, figure 3b), which were constructed, coupled and elaborated appropriately to two larger intermediates, enediyne fragment 10 and oligosaccharide fragment 17. These two domains were then coupled through a glycosidation reaction to afford the entire framework of the molecule in the required atomic spatial arrangements. This advanced intermediate was then transformed to synthetic calicheamicin 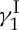 (1), identical in all respects (enantiomeric, chromatographic, spectroscopic and mass spectrometric) to the natural substance. A second synthesis of calicheamicin
(1), identical in all respects (enantiomeric, chromatographic, spectroscopic and mass spectrometric) to the natural substance. A second synthesis of calicheamicin 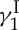 has been reported by the Danishefsky group [20].
has been reported by the Danishefsky group [20].
The calicheamicin 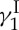 total synthesis endeavour proved delightfully rich in fundamental and applied knowledge. Thus, new synthetic strategies and technologies for the construction of the molecule’s unprecedented structural motifs were developed, and a series of analogues were designed, synthesized and tested for their ability to cleave double-stranded DNA and kill tumour cells. All in all, our synthetic studies with calicheamicin
total synthesis endeavour proved delightfully rich in fundamental and applied knowledge. Thus, new synthetic strategies and technologies for the construction of the molecule’s unprecedented structural motifs were developed, and a series of analogues were designed, synthesized and tested for their ability to cleave double-stranded DNA and kill tumour cells. All in all, our synthetic studies with calicheamicin 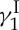 created the foundations that shaped the enediyne area of anti-tumour antibiotics [21]. This field continues to be of great interest to scientists and clinicians alike, with new enediynes, natural and designed, emerging from nature and the laboratory.
created the foundations that shaped the enediyne area of anti-tumour antibiotics [21]. This field continues to be of great interest to scientists and clinicians alike, with new enediynes, natural and designed, emerging from nature and the laboratory.
One of the most promising new leads from nature is uncialamycin, a scarce enediyne anti-tumour antibiotic recently isolated from a marine creature. Our first total syntheses of uncialamycin [22,23] that led to its full structural assignment are currently being optimized and exploited as a means to produce this natural product and its analogues in large quantities and as potential payloads for conjugation to antibodies. Such antibody drug conjugates (ADCs) have recently been hailed as potential ‘magic bullets’ for targeted cancer chemotherapy [24]. The first ADC drug to receive approval in the early 1990s for clinical use was gemtuzumab ozogamicin (Mylotarg; Wyeth/Pfizer), an antibody-linker–calicheamicin 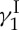 conjugate directed against acute myeloid leukaemia. Although withdrawn later owing to efficacy/safety concerns, Mylotarg proved inspirational and pathpointing. Today there are at least two ADC drugs on the market for cancer chemotherapy, brentuximab vedotin (Adcetris; Seattle Genetics and Millennium/Takeda; against advanced Hodgkin’s lymphoma) and trastuzumab emtansine (Kadcyla; Genentech/Roche; against late-stage HER2-positive breast cancer). Many other ADC drug candidates are currently in various stages of development [24].
conjugate directed against acute myeloid leukaemia. Although withdrawn later owing to efficacy/safety concerns, Mylotarg proved inspirational and pathpointing. Today there are at least two ADC drugs on the market for cancer chemotherapy, brentuximab vedotin (Adcetris; Seattle Genetics and Millennium/Takeda; against advanced Hodgkin’s lymphoma) and trastuzumab emtansine (Kadcyla; Genentech/Roche; against late-stage HER2-positive breast cancer). Many other ADC drug candidates are currently in various stages of development [24].
6. The total synthesis of Taxol
The legendary cancer curative properties of Taxol (paclitaxel) are matched by the intrigue of its discovery and development as an anti-cancer drug in the latter part of the twentieth century. Originally isolated from Taxus brevifolia (Pacific yew tree) and structurally characterized in the early 1970s, Taxol remained a scientific curiosity until its antimitotic mechanism of action as an anti-tumour agent was recognized in the early 1980s. The latter discovery gave momentum to its clinical development, and it became an approved drug in the early 1990s. Taxol is currently one of the most effective and widely used anti-cancer drugs for a variety of cancers, administered to patients either alone or in combination with other drugs. The natural scarcity of the molecule in its original source, coupled with the anticipated demand for the drug, created an urgency for its laboratory synthesis in the 1980s, one that was frustrated by the formidable challenge of the task owing to its molecular complexity. Indeed, numerous groups around the world embarked on its total synthesis at the time, and others continue to be intrigued to this day by its structure as a synthetic target. The importance and lure of Taxol did not escape us, and, in the early 1990s, we initiated a campaign to synthesize it, an endeavour that ended with the first published total synthesis of Taxol in 1994 [25].
The strategy developed for the synthesis of Taxol was based on the principle of convergency, meaning that a number of key building blocks had to be defined, constructed and coupled sequentially, and the resulting intermediates grown and elaborated towards the final target molecule. Depicted in retrosynthetic format in figure 4a, this strategy defined, through the strategic bond disconnections indicated, building blocks 22, 27 and 32 (figure 4b). These intermediates were constructed, coupled and elaborated, as outlined in figure 4b, through a series of key reactions as designated by the arrows. Thus, two [4+2] cycloadditions (Diels–Alder reactions) were employed to convert starting materials 18 and 19 and 23 and 24 to cyclohexene systems 21 and 26 through transition states 20 and 25, respectively. Each of these processes was notable for different reasons. The first led to the expected (from the rules of the Diels–Alder reaction) regioisomer, ring A (21), despite the severe steric congestion around the two adjacent tetrasubstituted (quaternary) carbon centres within this compound. The second [4+2] cycloaddition, leading, upon further rearrangement, to ring C (26), was impressive for the regiochemical exclusivity by which it proceeded, a consequence of the temporary boron tethering which oriented the two reactant partners properly in space, as shown in 25. Subsequent elaboration of 21 and 26 furnished required building blocks 22 and 27, respectively. Coupling of these key building blocks through a Shapiro reaction led to product 28, stereoselectively. Further elaboration of the latter compound gave bis-aldehyde 29, whose ring closure in the presence of freshly generated titanium metal afforded the desired ABC ring system of the growing molecule 30 through a process known as the McMurry reaction. This advanced intermediate was then subjected to further elaboration, leading to compound 31, which was coupled selectively to β-lactam 32 to give, after appropriate deprotections, synthetic Taxol (1), identical in all respects to the natural product. In the latter coupling reaction, the β-lactam served as a surrogate to the side chain of Taxol, as expected from the well-known chemistry of this structural motif. Indeed, the same kind of reactivity is manifested in the antibacterial mechanism of action of penicillin and other β-lactam antibiotics. Further strategic and experimental details of our total synthesis of Taxol can be found in the original publications and several reviews [19,25,26].
Figure 4.

Highlights of the total synthesis of Taxol: (a) in retrosynthetic format and (b) in forward synthetic format.
Besides our total synthesis, a number of other elegant total syntheses of Taxol have been reported by Holton et al. [27], Danishefsky et al. [28], Wender et al. [29], Mukaiyama et al. [30] and Kuwajima and co-workers [31]. Collectively, these accomplishments advanced the art and science of organic synthesis, enabled the design and synthesis of numerous analogues of Taxol, and facilitated biological investigations and drug discovery efforts in the area, including identification of biological tools and drug candidates. In addition to the methodological developments and facilitation of biology and medicine, the Taxol total syntheses served to demonstrate the sharp state of the art of total synthesis at the time and provided inspiration for further advancements to occur in the field.
7. The total synthesis of brevetoxin B
The long known ‘red tide’ phenomena, the first example of which is perhaps noted in the Bible, are often responsible for major catastrophes involving environmental damage, massive fish kills and poisoning of humans and other living creatures through seafood consumption. Two of the most notorious poisons associated with these menacing occurrences are the highly potent neurotoxin brevetoxin B (3, figure 5) and its sister molecule brevetoxin A.
Figure 5.

Highlights of the total synthesis of brevetoxin B: (a) in retrosynthetic format and (b) in forward synthetic format.
Produced by the dinoflagellate Karenia brevis, brevetoxin B (3) was isolated and structurally elucidated in 1981. Its molecular structure is a stunningly beautiful assembly of carbon, oxygen and hydrogen atoms arranged precisely in space in a ladder-like array of 11 rings, ranging in size from six- to eight-membered. Such structures were unprecedented at the time, and, as such, they provided challenge and inspiration to synthetic organic chemists whose drive to advance their science to higher levels of sophistication is often fuelled by discoveries of new structural motifs from nature. Indeed, this was our main motivation in entering the campaign of the total synthesis of brevetoxin B [32–34] and, later on, its related congener brevetoxin A [19,35].
The stunning molecular structure of brevetoxin B meant the lack of suitable methods for its construction, specifically its cyclic ether structural units of varying sizes. This state of affairs necessitated a search for such methods as a prerequisite before any serious attempt to develop a strategy for the synthesis of the molecule could be launched. This search was fruitful and led to a rich bounty of new synthetic methods for the construction of cyclic ethers, common structural motifs in natural and designed molecules of biological and medical importance. These synthetic technologies and strategies have been amply described in previous articles and will not be further commented on here, except for two that played pivotal roles in the synthesis of brevetoxin B and other ladder-like marine biotoxins. These are the regio- and stereospecific intramolecular opening of hydroxy epoxides carrying an olefinic bond adjacent to the epoxide carbon–oxygen bond that undergoes the initial nucleophilic attack to form tetrahydropyran systems (six-membered cyclic ether structural motifs), and the hydroxy dithioketal cyclization leading to oxocene systems (eight-membered cyclic ether structural motifs).
Armed with our newly developed synthetic technologies, we were able to devise a successful strategy towards our target molecule brevetoxin B [32,33], but not before an arduous odyssey of 12 years’ duration, full of unimaginable adventures and excitement [34]. Figure 5 summarizes the devised synthetic strategy both in retrosynthetic format (figure 5a), which defined the starting materials and key building blocks, and in the forward synthetic direction (figure 5b), which allowed the coupling and elaboration of the constructed building blocks to the final target molecule.
Thus, as shown in figure 5b, the total synthesis of brevetoxin B began with d-mannose (34), a readily available starting material that provided the appropriate chirality for reaching the target molecule in its naturally occurring enantiomeric form. This material was elaborated to hydroxy epoxide 35, which served admirably as a precursor to the next desired intermediate, tricyclic system 36, through an acid-catalysed regio- and stereoselective hydroxy epoxide opening according to our specifically developed conditions for tetrahydrofuran formation as mentioned above. The latter was then advanced to the IJK ring system aldehyde dithioketal 37 in waiting to be coupled to the larger ABCDEFG fragment (41), whose construction began with 2-deoxy-d-ribose (38), another readily available starting material possessing the correct chirality for our purposes, and proceeded through intermediates 39 and 40. Advanced key intermediates 37 and 41 were then coupled through a Wittig reaction, forming hydroxy dithioketal 42, whose ring closure under the action of silver perchlorate led first to ethylthio oxocene system 43 and thence, through appropriate chemical transformations, to the ABCDEFGHIJK ring system 44 containing the entire undecacyclic ring framework of brevetoxin B. Further elaboration of the latter precursor generated synthetic brevetoxin B (3), identical in all respects to the naturally occurring substance. Reported in 1995, the total synthesis of brevetoxin B [33,34] was followed by our total synthesis of brevetoxin A [35]. Subsequent to our work, the Nakata and Yamamoto groups accomplished a second and third total synthesis, respectively, of brevetoxin B [36,37], while the Crimmins group achieved a second total synthesis of brevetoxin A [38].
The brevetoxin B project proved delightfully enriching in knowledge and supply of this scarce biotoxin. Most importantly, it set the stage for more advances to occur in the field of ladder-like marine neurotoxins, whose family currently boasts over 50 members and continues to grow. Besides brevetoxins B and A, several other members of the class have been reached by a number of groups through total synthesis, including hemibrevetoxin, ciguatoxin 3C, gambierol, gymnosin A, breveral and the azaspiracids. These works have been reviewed by Nicolaou et al. [39]. Furthermore, large fragments of maitotoxin (see figure 6 for molecular structure), the largest member of the ladder-like family of marine biotoxins, have been synthesized [40].
Figure 6.
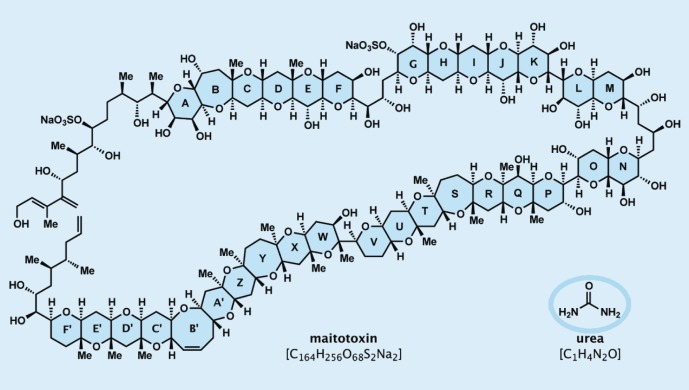
Molecular structures of maitotoxin and urea.
8. Future perspectives
Ever since its inception in 1828, organic synthesis has been on the march, advancing to new levels of performance and reach in terms of molecular complexity and diversity [16,41]. Its applications have been equally impressive and continue to expand into new domains, thereby increasing its impact on science and society. Thus, from the tiny molecule of urea (CH4N2O, see figure 6) containing a single carbon atom and no stereogenic site, synthetic organic chemists of our time dare to attempt the synthesis of giant molecules such as that of maitotoxin (C164H256O68S2Na2, see figure 6), the largest secondary metabolite discovered thus far from nature, boasting 164 carbon atoms and 99 sites of stereoisomerism [40]. And from dyes and pharmaceuticals, organic synthesis came to provide the foundation of a whole new generation of scientific endeavours and industries, including polymers and plastics. Endeavours in total synthesis provided numerous, more or less, complex biologically active molecules (natural or designed) for biological and pharmaceutical studies. Other synthetic efforts rendered available biomolecules such as nucleic acids, proteins and polysaccharides, and their smaller cousins oligonucleotides, peptides and carbohydrates. Fine chemicals used as fuels, pesticides and herbicides, diagnostics and medical devices, vitamins, perfumes, cosmetics, fabrics and all sorts of high-technology materials used in televisions, computers and other information technologies, and transportation and space machines are also the products of organic synthesis.
The latter applications became possible only because of the advances made in the field of organic synthesis. It is, therefore, of paramount importance to continue to advance this discipline for its own sake and to constantly imagine new areas for its application. The former objective is in the minds and hands of the practitioners of organic synthesis, those who devote their efforts to the discovery and invention of new synthetic reactions, methods and strategies. Indeed, their creativity and imagination is already aiming at new directions and ambitious goals. Among them are cascade reactions, C−H activation/functionalization, biomimetic synthetic strategies, new metal- and organocatalytic reactions and green chemistry. Ideally, the ultimate goal of organic synthesis practitioners should be to strive to elevate their art and science to the standards of nature and beyond in terms of efficiency, practicality and elegance.
The latter objective, that of applying wisely the power of organic synthesis to the benefit of other disciplines and society, can be best achieved through the ingenuity and imagination by those same practitioners of the art of synthesis or by other scientists and engineers whose needs may be met through the enabling means of organic synthesis. The most effective way, however, for achieving novel applications and products is through multi- and transdisciplinary research programmes involving scientists from different disciplines and with overlapping and complementary expertise. A perfect example of such collaborations is that currently practised among chemists, biologists and clinicians during the drug discovery and development process. As more academicians are attracted to this process, it is expected that new paradigms will emerge that will involve, in addition to chemists, biologists and clinicians, other specialists such as computational experts, bioinformaticians, engineers and logicians, among others. Their combined and integrated efforts should result in improved drug discovery and development practices with higher productivity, lower cost and higher odds of success in the clinic for drug candidates. In the meantime, other similar transdisciplinary programmes are being intensified, including collaborative research among chemists, physicists and materials scientists in the field of nanotechnology. In addition, one can also imagine several other grand challenges that can benefit from solutions provided through contributions of organic synthesis. These challenges include food production, energy sources and environmental protection through green chemistry and other means.
It is my sincere hope that with this short perspective I succeeded, at least partly, in explaining the essence, purpose and societal impact of organic synthesis to the broader readership of this journal of the Royal Society. The progress made in the field in its nearly two-century history is impressive, but considering the bewildering molecular complexity and diversity of molecules that nature can synthesize with such admirable elegance and efficiency, we must admit that our prowess and proficiency in this art is profoundly inadequate. We can only wonder what the founder of organic synthesis, Friedrich Wöhler, would think of its progress, the present state of the art and its prospects for the future. My suspicion is that, while he would be happy and content, he would urge us to keep reaching for higher levels of sophistication and into new pastures in search of further discoveries and applications of the new and the old.
Acknowledgements
My sincere thanks and deep appreciation go to my many students whose collaborative efforts led to the accomplishments described in this article and whose names are found in the references below and the papers cited therein. I also wish to express my gratitude to the various agencies, companies and benefactors, whose names are found in the original publications, for supporting our research programmes over the years. Last but not least, I am indebted to my teachers and mentors for their constant guidance and inspiration, and to my wife, Georgette, my daughter, Colette, my sons, Alex, Christopher and P. J., and my grandson, Nicolas, for their continuous support and unconditional love.
Funding statement
This work was partially supported by the US National Institutes of Health, The Skaggs Institute for Chemical Biology and the Cancer Prevention Research Institute of Texas (CPRIT).
Author profile

K. C. Nicolaou obtained his BSc degree at Bedford College and his PhD degree from University College, University of London, under the supervision of Peter Garratt and Franz Sondheimer (FRS). He subsequently carried out research as a postdoctoral fellow at Columbia and Harvard Universities, under the mentorship of Thomas J. Katz and E. J. Corey, respectively. During his independent career, he worked at the University of Pennsylvania, The Scripps Research Institute, the University of California, San Diego, and currently at Rice University, where he is the Harry C. and Olga K. Wiess Professor of Chemistry. Between 2004 and 2010, he served as the Director of the Chemical Synthesis Laboratory, of which he was the founder, at A*STAR, Singapore. His research activities focus on the discovery and development of new synthetic strategies and technologies, and their applications to the total synthesis of natural and designed molecules of biological and medical importance. K. C. Nicolaou was elected as a Foreign Member of the Royal Society in 2013.
References
- 1.Nicolaou KC. 2013. The emergence of the structure of the molecule and the art of its synthesis. Angew. Chem. Int. Ed. 52, 131–146 (doi:10.1002/anie.201207081) [DOI] [PubMed] [Google Scholar]
- 2.Nicolaou KC, Montagnon T. 2008. Molecules that changed the world. Weinheim, Germany: Wiley-VCH Publishers [Google Scholar]
- 3.Rocke AJ. 1984. Chemical atomism in the nineteenth century: from Dalton to Cannizzaro. Columbus, OH: Ohio State University Press [Google Scholar]
- 4.Nobelprize.org All Nobel Prizes in Chemistry. See http://www.nobelprize.org/nobel_prizes/chemistry/laureates/.
- 5.Pauling L. 1939. The nature of the chemical bond. New York, NY: Cornell University Press [Google Scholar]
- 6.Benfey OT, Morris PJT. 2001. Robert Burns Woodward. Philadelphia, PA: Chemical Heritage Foundation [Google Scholar]
- 7.Eschenmoser A. 2011. Etiology of potentially primordial biomolecular structures: from vitamin B12 to the nucleic acids and an inquiry into the chemistry of life’s origin: a retrospective. Angew. Chem. Int. Ed. 50, 12 412–12 472 (doi:10.1002/anie.201103672) [DOI] [PubMed] [Google Scholar]
- 8.Corey EJ, Cheng X-M. 1989. The logic of chemical synthesis. New York, NY: Wiley [Google Scholar]
- 9.Corey EJ, Czakó B, Kürti L. 2007. Molecules and medicine. Weinheim, Germany: Wiley [Google Scholar]
- 10.Corey EJ, Kürti L. 2010. Enantioselective chemical synthesis. Maylene, AL: Direct [Google Scholar]
- 11.Nicolaou KC, Sorensen EJ. 1996. Classics in total synthesis. Weinheim, Germany: VCH Publishers [Google Scholar]
- 12.Nicolaou KC, Snyder SA. 2003. Classics in total synthesis II. Weinheim, Germany: Wiley-VCH Publishers [Google Scholar]
- 13.Nicolaou KC, Chen JS. 2011. Classics in total synthesis III. Weinheim, Germany: Wiley-VCH Publishers [Google Scholar]
- 14.Nicolaou KC, Nilewski C. In press. Organic synthesis. In Discoveries in modern science: exploration, invention, technology (ed. Trefil J.). Woodbridge, CT: Macmillan Reference USA [Google Scholar]
- 15.Nicolaou KC, Hale CRH. In press. The endeavor of total synthesis and its impact on chemistry, biology and medicine. Nat. Sci. Rev. [Google Scholar]
- 16.Nicolaou KC, Hale CRH, Nilewski C, Ioannidou HA. 2012. Constructing molecular complexity and diversity: total synthesis of natural products of biological and medicinal importance. Chem. Soc. Rev. 41, 5185–5238 (doi:10.1039/c2cs35116a) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicolaou KC, Hummel CW, Pitsinos EN, Nakada M, Smith AL, Shibayama K, Saimoto H. 1992. Total synthesis of calicheamicin γ1I. J. Am. Chem. Soc. 114, 10082–10084 (doi:10.1021/ja00051a063) [Google Scholar]
- 18.Nicolaou KC. 1993. The battle of calicheamicin γ1I. Angew. Chem. Int. Ed. Engl. 32, 1377–1385 (doi:10.1002/anie.199313773) [Google Scholar]
- 19.Nicolaou KC, Hale CRH, Nilewski C. 2012. A total synthesis trilogy: calicheamicin γ1I, Taxol, and brevetoxin A. Chem. Rec. 12, 407–441 (doi:10.1002/tcr.201200005) [DOI] [PubMed] [Google Scholar]
- 20.Hitchcock SA, Boyer SH, Chu-Moyer MY, Olson SH, Danishefsky SJ. 1994. A convergent total synthesis of calicheamicin γ1I. Angew. Chem. Int. Ed. Engl. 33, 858–862 (doi:10.1002/anie199408581) [Google Scholar]
- 21.Nicolaou KC, Smith AL, Yue EW. 1993. Chemistry and biology of natural and designed enediynes. Proc. Natl Acad. Sci. USA 90, 5881–5888 (doi:10.1073/pnas.90.13.5881) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicolaou KC, Zhang H, Chen JS, Crawford JJ, Pasunoori L. 2007. Total synthesis and stereochemistry of uncialamycin. Angew. Chem. Int. Ed. 46, 4704–4707 (doi:10.1002/anie.200700917) [DOI] [PubMed] [Google Scholar]
- 23.Nicolaou KC, Chen JS, Zhang H, Montero A. 2008. Asymmetric synthesis and biological properties of unicialamycin and 26-epi-uncialamycin. Angew. Chem. Int. Ed. 47, 185–189 (doi:10.1002/anie.200704577) [DOI] [PubMed] [Google Scholar]
- 24.Sliwkowski MX, Mellman I. 2013. Antibody therapeutics in cancer. Science 341, 1192–1198 (doi:10.1126/science.1241145) [DOI] [PubMed] [Google Scholar]
- 25.Nicolaou KC, et al. 1994. Total synthesis of Taxol. Nature 367, 630–634 (doi:10.1038/367630a0) [DOI] [PubMed] [Google Scholar]
- 26.Nicolaou KC, Guy RK. 1995. The conquest of Taxol. Angew. Chem. Int. Ed. Engl. 34, 2079–2090 (doi:10.1002/anie.199520791) [Google Scholar]
- 27.Holton RA, et al. 1994. First total synthesis of Taxol. 2. Completion of the C and D rings. J. Am. Chem. Soc. 116, 1599–1600 (doi:10.1021/ja00083a067) [Google Scholar]
- 28.Danishefsky SJ, et al. 1996. Total synthesis of baccatin III and Taxol. J. Am. Chem. Soc. 118, 2843–2859 (doi:10.1021/ja952692a) [Google Scholar]
- 29.Wender PA, et al. 1997. The pinene path to taxanes. 6. A concise stereocontrolled synthesis of Taxol. J. Am. Chem. Soc. 119, 2757–2758 (doi:10.1021/ja963539z) [Google Scholar]
- 30.Mukaiyama T, Shiina I, Iwadare H, Sakoh H, Tani Y, Hasegawa M, Saitoh K. 1997. Asymmetric total synthesis of Taxol. Proc. Jpn. Acad. B 73, 95–100 (doi:10.2183/pjab.73.95) [Google Scholar]
- 31.Morihira K, Hara R, Kawahara S, Nishimori T, Nakamura N, Kusama H, Kuwajima I. 1998. Enantioselective total synthesis of Taxol. J. Am. Chem. Soc. 120, 12 980–12 981 (doi:10.1021/ja9824932) [Google Scholar]
- 32.Nicolaou KC, Theodorakis EA, Rutjes FPJT, Tiebes J, Sato M, Untersteller E, Xiao X-Y. 1995. Total synthesis of brevetoxin B. I. CDEFG framework. J. Am. Chem. Soc. 117, 1171–1172 (doi:10.1021/ja00108a051) [Google Scholar]
- 33.Nicolaou KC, Rutjes FPJT, Theodorakis EA, Tiebes J, Sato M, Untersteller E. 1995. Total synthesis of brevetoxin B. II. Completion. J. Am. Chem. Soc. 117, 1173–1174 (doi:10.1021/ja00108a052) [Google Scholar]
- 34.Nicolaou KC. 1996. The total synthesis of brevetoxin B: a twelve-year odyssey in organic synthesis. Angew. Chem. Int. Ed. Engl. 35, 589–607 (doi:10.1002/anie.199605881) [Google Scholar]
- 35.Nicolaou KC, Yang Z, Shi G-Q, Gunzner JL, Agrios KA, Gärtner P. 1998. Total synthesis of brevetoxin A. Nature 392, 264–269 (doi:10.1038/32623) [DOI] [PubMed] [Google Scholar]
- 36.Matsuo G, Kawamura K, Hori N, Matsukura H, Nakata T. 2004. Total synthesis of brevetoxin-B. J. Am. Chem. Soc. 126, 14374–14376 (doi:10.1021/ja0449269) [DOI] [PubMed] [Google Scholar]
- 37.Kadota I, Takamura H, Nishii H, Yamamoto Y. 2005. Total synthesis of brevetoxin B. J. Am. Chem. Soc. 127, 9246–9250 (doi:10.1021/ja051171c) [DOI] [PubMed] [Google Scholar]
- 38.Crimmins MT, Zuccarello JL, Ellis JM, McDougall PJ, Haile PA, Parrish JD, Emmitte KA. 2008. Total synthesis of brevetoxin A. Org. Lett. 11, 489–492 (doi:10.1021/ol802710u) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicolaou KC, Frederick MO, Aversa RJ. 2008. The continuing saga of the marine polyether biotoxins. Angew. Chem. Int. Ed. 47, 7182–7225 (doi:10.1002/anie.200801696) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicolaou KC, Aversa RJ. 2011. Maitotoxin: an inspiration for synthesis. Isr. J. Chem. 51, 359–377 (doi:10.1002/ijch.201100003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicolaou KC, Vourloumis D, Winssinger N, Baran PS. 2000. The art and science of total synthesis at the dawn of the twenty-first century. Angew. Chem. Int. Ed. 39, 44–122 (doi:10.1002/(SICI)1521-3773(20000103)39:1<44::AID-ANIE44>3.0.CO;2-L) [PubMed] [Google Scholar]