

Abstract
Elevated levels of low density lipoprotein cholesterol (LDL-C) in plasma are a major contributor to cardiovascular disease (CVD), which is the leading cause of death worldwide. Genome–wide association studies (GWAS) have identified 95 loci that associate with control of lipid/cholesterol metabolism. Although GWAS results are highly provocative, direct analyses of the contribution of specific allelic variations in regulating LDL-C has been challenging due to the difficulty in accessing appropriate cells from affected patients. The primary cell type responsible for controlling cholesterol and lipid flux is the hepatocyte. Recently we have shown that cells with hepatocyte characteristics can be generated from human induced pluripotent stem cells (iPSC). This finding raises the possibility of using patient–specific iPSC–derived hepatocytes to study the functional contribution of GWAS loci in regulating lipid metabolism. To test the validity of this approach we produced iPSCs from a patient with mutations in the Low density lipoprotein receptor(LDLR) gene that result in familial hypercholesterolemia (FH). Conclusion: We demonstrate that 1) hepatocytes can be efficiently generated from FH iPSCs, 2) in contrast to control cells FH iPSC–derived hepatocytes are deficient in LDL–C uptake, 3) control but not FH iPS cell–derived hepatocytes increase LDL uptake in response to lovastatin, and 4) FH iPSC–derived hepatocytes display a marked elevation in secretion of lipidated ApoB-100. Cumulatively, these findings demonstrate that FH iPSC–derived hepatocytes recapitulate the complex pathophysiology of FH in culture. These results also establish that patient specific iPSC–derived hepatocytes could be used to definitively determine the functional contribution of allelic variation in regulating lipid and cholesterol metabolism and could potentially provide a platform for the identification of novel treatments of CVD.
Keywords: induced pluripotent stem cells, hepatocyte differentiation, statins, LDL-cholesterol
Introduction
A study of cardiovascular disease in the United States revealed that approximately 1 in 3 (79 million) American adults suffer from heart disease with ∼16 million specifically afflicted with coronary artery disease (CAD)1. The level of circulating LDL-C within an individual has been functionally associated with CAD and meta-analyses of patients that are aggressively treated with LDL-C lowering drugs revealed that treatment reduces the incidence of heart attacks and stroke2. Although environmental factors contribute to plasma LDL–C concentration, GWAS studies performed on >100,000 patients have identified genetic variants at 95 loci that are closely associated with cholesterol and lipid levels linked to CAD3. Some of these genetic variants are associated with genes encoding proteins with known roles in regulating cholesterol metabolism including Low density lipoprotein receptor(LDLR), Low density lipoprotein receptor associated protein 1(LDLRAP1), and Scavenger receptor class B, member 1(SCARB1) genes. In addition to identifying genes with known roles in controlling cholesterol flux, GWAS uncovered many novel loci whose contribution to CAD is not understood.
Linking genetic findings to biological mechanism historically has proven to be a challenge. In some cases the mouse has provided a suitable system to relate genetic variation to the pathophysiology of disease; however, the usefulness of the mouse is tempered by differences from humans in terms of physiology, metabolism, and genetics, and this is exacerbated when complex traits are being analyzed. Cell culture models can also be useful; however, cholesterol metabolism is predominantly controlled by hepatocytes and obtaining primary liver cells from patients would require a liver biopsy. Moreover, when primary hepatocytes are cultured, the cells quickly dedifferentiate and lose key liver functions rendering them unsuitable for detailed metabolic studies. Recently, it has been shown that human induced pluripotent stem cells (hiPSCs) can differentiate into cells that are functionally similar to hepatocytes4-6. Because hiPSCs can be reprogrammed from easily accessible somatic cell types, such as skin fibroblasts, this raises the possibility of using hiPSCs from GWAS patients as a source of hepatocytes to study the role of specific allelic variants in regulating cholesterol metabolism. In addition, the availability of hepatocytes derived from patients with inborn errors in hepatic metabolism could provide a platform for drug discovery. While, the use of hiPSC–derived hepatocytes to recapitulate metabolic liver disease in culture is conceptually appealing, direct evidence that demonstrates the validity of such an approach is scarce7. Importantly, although iPSC–derived hepatocyte-like cells can be generated with high efficiency, the resulting cells fail to express the complete repertoire of proteins found in adult primary hepatocytes and do not fully silence expression of fetal hepatocyte mRNAs such as AFP {Si-Tayeb et al., 2010, Hepatology, 51, 297-305}. These observations have raised questions over the credibility of using iPSCs to reliably study hepatic dysfunction8. We, therefore, attempted to determine the feasibility of using hiPSCs to study genetic variants that could contribute to dysregulation of cholesterol flux by producing hepatocytes from hiPSCs that were generated from a familial hypercholesterolemia (FH) patient with defined mutations.
Experimental Procedures
A detailed description of procedures used has been provided in the online supplemental information. Procedures used for the generation of iPS cells and differentiation of pluripotent stem cells to hepatocytes have been described previously4,14. All culture of human ES and generation of iPS cells was approved by the MCW Human Stem Cell Research Oversight Committee (hSCRO approval #09-005) and all animal procedures were approved by the Medical College of Wisconsin's IACUC.
Results
Generation of iPSCs from ‘JD’ fibroblasts
FH is an autosomal dominant dyslipidemia caused by mutations in the LDLR gene that result in severely elevated plasma LDL–C levels and premature cardiovascular disease9. The liver is central to the pathogenesis of FH and homozygous FH patients are successfully treated by liver transplant. Although hepatocytes are the key cells that control cholesterol flux, LDLR mutations have primarily been studied using fibroblasts9. Such studies revealed that LDLR–deficient fibroblasts had an impaired capacity to internalize LDL, which gave rise to the paradigm that the level of LDL–C in serum is determined by the rate of LDL clearance10. However, modifications to this model have recently been proposed based on evidence suggesting that FH patients often possess profoundly elevated hepatic VLDL production11. Given the extensive understanding of FH and the fact that single nucleotide polymorphisms (SNPs) have been identified in the vicinity of the LDLR gene, we felt that hepatocytes derived from FH hiPSCs would offer an ideal model to define the feasibility of using iPSCs to study genetic variations that could impact complex hepatic metabolism.
The generation of iPS cells from a patient with early onset atherosclerotic disease with hypercholesterolemia has been described previously7; however, the genetic lesion was undefined. In addition, this study by Rashid and colleagues was designed only to test whether cells derived from LDLR–deficient iPSCs could internalize LDL-cholesterol. However, LDLR–mediated uptake of LDL is not a hepatocyte-specific process and most cells use this pathway to internalize cholesterol. The goal of the current study was therefore to test the feasibility of using iPSC–derived hepatocytes to study complex metabolic disorders that specifically impact hepatocyte function. To develop a clearer understanding of the pathophysiology of FH iPSC–derived hepatocytes, we reprogrammed fibroblasts from ‘JD’, a 14-year-old boy with cutaneous xanthomatosis and advanced cardiovascular disease12. The choice to generate ‘JD’ hiPSCs was considered historically relevant because Brown, Goldstein and colleagues, in establishing the LDLR paradigm, extensively studied ‘JD’ fibroblasts. We produced several ‘JD’ iPS cell lines by transducing primary fibroblasts with lentiviral vectors encoding the transcription factors OCT4, SOX2, NANOG, and LIN2813 and demonstrated that they expressed characteristic markers of pluripotency (Fig. 1a). In each hiPSC line we confirmed the retention of the ‘JD’ LDLR mutations (Fig. 1b, Supplemental Fig. 1), established that each had a normal karyotype (Fig. 1c), and determined that each ‘JD’ hiPSC line could differentiate into derivatives of all three germ layers using teratoma assays (Fig. 1d).
Fig. 1. Generation of induced pluripotent stem cells from ‘JD’ fibroblasts.
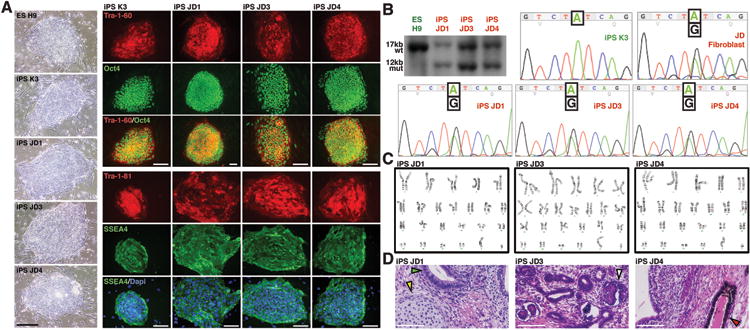
(a) Micrographs showing colony morphology and expression of characteristic markers of pluripotency (Tra-1-60, OCT4, Tra-1-81, SSEA4) identified by immunocytochemistry in huESCs (H9), control hiPSC (K3), and three independently derived ‘JD’ iPSC lines (JD1, JD3, JD4). (b) Southern blot analysis (upper left panel) using a probe from LDLR exon 18 to identify a 17kb wild-type BamH1 fragment (wt) in control cells and an additional 12kb mutant fragment (mut) in ‘JD’ hiPSCs that represents the maternal allele. DNA sequencing revealed the presence of the paternal A-G transition exclusively in ‘JD’ fibroblasts and ‘JD’ iPSCs. (c) ‘JD’ hiPSCs retain a normal chromosomal arrangement as revealed by karyotyping. (d) Teratomas generated in immunocompromised mice from ‘JD’ hiPSCs contained cell types representative of all germ layers (arrowheads). Scale bars = 100μM.
‘JD’ iPS cells can be induced to form hepatocytes with high efficiency and reproducibility
Using a previously described protocol (Fig. 2a), which we had shown could generate functional hepatocyte–like cells (referred to here as hepatocytes)4,14, we demonstrated that each ‘JD’ hiPSC clone was capable of directed differentiation toward a hepatic fate. On day 20 of differentiation, the morphology of both control hiPSCs– and ‘JD’ hiPSC–derived cells was indistinguishable and closely resembled that of hepatocytes including the presence of lipid vesicles, a high cytoplasmic to nuclear ratio, granular cytoplasm and prominent nucleoli (Fig.2b). In addition, the differentiated cells expressed hepatocyte markers including HNF4a and Albumin (Fig. 2c). Flow cytometric analyses of hepatocytes from both control and ‘JD’ hiPSCs confirmed that the cells differentiated into asialoglycoprotein receptor (ASGPR1) positive hepatocytes with comparable efficiency (Fig. 2d). Only cells expressing high levels of ASGPR were counted to avoid the possibility of counting false negatives. Finally, hepatocytes derived from control hESCs or hiPSCs as well as ‘JD’ hiPSCs were found to express hepatic mRNAs at similar levels, whereas expression of each of these mRNAs was not detected in undifferentiated hESCs (Fig. 2e). Based on these data we conclude that ‘JD’ iPSCs could be directed to form cells with hepatocyte characteristics at efficiencies that were comparable to huESCs or control hiPSCs.
Fig. 2. Hepatocyte–like cells can be efficiently derived from ‘JD’ hiPSCs.
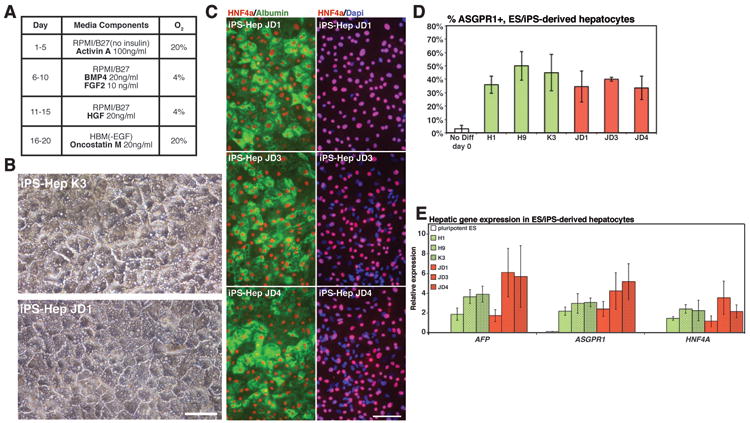
(a) Schematic showing procedure used to generate hepatocytes from human pluripotent stem cells. (b) Micrographs showing morphology of hepatocytes derived from WT and ‘JD’ hiPSCs. (c) Immunocytochemistry revealing the presence of HNF4a in the nucleus (red) and Albumin in the cytoplasm (green) of ‘JD’–hiPSC–derived hepatocytes. Nuclei are identified by DAPI staining (blue). (d) Bar graph illustrating the efficiency and reproducibility (n=3 independent experiments) of generating ASGPR positive hepatocytes from control (H1, H9, K3) and ‘JD’ (JD1, JD3, JD4) pluripotent stem cells. No significant difference was observed between control and ‘JD’ cells (ANOVA, p>0.05). (f) Bar graph showing result of real time qRT-PCR analyses of characteristic hepatocyte mRNAs (AFP, ASGPR1, HNF4A) in undifferentiated pluripotent stem cells and in hepatocytes generated from control (H1, H9, K3) and ‘JD’ (JD1, JD3, JD4) pluripotent stem cells. No significant difference was observed between control and ‘JD’ cells (ANOVA, p>0.05). Scale bars = 100μM.
‘JD’ iPSC–derived hepatocytes show deficiencies in LDL-C uptake and response to Lovastatin treatment
The FH associated with ‘JD’ is a consequence of compound heterozygosity at the LDLR locus. ‘JD’ inherited a maternal allele containing a 5kb deletion spanning part of exon 13 and all of exons 14 and 15 that results in the absence of functional protein12. The inherited paternal allele contains an A>G transition within exon 17, which encodes a tyrosine>cysteine substitution at residue 807 in the LDLR cytoplasmic domain resulting in a mutant protein that can still bind LDL–C, but is inefficiently internalized12. The combination of these two alleles encoded by the ‘JD’ genome therefore effectively results in homozygous loss of LDLR function. We first measured whether ‘JD’ hiPSC–derived hepatocytes exhibited the expected deficiencies in LDL–C uptake. After 3.5 hours incubation with fluorescently-labeled LDL–C particles (FL–LDL), control hiPS–derived hepatocytes contained intense fluorescence staining extending from a perinuclear location throughout the cytoplasm (Fig. 3a). In contrast, cytoplasmic fluorescence within ‘JD’ hiPSC-derived hepatocytes was reduced (Fig. 3a, Supplemental Fig. 2) and we observed intense clusters of staining at the cell surface, which is consistent with trapping of FL-LDL by the paternally encoded mutant LDLR. These results therefore confirm that ‘JD’ encoded LDLR alleles are defective as has been described in the studies of ‘JD’ fibroblasts.
Fig. 3. ‘JD’ hiPSC–derived hepatocytes exhibit deficiencies in uptake of LDL–C and in their response to lovastatin.
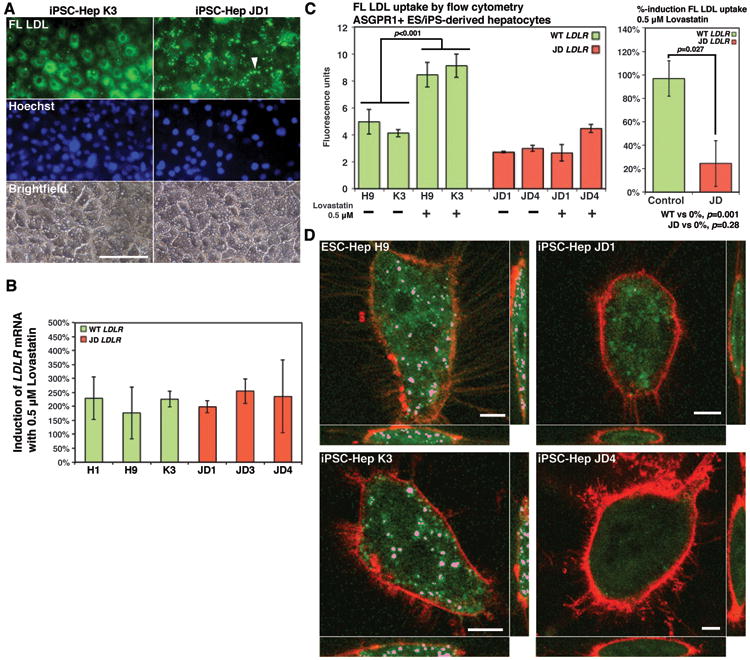
(a) Micrographs showing localization of FL LDL (green) after incubation of control and ‘JD’ iPS cell–derived hepatocytes with BODIPY labeled LDL for 3.5 hours. Note clustering of FL LDL specifically on the surface of ‘JD’ cells (arrow). Staining with Hoechst dye identified nuclei (blue) and cell distribution is shown by bright field images. Scale bars = 100μM. (b) Bar graph shows percent increase in LDLR mRNA following lovastatin treatment of control (green bars) and ‘JD’ (red bars) hepatocytes. No statistically significance difference in the level of induction was observed between control and ‘JD’ cells (ANOVA p>0.05). (c) Bar graph (left panel) showing a significant (Student's t-test p<0.05) increase in FL LDL uptake was observed in response to lovastatin treatment by control hepatocytes (green bars) but not by ‘JD’ hepatocytes (red bars). Right bar graph shows that the differential response to lovastatin treatment between control (green bar) and ‘JD’ (red bar) hepatocytes is significant (Student's t-test, p<0.05). (d) Volume render images obtained by confocal microscopy showing that uptake of FL LDL (pink) could be identified in endosomes of lovastatin–treated control (ES-Hep H9, iPS-Hep K3) hepatocytes but not in ‘JD’ hepatocytes. Plasma membranes were identified using FM® 4-64 (Molecular Probes) (red). Scale bars = 20μM.
In addition to probing GWAS phenotypes, patient–specific hiPSC–derived hepatocytes could provide a platform to identify cholesterol lowering pharmaceuticals; however, again proof–of–feasibility experiments have not been described. Lovastatin is a hepatoselective lipid-lowering drug whose activity is conferred by oxidation of the lactone prodrug to its β-hydroxy acid form, which then inhibits HMG-CoA reductase. Since activation of the pro-drug is hepatocyte-specific, in vitro studies using lovastatin ubiquitously employ biochemically activated lovastatin β-hydroxy acid rather than the lactone prodrug. Under normal circumstances, the response of the hepatocyte to HMG-CoA reductase inhibition is to increase expression of the LDLR gene resulting in enhanced LDL–C uptake. Importantly, because this drug manifests its activity primarily through increasing LDLR, lovastatin is ineffective in FH patients that encode defective LDLR alleles. We therefore examined the response of both control– and ‘JD’–derived hepatocytes to lovastatin treatment (Figs. 3b–d). When either control or ‘JD’ hepatocytes were treated for 24 hours with 0.5 μM lovastatin lactone we observed a significant induction of LDLR mRNA (p=0.003(control), p=0.011(‘JD’), Fig. 3b) and the extent of induction was similar regardless of genotype (Fig 3b). In addition, both control and ‘JD’ hepatocytes expressed similar levels of enzymes involved in oxidative metabolism of lovastatin lactone (CYP 3A4, CES1, CES2, PON2 and PON3; Supplemental Fig. 3). Induction of LDLR gene expression is predominantly regulated through proteolytic activation of SREBP2 (encoded by SREBF2); however, it has also been reported that hepatocyte expression of SREBF2 mRNA is increased in response to Lovastatin treatment. qRT-PCR analyses revealed modest increases in expression of SREBF2 mRNA following lovastatin treatment of both control and ‘JD’ hepatocytes (Supplemental Fig. 4). Although SREBP1 has also been implicated in regulating cholesterol metabolism, no significant difference in SREBF1 mRNA levels was detected. Cumulatively, these data demonstrate that both control and ‘JD’ hepatocytes responded appropriately to statin treatment and that the pluripotent stem cell–derived hepatocytes were capable converting the pro-drug to an active form. We next measured the impact of lovastatin on LDL–C uptake. To ensure that flow cytometery could quantitatively measure LDL–C uptake, we first measured the level of FL-LDL uptake in control hESC-derived hepatocytes over time. We found that FL-LDL uptake tripled over a period of one hour, increasing linearly through 30 minutes (Supplemental Fig. 5). We therefore used a 30-minute incubation with FL–LDL in all further analyses, which ensured that all measurements were in the linear range. In control stem cell–derived hepatocytes, flow cytometery revealed that the increase in LDLR mRNA levels in response to lovastatin treatment translated to a 99.1% increase in FL–LDL uptake compared with untreated cells (p<0.001, Fig. 3d). In contrast to control cells, no significant change in FL–LDL uptake was observed between treated and untreated ‘JD’ hiPSC-derived hepatocytes (Fig. 3c). LDL–C uptake by hepatocytes is divided into a high-affinity, low-volume mechanism mediated by the LDLR, and a low-affinity, high-volume mechanism controlled independently. We, therefore, also examined the distribution of FL–LDL internalized by control and ‘JD’ hiPSC–derived hepatocytes after lovastatin treatment using confocal microscopy (Fig. 3d, Supplemental Fig 2). In control cells, FL-LDL was identified within distinct subcellular foci consistent with transport of the FL-LDL to endosomes via clathrin–mediated endocytosis. In contrast, ‘JD’ hiPSC–derived hepatocytes exhibited no endosomal localization of FL-LDL, although relatively low levels of fluorescence were uniformly distributed throughout the ‘JD’ cell cytoplasm. Cumulatively, these data demonstrate that hiPSC–derived hepatocytes can be used effectively to identify lipid lowering pharmaceuticals and that the ‘JD’ hiPSC–derived hepatocytes accurately reflect the pathophysiology of FH.
Hepatocytes derived from ‘JD’ iPSCs show highly elevated secretion of lipidated ApoB-100
Several studies have supported a view that loss of LDLR receptor function not only results in reduced LDL–C uptake but also significantly increases production of VLDL/LDL by the hepatocyte and it has been argued that enhanced VLDL/LDL secretion may be the predominant etiology of hypercholesterolemia11. The proposal that LDLR deficiency results in enhanced LDL catabolism remains controversial because of conflicting results obtained from multiple patient and animal studies15-18. One problem is that direct study of LDL catabolism in FH patients has been somewhat limited because of the difficulty in obtaining primary LDLR–deficient human hepatocytes. Additionally, studies using human hepatocellular carcinoma cells (e.g. HepG2) are confounded because these cells are deficient in lipoprotein production, requiring investigators to use transient incubation with exogenous lipids or drugs to coax them toward a more physiologically normal state. Using ELISA to detect lipidated ApoB-100, we confirmed that hepatocytes derived from both control and ‘JD’ hESC/hiPSC actively secrete VLDL/LDL (Fig. 4a). Strikingly, ‘JD’ iPS-derived hepatocytes displayed an approximate 8–fold increase in the level of secreted ApoB-100 compared with hepatocytes derived from 3 genetically independent control pluripotent stem cell lines across 3 independent differentiation experiments (JD: 1484 ng/ml, Control:173 ng/ml, p<0.001). When we controlled for the efficiency of hepatocyte differentiation by normalizing secreted lipidated ApoB-100 concentration to human albumin concentration, similar results were obtained (‘JD’: 6034 ng/ml, Control: 1123 ng/ml, p<.001). Continued sampling from ES/iPS-derived hepatocyte cultures beyond day 20 of differentiation revealed that secretion of lipidated ApoB-100 is maintained for at least 7 days and that the elevated ApoB-100 concentration associated with the ‘JD’ background is preserved throughout this period (Fig. 4b). Previous reports studying rodent hepatocytes have documented that increases in VLDL/LDL secretion in Ldlr–/– hepatocytes is determined by the amount of ApoB that circumvents post-translation degradation rather than by changes in gene expression17. Consistent with this finding, no significant difference in APOB mRNA levels was observed between control and ‘JD’ hepatocytes (p=0.54, Fig. 4c).
Fig. 4. ApoB-100 secretion is elevated in ‘JD’ iPSC-derived hepatocytes.
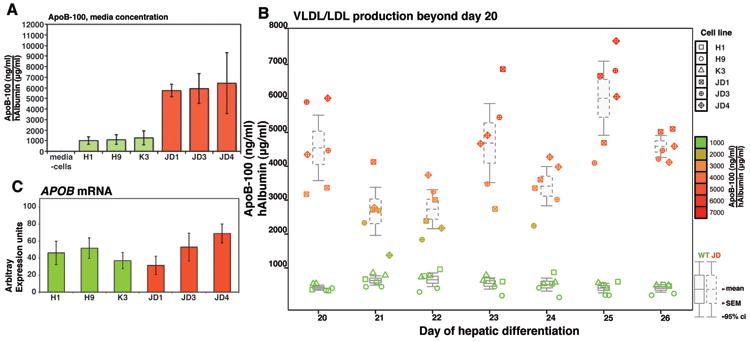
(a) Bar graphs showing concentration of lipidated ApoB secreted into the medium by control (H1, H9, and K3) (green boxes) or ‘JD’ (JD1, JD3, JD4) (red boxes) iPSC-derived hepatocytes as determined by ELISA (n=3 independent differentiations). No significant difference was observed within control or mutant groups; however, secreted ApoB-levels were significantly elevated in ‘JD’ compared to control hepatocytes (p<0.05 Tukey-Kramer post hoc multiple comparison test). (b) Box and whisker plot showing that elevated ApoB production is maintained in hepatocytes derived from ‘JD’ iPSCs compared to control hepatocytes after extended culture (days 20 – 26) of the differentiated cells. (c) Bar graph showing level of APOB mRNA in hepatocytes derived from control (H1, H9, and K3) (green boxes) or ‘JD’ (JD1, JD3, JD4) (red boxes) pluripotent stem cells as determined by qRT-PCR analyses. APOB mRNA levels were not significantly different between cell lines (ANOVA, p>0.05).
Discussion
The idea of using hiPSCs to model diseases in culture is not novel19-21. Rashid and colleagues made a significant advance in generating iPSCs from patients with several liver disorders including alpha-1 anti-trypsin deficiency (A1ATD), glycogen storage disease type 1a (GSD1a), familial hypercholesterolemia (FH), Crigler-Najjar syndrome type 1, and hereditary tyrosinemia. However, due to the large number of disease specific lines that were generated, a detailed characterization of each was beyond the scope of that study. With regard to FH, Rashid and colleagues limited their analysis to the ability of differentiated FH iPSCs to internalize LDL7. The LDLR is ubiquitously expressed and so determining LDL uptake, while important, does not address the pathophysiology of FH, which is primarily a consequence of deficiencies in both catabolism and metabolism of cholesterol specifically by the hepatocyte. Whether patient-specific iPSCs could be used to faithfully recapitulate complex metabolic disorders associated with hepatocyte function therefore remained unaddressed8. Several caveats that can affect efficiency of using iPSCs to study complex metabolic disorders need to be considered. For example, although the generation of hiPSCs from somatic cells can be relied upon, the procedure yields iPSC populations that are heterogeneous in nature22. In addition, the generation of iPS cells has been found to result in the presence of somatic mutations within the iPSC population23,24 and some iPSCs retain aspects of the epigenetic profile of donor cells25,26, both of which could impact the interpretation of data generated from iPSC–derived hepatocytes. Perhaps most importantly, however, hepatocytes derived from iPSCs fail to express the full repertoire of genes encoding proteins associated with mature hepatocyte function. The fact that not all hepatocyte mRNAs are expressed is especially concerning given that lipid and cholesterol homeostasis is strictly dependent upon a multitude of interactions that involve metabolic enzymatic activity, gene expression, and protein trafficking. To determine the feasibility of using iPSCs to model metabolic liver disease we therefore chose to focus on a well–defined mutation that was inherited in Mendelian fashion. To control for variations associated with reprogramming we performed our analyses on multiple independent ‘JD’ iPSC clones and we compared our data to genetically distinct ES cell and iPSC lines. We believe our data convincingly show that key features of FH in cultures of ‘JD’ iPSC–derived hepatocytes can be recapitulated and therefore conclude that it will be feasible to use patient–specific iPSCs to elucidate the functional contribution of allelic variations that potentially affect control of cholesterol and lipid flux. While some genetic variations may manifest through hepatocyte–independent processes, given the central role of the liver in control of serum lipid and cholesterol levels, it seems likely that the majority of functional polymorphisms will affect hepatocyte metabolism. Although all of this is encouraging, in other studies we have found variations in differentiation efficiency exist amongst huESCs and hiPSCs, which add a significant complication to experimental interpretation. It is, therefore, important to note that all of the pluripotent stem cells used in the current study were chosen because they displayed a similar efficiency in their capacity to generate hepatocytes and we believe that this is an important variable to consider if patient specific iPSCs are to be used to probe disease mechanisms.
As expected the ‘JD’ hepatocytes exhibited reduced LDL uptake, however, the most striking change was a reproducible increase in ApoB-100/VLDL secretion, which is consistent with several studies suggesting that plasma LDL-C concentrations may be significantly impacted by the VLDL production rate in FH patients15,27. The evidence describing the relationship between LDLR mutations and LDL-C catabolism by hepatocytes has in some cases been contradictory. Loss of functional LDLR in primary mouse hepatocytes can result in elevated hepatic secretion of ApoB-10017, which is exacerbated in Ldlr–/– hepatocytes that overexpress sterol regulatory element binding protein-1a (SREBP1a)16. However, in other studies ApoB-100 production was unaffected in Ldlr–/– mice18 and similar results were obtained in the LDLR defective WHHL rabbit28. The reason for such discrepancies remain obscure but they may reflect variations in animal models being studied, inherent differences in response of cultured hepatocytes compared to liver cells in a more complex in vivo environment, and perhaps most importantly, the nature of the LDLR mutations present in each model and whether or not these mutations encode proteins that are functionally null or inert. The mechanisms relating LDLR function to Apo-B100/VLDL secretion are complex; however, it has been proposed that variances in intracellular pools of cholesterol may impact ApoB-100 presecretory degradation17,29. Although our data are consistent with this view, preliminary analyses of changes in mRNA levels (not shown) in ‘JD’ hepatocytes have revealed that diverse aspects of cholesterol metabolism, catabolism and transport may be coordinately regulated at the level of gene expression and appear tightly linked to cholesterol flux. We believe that future analyses of iPSC–derived hepatocytes from FH patients with distinct LDLR alleles will likely enhance our understanding of the molecular mechanisms that link LDLR function to LDL catabolism.
Finally, treatment of elevated cholesterol levels has relied heavily on the use of statins that inhibit HMG-CoA reductase activity. Statins act both by reducing cholesterol synthesis and elevating cholesterol uptake by increasing expression of the LDLR in hepatocytes. Although statins can be highly efficacious, there is a surprisingly wide variation of effectiveness between individuals, with >20% of patients showing a poor response to statin treatment30. The pharmacogenetics of statin action are highly complex and involve a large repertoire of regulators and unsurprisingly several polymorphisms have been described that are associated with poor responders31. We propose that the generation of hepatocytes from hiPSCs from individuals that exhibit a differential statin response and display elevated lipid/cholesterol levels could be valuable in the search for novel cholesterol lowering drugs. In this regard, our finding that control hiPSC–derived hepatocytes could respond to lovastatin treatment by effectively increasing LDL–C uptake is extremely encouraging if one is to consider using iPSC–derived hepatocytes as a platform for drug discovery. As an alternative to drug screens, it has been proposed that gene therapy could be applied to iPSCs thereby providing an exogenous supply of ‘repaired’ hepatocytes that could potentially be used to reverse at least a subset of metabolic liver disorders32. Although there are many significant hurdles that need to be overcome before iPSC–derived hepatocytes could be used as a therapeutic cell source, precise genome editing through zinc finger or TALEN technologies32,33 could be valuable in confirming whether a given SNP is associated with a specific functional consequence in iPSC–derived hepatocytes.
In conclusion, our data show that if appropriate controls are included, hepatocytes derived from ‘JD’ iPS cells recapitulate key aspects of FH in culture, which supports the proposal that patient specific iPSC–derived hepatocytes will become an important tool to dissect the contribution of GWAS lipid loci in controlling hepatic lipid and cholesterol metabolism. In addition, our finding that the hiPSC–derived hepatocytes are competent to respond to hepatoselective pharmaceuticals implies that patient specific iPS cell–derived hepatocytes will facilitate the identification of drugs that can treat inborn errors of liver metabolism.
Supplementary Material
Supplemental Fig. 1 Genotype of the LDLR gene in control and ‘JD’ pluripotent stem cells. (a) PCR of genomic DNA using primers that span deletion FH381 in the LDLR gene yields the expected 174 bp amplicon exclusively in JD iPSCs (JD1, JD3, JD4). This product is not amplified from control cells (H1 and H9 ESC lines and K3 iPSC line) due to the presence of 5kb of intervening sequence that lies between the amplification primers. (b) DNA sequence analyses confirmed the presence of the ‘JD’ paternal LDLR mutation (FH382, A-G transition) in genomic DNA from ‘JD’ fibroblasts and iPSCs, whereas only the wild type SNP was detected in control pluripotent stem cells (H1, H9 and K3).
{kind=link}
Supplemental Fig. 2 FL-LDL uptake in ‘JD’ iPSC derived hepatocytes. Following differentiation and overnight incubation with 0.5μM lovastatin, control and ‘JD’ stem cell-derived hepatocytes were incubated for 30 mins with FL-LDL. Cells were fixed and the location of FL-LDL was identified by confocal microscopy. The cell location and plate orientation was marked and hepatocyte transcription factor HNF4a (red) was identified in the nuclei of the same cells by immunocytochemistry. Scale bar = 50μM.
{kind=link}
Supplemental Fig. 3 qRT-PCR analyses of mRNAs encoding proteins involved in the activation of lovastatin lactone. Bar graphs showing that no significant difference in the level of mRNAs encoding carboxyesterases 1 and 2 (CES1, CES2), paraoxonases 2 and 3 (PON2, PON3), or CYP3A4 were detected between control (green bars) and ‘JD’ (red bars) hepatocytes. Error bars = ±SEM, n = 3 independent differentiations. Significance determined by 2-sided t-test, independent samples.
{kind=link}
Supplemental Fig. 4 qRT-PCR analyses of mRNAs encoding sterol response element binding proteins (SREBP) (a) Bar graphs showing relative levels of SREBP1 and SREBP2 mRNAs in control (green bars) and ‘JD’ (red bars) hepatocytes that were determined by qRT-PCR. No significant difference in mRNA levels was identified between control and ‘JD’. (b) Bar graphs showing levels of SREBP1 and SREBP2 mRNA in control (green bars) and ‘JD’ (red bars) hepatocytes following lovastatin treatment. All values were normalized to the levels of HNF4a mRNA to account for subtle variances in differentiation efficiency. Graphs are presented as the change in mRNA level found in lovastatin treated cells compared to untreated cells. Error bars = ±SEM, n = 3 independent differentiations for all samples except H9 +lovastatin and JD1 +lovastatin, where n=2. Significance determined by 2-sided t-test, independent samples.
{kind=link}
Supplemental Fig. 5 Rate of Fl-LDL uptake in iPSC– and ESC–derived hepatocytes. Hepatocytes generated from H1 (open triangles) or H9 (solid squares) ESCs were washed and labeled with FL-LDL for the indicated time at 37 °C at which point cells were treated with heparin and collected for flow cytometry. Error bars = SD, n = 2 biological replicates per cell line per time point.
{kind=link}
Acknowledgments
Authors would like to thank Dr. Paula North for analyses of teratomas, Tom Wagner for technical support and Brian Link for advice with confocal analyses.
Funding for this project was provided by National Institutes of Health to S.A.D. (DK55743, DK087377, HL094857, HG006398), M.A.C. (F30 DK091994) and F.K.N. (F31 AA019874), JDRF Fellowship (3-2010-497) to J.C., and additional support from Advancing a Healthier Wisconsin Fund, the Marcus Family, Phoebe R. and John D. Lewis Foundation, the Sophia Wolf Quadracci Memorial Fund, and the Dr. James Guhl Memorial Fund.
References
- 1.Rosamond W, Flegal K, Friday G, et al. Heart disease and stroke statistics--2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 2.Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Si-Tayeb K, Noto FK, Nagaoka M, et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sullivan GJ, Hay DC, Park IH, et al. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010;51:329–335. doi: 10.1002/hep.23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song Z, Cai J, Liu Y, et al. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res. 2009;19:1233–1242. doi: 10.1038/cr.2009.107. [DOI] [PubMed] [Google Scholar]
- 7.Rashid ST, Corbineau S, Hannan N, et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest. 2010;120:3127–3136. doi: 10.1172/JCI43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soto-Gutierrez A, Tafaleng E, Kelly V, et al. Modeling and therapy of human liver diseases using induced pluripotent stem cells: how far have we come? Hepatology. 2011;53:708–711. doi: 10.1002/hep.24143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 11.Sniderman AD, De Graaf J, Couture P, et al. Regulation of plasma LDL: the apoB paradigm. Clin Sci (Lond) 2010;118:333–339. doi: 10.1042/CS20090402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis CG, Lehrman MA, Russell DW, et al. The J.D. mutation in familial hypercholesterolemia: amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell. 1986;45:15–24. doi: 10.1016/0092-8674(86)90533-7. [DOI] [PubMed] [Google Scholar]
- 13.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 14.Delaforest A, Nagaoka M, Si-Tayeb K, et al. HNF4A is essential for specification of hepatic progenitors from human pluripotent stem cells. Development. 2011;138:4143–4153. doi: 10.1242/dev.062547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tremblay AJ, Lamarche B, Ruel IL, et al. Increased production of VLDL apoB-100 in subjects with familial hypercholesterolemia carrying the same null LDL receptor gene mutation. J Lipid Res. 2004;45:866–872. doi: 10.1194/jlr.M300448-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Horton JD, Shimano H, Hamilton RL, et al. Disruption of LDL receptor gene in transgenic SREBP-1a mice unmasks hyperlipidemia resulting from production of lipid-rich VLDL. J Clin Invest. 1999;103:1067–1076. doi: 10.1172/JCI6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Twisk J, Gillian-Daniel DL, Tebon A, et al. The role of the LDL receptor in apolipoprotein B secretion. J Clin Invest. 2000;105:521–532. doi: 10.1172/JCI8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Millar JS. Normal Production Rate of Apolipoprotein B in LDL Receptor-Deficient Mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:989–994. doi: 10.1161/01.atv.0000018304.30943.06. [DOI] [PubMed] [Google Scholar]
- 19.Park IH, Arora N, Huo H, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebert AD, Yu J, Rose FFJ, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soldner F, Hockemeyer D, Beard C, et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mikkelsen TS, Hanna J, Zhang X, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gore A, Li Z, Fung HL, et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayshar Y, Ben-David U, Lavon N, et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 25.Kim K, Zhao R, Doi A, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol. 2011;29:1117–1119. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Millar JS, Maugeais C, Ikewaki K, et al. Complete deficiency of the low-density lipoprotein receptor is associated with increased apolipoprotein B-100 production. Arterioscler Thromb Vasc Biol. 2005;25:560–565. doi: 10.1161/01.ATV.0000155323.18856.a2. [DOI] [PubMed] [Google Scholar]
- 28.Hornick CA, Kita T, Hamilton RL, et al. Secretion of lipoproteins from the liver of normal and Watanabe heritable hyperlipidemic rabbits. Proc Natl Acad Sci U S A. 1983;80:6096–6100. doi: 10.1073/pnas.80.19.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson GR, Naoumova RP, Watts GF. Role of cholesterol in regulating apolipoprotein B secretion by the liver. J Lipid Res. 1996;37:439–447. [PubMed] [Google Scholar]
- 30.Voora D, Shah SH, Reed CR, et al. Pharmacogenetic predictors of statinmediated low-density lipoprotein cholesterol reduction and dose response. Circ Cardiovasc Genet. 2008;1:100–106. doi: 10.1161/CIRCGENETICS.108.795013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voora D, Shah SH, Spasojevic I, et al. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J Am Coll Cardiol. 2009;54:1609–1616. doi: 10.1016/j.jacc.2009.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yusa K, Rashid ST, Strick-Marchand H, et al. Targeted gene correction of alpha(1)-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011 doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hockemeyer D, Wang H, Kiani S, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1 Genotype of the LDLR gene in control and ‘JD’ pluripotent stem cells. (a) PCR of genomic DNA using primers that span deletion FH381 in the LDLR gene yields the expected 174 bp amplicon exclusively in JD iPSCs (JD1, JD3, JD4). This product is not amplified from control cells (H1 and H9 ESC lines and K3 iPSC line) due to the presence of 5kb of intervening sequence that lies between the amplification primers. (b) DNA sequence analyses confirmed the presence of the ‘JD’ paternal LDLR mutation (FH382, A-G transition) in genomic DNA from ‘JD’ fibroblasts and iPSCs, whereas only the wild type SNP was detected in control pluripotent stem cells (H1, H9 and K3).
Supplemental Fig. 2 FL-LDL uptake in ‘JD’ iPSC derived hepatocytes. Following differentiation and overnight incubation with 0.5μM lovastatin, control and ‘JD’ stem cell-derived hepatocytes were incubated for 30 mins with FL-LDL. Cells were fixed and the location of FL-LDL was identified by confocal microscopy. The cell location and plate orientation was marked and hepatocyte transcription factor HNF4a (red) was identified in the nuclei of the same cells by immunocytochemistry. Scale bar = 50μM.
Supplemental Fig. 3 qRT-PCR analyses of mRNAs encoding proteins involved in the activation of lovastatin lactone. Bar graphs showing that no significant difference in the level of mRNAs encoding carboxyesterases 1 and 2 (CES1, CES2), paraoxonases 2 and 3 (PON2, PON3), or CYP3A4 were detected between control (green bars) and ‘JD’ (red bars) hepatocytes. Error bars = ±SEM, n = 3 independent differentiations. Significance determined by 2-sided t-test, independent samples.
Supplemental Fig. 4 qRT-PCR analyses of mRNAs encoding sterol response element binding proteins (SREBP) (a) Bar graphs showing relative levels of SREBP1 and SREBP2 mRNAs in control (green bars) and ‘JD’ (red bars) hepatocytes that were determined by qRT-PCR. No significant difference in mRNA levels was identified between control and ‘JD’. (b) Bar graphs showing levels of SREBP1 and SREBP2 mRNA in control (green bars) and ‘JD’ (red bars) hepatocytes following lovastatin treatment. All values were normalized to the levels of HNF4a mRNA to account for subtle variances in differentiation efficiency. Graphs are presented as the change in mRNA level found in lovastatin treated cells compared to untreated cells. Error bars = ±SEM, n = 3 independent differentiations for all samples except H9 +lovastatin and JD1 +lovastatin, where n=2. Significance determined by 2-sided t-test, independent samples.
Supplemental Fig. 5 Rate of Fl-LDL uptake in iPSC– and ESC–derived hepatocytes. Hepatocytes generated from H1 (open triangles) or H9 (solid squares) ESCs were washed and labeled with FL-LDL for the indicated time at 37 °C at which point cells were treated with heparin and collected for flow cytometry. Error bars = SD, n = 2 biological replicates per cell line per time point.