

Abstract
Polycyclic aromatic hydrocarbons (PAHs) are among the most prevalent environmental pollutants and result from the incomplete combustion of hydrocarbons (coal and gasoline, fossil fuel combustion, byproducts of industrial processing, natural emission, cigarette smoking, etc.). The first phase of xenobiotic biotransformation in the PAH metabolism includes activities of cytochrome P450 from the CYP1 family and microsomal epoxide hydrolase. The products of this biotransformation are reactive oxygen species that are transformed in the second phase through the formation of conjugates with glutathione, glucuronate or sulphates. PAH exposure may lead to PAH-DNA adduct formation or induce an inflammatory atherosclerotic plaque phenotype. Several genetic polymorphisms of genes encoded for enzymes involved in PAH biotransformation have been proven to lead to the development of diseases. Enzyme CYP P450 1A1, which is encoded by the CYP1A1 gene, is vital in the monooxygenation of lipofilic substrates, while GSTM1 and GSTT1 are the most abundant isophorms that conjugate and neutralize oxygen products. Some single nucleotide polymorphisms of the CYP1A1 gene as well as the deletion polymorphisms of GSTT1 and GSTM1 may alter the final specific cellular inflammatory respond. Occupational exposure or conditions from the living environment can contribute to the production of PAH metabolites with adverse effects on human health. The aim of this study was to obtain data on biotransformation and atherosclerosis, as well as data on the gene polymorphisms involved in biotransformation, in order to better study gene expression and further elucidate the interaction between genes and the environment.
Keywords: atherosclerosis, biotransformations, genetic polymorphism, polycyclic aromatic hydrocarbons
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are among the most prevalent environmental pollutants known to be involved in carcinogenesis (1). These are aromatic hydrocarbons with 3 and more aromatic rings. The best known PAHs are naphthalene, benz(a)pyrene (b[A]P), phenantrene, and anthracite. PAHs mainly result from fossil fuel combustion, as byproducts of industrial processing, human activity, industrial emission, and natural emission (2,3). The main channels through which PAHs enter the environment are coal production and processing, crude oils, natural gases, production of heavy and light metals, and waste incineration.
There are over a hundred of various PAHs, but in practice, only six to sixteen are interesting for analyses and monitoring (4).
Environmental sources of PAH exposure and bioaccumulation
PAHs can be found as air pollutants associated with dust particles, in water, and in the soil and sediments. Air streaming can extend PAHs to large distances and subsequently return them to the soil through precipitation. In the soil, we find them bound to bigger particles, from where some evaporate in the air, while others penetrate into deeper layers contaminating underground water (5). The general population is exposed to PAHs from the environment through small-sized respirable particles, water and food, as well as from the occupational environment (6). The most widespread manner of PAH input is the inhalation of exhaust gases, tobacco smoke, and the usage of products and materials containing PAHs (7). Contrary to popular opinion, alimentary exposure causes a higher rate of human exposure to PAHs than air pollutants do (8). PAH bioaccumulation affects mostly adipose tissue, kidneys, and the liver (9). Smaller amounts may accumulate in the spleen, suprarenal glands, and ovaries. PAHs are excreted in urine or bile in the form of glucoconjugates, sulfoconjugates, and gluthatione-conjugates.
Biomolecular and patho-biochemical effects of PAH exposure in humans
PAHs accumulation may have an effect on human health. These compounds have toxic, mutagenic, and teratogenic effects. The PAH metabolism in the first phase of xenobiotic biotransformation includes cytochrome P450 from the CYP1 family and microsomal epoxide hydrolase, which may lead to the production of reactive oxygen species (ROS) (Figure 1). Upon chemically binding to DNA, reactive oxygen species give rise to DNA adducts with very different structures and biological activities (10). Lipids and proteins can also be affected by PAH metabolites. Some subgroups of PAHs and halogenated aromatic hydrogen carbons may even pass the placental barrier (11), which can have a negative impact on fetal cognitive development and cause undesirable consequences to fetal health. PAH exposure also showed to have an important role in atherosclerotic ethiopathology, particularly through the PAH-induced activities of biotransformation enzymes. PAHs from particulate matter (bacterial contaminants, transition elements, salts, and carbonaceous material) may increase monocyte cell adhesion to human aortic endothelia as well as the attenuation of cytokines (12). The variation of genes that encode enzymes involved in PAH-biotransformation may ultimately alter enzyme activity and consequently be involved in the pathobiochemistry of different disorders.
FIGURE 1.
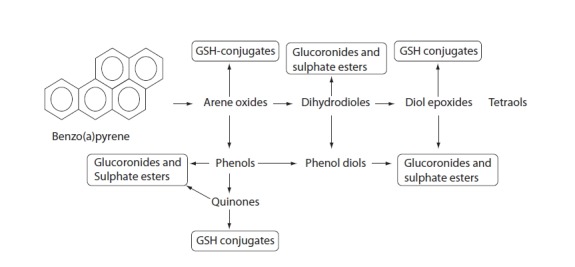
Biotransformation of benzo[a]pyrene.
PAHs are metabolized in first phase of xenobiotic biotransformation to form several phenols (hydroxy derivatives), phenol diols, dihydrodiols, quinones, and reactive diol-epoxide’s enantiomers. The reactive species are then conjugated to GSH-conjugates glucuronides and sulphate esters to enable better excretion and detoxification of metabolized xenobiotics owing to increased hydrophilicity.
Atherogenesis and PAH exposure
Many sources from the literature have presented the relationships between genetic polymorphisms of gene-encoded enzymes involved in PAH biotransformation and carcinogenesis. The data on atherogenesis are not as abundant.
Atherosclerosis is a process that starts early in a person’s life, but in adults, it is enhanced by various factors. Besides the well-established risk factors involved in atherogenesis (age, sex, hyperlipidemia, hypertension, diabetes mellitus, obesity, lack of physical activity and heredity) (13), many other environmental factors contribute to atherogenesis.
The study of interactions between genes and the environment will surely lead to a better understanding of numerous diseases. In this case, exposure to pollutants like PAHs induces specific gene expressions that lead to the biosynthesis of enzymes, which in turn may alter cellular metabolic processes. The diversity of environmental factors and the individual human sensitivity to exposure to these factors may be crucial in the mechanisms responsible for atherogenesis. The polymorphisms of genes that encode enzymes of PAH biotransformation may alter the final specific cellular respond. Thus far, two theories on the mechanisms through which PAHs are involved in atherosclerotic processes have been postulated: 1) formation of DNA-adducts, and 2) involvement of PAHs and its metabolites in the inflammatory processes of atherogenesis (14,15). These theories are mostly based on in vitro and animal model experiments.
A summary of the data obtained in the fields of biotransformation and atherosclerosis as well as of the data on gene polymorphisms involved in biotransformation may provide us with a clearer idea of the interactions between genes and the environment. The general aim of this review is to present the substantial data on PAH exposure as a factor in the development of atherosclerosis. The focus points of this paper include: 1) activation of PAH biotransformation, biotransformation mechanisms, production of specific metabolites that may increase the risk of atherosclerosis; 2) the most frequent genetic polymorphisms of genes involved in PAH biotransformations and atherosclerosis presented in biomedical literature; 3) the influence of certain common genetic variants of genes involved in xenobiotic biotransformations on the development of coronary artery disease (CAD); 4) an overview of different population studies related to the association of common genetic polymorphisms and atherosclerosis; and 5) gene–environment mechanisms involved in the process of atherosclerosis development.
PAH biotransformation
The function of the receptor aryl hydrocarbon (AhR)
PAHs such as (B(a)P) are known as potent ligands of AhR. These molecules are mediators for increasing intracellular calcium concentrations (16). AhR recognizes a large number of xenobiotics, such as PAHs and dioxins, and activates several metabolic and detoxification pathways (17). AhR acts as transcriptional factor and mediates intracellular response through regulating genetic expression. AhR induces cytochrome P450 (Cyp)1a1 expression, which corresponds well with the induction of apoptosis and mutagenesis (18). After entering into the cell, PAHs bind to the AhR and the complex PAH-AhR is translocated into the nucleus. The expression of the CYP1A1 gene is enhanced by the xenobiotic responsive element (XRE) from the promoter region of the CYP1A1 gene, the aryl hydrocarbon nuclear translocator (ARNT), the xenobiotic responsive element (XRE) and AhR (19,20).
PAHs are metabolized in enzymatic reactions catalyzed by several different enzymes, including cytochrome P450 (CYP), epoxyde hydrolase (EPH), glutathione transferase (GST), UDP-glucuronosyl transferase (UGT), and sulfotransferase (SULT) (21).
First phase of PAH metabolism
The most abundant isoforms of CYP induced by non-halogenated PAHs are those from the CYP1A subfamily (22). Specific CYP families may be induced by different chemicals as presented in Table 1.
TABLE 1.
Different CYP families induced by different chemical inducers (19).
| Inducer | Source of inducer | Cyp subfamily induced |
|---|---|---|
| Nonhalogenated polycyclic aromatic hydrocarbons | Petroleum derivatives and coal-tar constituents | CYP1A |
| Polyhalogenated aromatic hydrocarbons dioxins | Forest fires, volcano activities, industrial processes, waste incretion | Predominately CYP1A or predominately CYP2B or mixed |
| DDT, dieldrin, chlordane, and mirex | Chlorinated hydrocarbon pesticides | CYP2B and to a lesser extent the CYP3A |
CYP - cytochrome P450; DDT - dichlorodiphenyltrichloroethane
After exposure, PAHs are metabolized to form several phenols (hydroxy derivatives), dihydrodiols, quinones, and reactive diol-epoxide enantiomers (Figure 1) that have the capacity to bind DNA and form PAH–DNA adducts (1,23).
Second phase of PAH metabolism
Covalent additions of sugars, amino acids and peptides, as endogenous ligands, are the major reactions involved in the second phase of xenobiotic transformation (24).
The enzymes from different superfamilies enable conjugation (e.g., with glutathione or glucuronic acid). Several polar compounds are produced and thereby the excretion of chemicals is facilitated by the formation of products with more polar groups. Enzyme superfamilies involved in conjugation reactions are different transferases like SULT, UGT, GST, and N-acetyltransferases (NAT) (25,26). Beside transferases, oxidoreductases NAD(P)H:quinone oxidoreductase (NQO) and NAD(P)H: menadione reductase (NMO), hydrolases such as EPH are also involved in the second phase of the xenobiotic metabolism (25,26). The gene expression of drug metabolizing enzyme (DMEs) genes provides the various isoforms of the enzymes with different induction and leads to inhibition by xenobiotics, different substrate specificity, and different expression (27). Conjugation with DMEs in the second phase of xenobiotic metabolism enables better excretion and detoxification of metabolized xenobiotics owing to increased hydrophilicity (28). Sometimes, the second phase produces activated metabolites with increased toxicity (29). The expression of the second phase can be induced by numerous structurally unrelated chemicals.
Human GSTs in the second phase of the PAH metabolism
Human GSTs are divided into three main families: cytosolic, mitochondrial, and membrane-bound microsomal (30). The cytosolic GST (cGSTs) super-family, which exists predominantly in the liver but is also widely distributed into various tissues, encompasses a number of isoenzymes. Glutathione is a cytosolic nucleophilic tripeptide that conjugates epoxides and other reactive intermediates via GSTs and is used in the prevention of oxidative stress (31). Oxidative stress is well-known to be the basis of atherosclerotic pathology.
cGSTs alpha (GSTA), mu (GSTM), pi (GSTP), sigma (GSTS), omega (GSTO), zeta (GSTZ) and theta (GSTT) are the most ubiquitous isoforms(32). Isoenzymes are grouped according to substrate specificity, amino acid sequence, and immunological cross-reactivity (30).
CYP1A1 and GSTs µ and θ as major factors in PAH-biotransformations and their influence on atherogenesis
CYP1A1 and atherogenesis
The enzyme CYP1A1, which is encoded by the CYP1A1 gene, is the most important in the monooxygenation of lipofilic substrates such as PAHs. The most common PAH, as a substrate for CYP1A1 in this subgroup, is B(a)P, a carcinogen compound found in tobacco smoke (33). As one of the main enzymes for detoxification, CYP1A1 is involved in the mechanisms of carcinogenesis and atherogenesis induced by PAHs from cigarette smoke (34).
There are at least two theories that explain the involvement of PAHs in atherogenesis. PAH-DNA adducts are known to cause DNA mutations involved in carcinogenesis. It is hypothesized that there is an interrelationship between PAH exposure and PAH-DNA adducts in aortae. Chemically induced arterial DNA damage may induce mutations that lead to cellular transformations into proliferative clones (14).
On the other hand, the inflammatory atherosclerotic plaque phenotype can be induced by PAHs independently of the formation of their DNA-adducts. A study performed on an animal model of apoE-KO mice clearly demonstrated this (15).
In vitro examination showed that CYP1A1 activates PAHs. The suppression of LXR-mediated (liver X-receptor) signal transductions ensures PAHs direct involvement in atherosclerotic processes (35). LXR are ligand-gated transcriptional factors that belong to a superfamily of nuclear receptors. These receptors act as key regulators of lipid metabolism and inflammation (36). The regulation of the lipid metabolism by LXR is provided by the regulation of intestinal cholesterol absorption through the ATP-binding cassette (ABC) gene family (37). On the other hand, the regulation of inflammatory events is evident from an animal model experiment which showed that B[a]P induces monocyte chemo-attractant protein (MCP-1) gene expression in the aortic tissue of ApoE-/- mice (38). Monocytes travel to damaged areas (atherosclerotic lesions) facilitated by MCP-1. In addition, CYP1A1 is also involved in the production of the arachidonic acid-derived vasoactive substance which also may be involved in proinflammatory processes (39).
GSTM1 and GSTT1 convert electrophilic pro ducts of the first phase of PAH biotransformation
GSTM1 and GSTT1 are enzymes from the human cytosolic GSTs superfamily, which contains 8 distinct classes (40). These two enzymes are encoded by the GSTM and GSTT genes, respectively. The main role of GSTM1 lies in the detoxification of electrophilic xenobiotics such as benzopyrene diol-epoxide, the products of CYP1A1, as well as other xenobiotic and environmental pollutants (41). The main role of the GSTT1 isoform is the conjugation of oxidized lipids and halogenated compounds. The safe removal of toxins by conjugation with glutathione saves cells from oxidative damage and DNA mutation, potentially altering the rate of cellular sensibility. However, recent findings have shown that GSTs may act via non-enzymatic pathways and be involved in cell signaling through MAP kinase, which includes DNA damage signaling (42).
Mapping CYP1A1, GSTM1 and GSTT1 genes and their common genetic variants
CYP1A1 gene
The human gene CYP1A1 locus has been mapped on chromosome 15q24.1, which is composed of 7 exons (43). Several common genetic polymorphisms of the CYP1A1 gene (Table 2) have been described in the literature and designated as m1, m2 m3 and m4. The polymorphism Cyp1A1-Msp I or m1 represents 3801 T>C substitution in the 3’ non-coding region of the CYP1A1 gene (rs4646903), which determines translation and mRNA stability (43,44). Nucleotide substitution 2455 A>G (m2) results in amino acid substitution, Ile462Val (rs1048943) (44). The m3 variant symbolizes nucleotide substitution 3205 T>C in intron 7 (rs4986883) (43,45), while nucleotide substitution 42453 C>A represents variant m4, which results in the amino acid substitution Thr>Asp, (rs1799814) (46). Investigations of CYP1A1 gene polymorphisms mostly indicated their causal role in carcinogenesis, while data on the association between polymorphisms and atherogenesis are lacking. The CYP1A1-Msp I genetic polymorphism is mostly presented in the literature as an enhancer of human predisposition to CAD.
TABLE 2.
The most common CYP1A1, GSTM, and GSTT polymorphisms.
| Gene | Polymorphism | The main observations related to polymorphism (association) |
|---|---|---|
| CYP1A1 | m1 = 3801 T>C | Lung cancer, CAD |
| m2 = 2455 A>G; Ile>Val | Lung cancer, breast cancer | |
| m4 = 2453 C>A; Thr>Asp | Lung cancer | |
|
| ||
| GSTM1 | deletion | Oxidative stress, decreased enzyme activities, cancer, CAD |
| GSTT1 | deletion | |
CYP - cytochrome P450; GSTM1 - glutathione S-transferase M1; GTST1 - glutation S-transferase T1
GSTM1 and GSTT1 genes
The GSTM1 and GSTT1 loci have been mapped on chromosome 1p13.3 and 22q11.2, respectively. μ (GSTM1) and θ (GSTT1), as members of the GST family, most commonly exhibit deletion polymorphisms (Table 2). The complete deletion of the gene (homozygotes) is a consequence of the absolute absence of both alleles (‘null’ alleles-GSTM1*0 or GSTT1*0), which results in a deficit of enzyme activity. Heterozygotes with only one ‘null’ allele usually have reduced enzyme activity (47–49).
As GSTT1 and GSTM1 play a role in the deactivation of reactive oxygen species, which are involved in cellular inflammation and degenerative disorders, it has been shown that polymorphic isoforms with reduced enzyme activity might contribute to these processes in the cell (50). Genetic polymorphisms can increase or decrease the sensibility of an organism to cancerogenesis and the inflammatory processes involved in the development of atherosclerosis (30).
The influence of CYP1A1, GSTM1 and GSTT1 genetic variants on the development of CAD
An overview of population studies
Data on the association of atherosclerosis and CYP1A1, GSTM1 and GSTT1 polymorphisms are lacking and controversial due to aspects such as study design, population, and number of subjects examined. Several case–control studies have observed the association between the genetic polymorphisms CYP1A1, GSTT1 and GSTM1 and CAD. An overview of these studies is presented in Table 3.
TABLE 3.
An overview of different population studies related to association of common CYP1A1 and GST genetic polymorphisms with CAD.
| Author | Year | Origin | Study design | Polymorphism | Subjects | Major observation |
|---|---|---|---|---|---|---|
| Taspinar et al. (51) | 2012 | Turkish | Case-control | CYP1A1-m1 GSTT1 deletion |
132 CAD 151 control |
8.907-fold increased CAD risk in subjects with polymorphisms |
| Moon et al. (39) | 2007 | Korean | Case-control | CYP1A1-m1 GSTT1 deletion |
353 CI 376 control |
GSTM1 ‘null’ genotype increased the relative risk for the CI in the subjects with the CYP1A1- m1-C allele |
| Wang et al. (52) | 2002 | Australian | Cross-sectional | C of allele CYP1A1 –m1 |
701 TVD | C of allele CYP1A1 Msp I polymorphism had an increased risk for triple-vessel disease |
| Nørskov et al. (54) | 2011 | Danish | Meta-analysis | GSTT1 deletion GSTM1 deletion |
23 059 GP 4930 IHD 2086 ICD |
No association between polymorphisms and markers of inflammation or oxidation in the general population |
| Bazo et al. (55) | 2011 | Brazilian | Case-control | GSTT1 deletion GSTM1 deletion |
299 CAD 101 control |
No association between polymorphisms and coronary atherosclerosis CAD |
| Maciel et al. (56) | 2009 | Brazilian | Cross-sectional | GSTT1 deletion GSTM1 deletion |
1577 GP White, Black and Mulatto | Significant association of TGs, HDL-cholesterol and the TG/HDL ratio with polymorphisms |
| Manfredi et al. (57) | 2009 | Italian | Case-only | GSTT1 deletion GSTM1 deletion |
231 DM | Significant association of both ‘null’ genotypes with an increased risk of CAD, especially among smokers |
| Manfredi et al. (58) | 2007 | Italian | Cross-sectional | GSTT1 deletion GSTM1 deletion |
222 consecutive smokers | Significant association of both ‘null’ genotypes with an increased risk of CAD and number of stenosis vessels |
| Hayek et al. (59) | 2006 | Caucasian-UK | Case-control | GSTT1 deletion | 773 DM 2592 no DM |
No association between genotype and CAD risk, but higher CRP, oxLDL and smaller LDL particles with functional GSTT-1 |
| Miller et al. (60) | 2003 | Afro-American & Caucasian Cross-sectional | GSTM1 deletion | 989 GP | Higher CRP, FBG, vWF, ICAM-1, VCAM-1 and lower albumin in smokers with ‘nul’ genotype |
CAD - coronary artery disease; CI - cerebral infarction; CRP - C-reactive protein; DM - diabetes mellitus; ICAM-1 - Intercellular adhesion molecule 1; TVD - triple-vessel disease; GP - general population; UK - United Kingdom; VCAM-1 - vascular cell adhesion molecule 1
A very high predisposition for CAD was observed in Turkish CAD patients in comparison to healthy controls with the CYP1A1-m1 variant and GSTT1‘null’ genotypes after adjustment for risk factors (51).
An association between the CYP1A1-m1 variant and cerebral infarction (CI) was found between case and control Korean subjects (with or without cerebral infarction). Increased relative risk for CI was observed in individuals with the CYP1A1-m1 variant and GSTM1‘null’ genotype. (39).
The CYP1A1-m1 variant was also shown in Australian subjects as a significant factor for increasing the risk of triple-vessel disease (three major epicardial coronary arteries with 50% luminal obstruction) in light smokers. This relation was not present in heavy smokers probably due to excessive toxic exposure, which makes genetic effects irrelevant (52,53).
A meta-analysis in Denmark, which included subjects from general population studies and case-control studies with case subjects suffering from ischemic heart disease and ischemic cerebrovascular disease, did not show an association between the GSTT1 and GSTM1 genotypes and markers of inflammation or oxidation (54).
Two Brazilian studies investigated the association of GSTM1 GSTT1 polymorphisms, CAD, and plasma lipid parameters (55,56). There was no association with CAD in the first case-control study (55), while a significant association was observed for lipid parameters and genotype distribution of GSTM1 and GSTT1 deleted deletion polymorphisms in patients who underwent coronary angiography (56).
Two studies performed on Italian subjects who underwent coronary angiography showed an association between GST polymorphisms and CAD in smokers (57,58). Type 2 diabetes patients who smoked had a significantly higher risk of suffering from CAD if they were carriers of GSTM1 and GSTT1’null’ genotypes (59). Consecutive smokers with combined GSTM1’null’GSTT1’null’ genotypes also had a higher risk of CAD and significantly higher number of stenosis vessels, after adjustment for risk factors (58). A case control study that included Caucasian subjects with diabetes and non-diabetics showed no association between the GSTT 1 genotype and CAD risk (59). Indeed, diabetics with functional GSTT1 had higher levels of C-reactive protein (CRP) and oxidated-LDL (Ox-LDL) as well as smaller LDL particles when compared to GSTT1’null’ individuals, while smokers with functional GSTT-1 had a higher CAD risk when compared to non-smokers (59).
The highest mean levels of CRP, fibrinogen, von Willebrand factor, ICAM-1, and VCAM-1 as well as the lowest mean levels of albumin were observed in a cross-sectional study performed on a biracial cohort (African-American and Caucasian), in subjects with the GSTM1 ‘null’ genotype who smoked a pack of cigarettes per day for 20 years (60).
Limiting data aside, it is clear that GSTM1 and GSTT1 deletion polymorphisms alter the effect of smoking on endothelial function, as well as the processes of inflammation and hemostasis (60).
The association between GST polymorphisms and alterations in lipid concentrations
Although peculiar, the association between GST polymorphisms and lipid concentrations can be explained. Maciel et al. (56) hypothesized that GTSs are responsible for hypertriglyceridemia and decrease of HDL-cholesterol plasma concentrations by inhibiting the peroxisome proliferator-activated receptor-y (PPARy). This is due to the role of GST activity in the formation of prostaglandin J 12 (PgJ12)-glutathione conjugate responsible for PgJ12 activation of PPAR-y dependent transcription (56). The formation of this conjugate is thereby involved in the sequestering of the ligands in the cytosol away from their nuclear target - PPARy. The conjugation of PgJ12 disables the PPAR-y regulation of fatty acid metabolism and therefore attenuates the favorable effects of PPAR-y on lipoprotein metabolism, adipocyte function, insulin sensitivity, and vascular structure (61).
Interaction of other atherosclerosis risk factors with genetic polymorphisms of enzymes involved in the biotransformation of PAH
In the environment, PAHs are biologically inert. In the human organism, they induce enzyme activities which convert them into reactive intermediates. Therefore environmental exposure to PAHs negatively impacts human health. As mentioned previously, it is evident that the literature data on the genetic polymorphisms of CYP1A1, GSTT1, and GSTM1 demonstrated a relationship between these polymorphisms and smoking and the development of malignant diseases and atherosclerosis. The main substrates for the biotransformation by enzymes mentioned earlier are PAHs from cigarette smoke and their metabolites (62). Cigarette smoking causes DNA alterations in the heart and blood vessels, which leads to the development of CAD. The individual variability of CAD development in cigarette smokers results from the genetic polymorphisms of enzymes involved in the xenobiotic metabolism (58). It has also been proven that several genetic variants interact with smoking to increase lipid peroxidation, thus providing an even stronger atherogenic effect (59).
Apart from cigarette smoke, PAHs can be found in crude oil, natural gases, during the production of heavy and light metals, waste incineration, etc. Urinary 1-hydroxypyrene (1-HP) was used as a biomarker of occupational and environmental PAH exposure in several studies. A review of studies conducted on Taiwanese and Chinese workers in the petrochemical industry showed the highest concentrations of urinary 1-HP in coke-oven workers (63). Exposure of city policemen to the city air in downtown Prague induced genetic damage and increased DNA adducts even in non-smokers (64). Some human studies showed that survival depends not only on traditional risk factors for atherosclerosis, but also on the molecular endpoint, genetic polymorphisms, and their combinations (65).
For example, subjects with double ‘null’ GSTM1/GSTT1 polymorphisms are more susceptible to the development of atherosclerosis, while smoking significantly increases the number of stenosis vessels in subjects with a positive genotype. On the contrary, the association of GSTM1 deletion and molecular damage in atherosclerosis depends on different factors and includes other polymorphisms (gene-gene interactions), life style habits, and the influence of the environment (58,66).
In general, the overview of population studies has shown that CYP 1A1-Msp I polymorphisms and the deletion polymorphisms of GSTT1 and GSTM1 may be involved in the pathogenesis of cardiovascular disorders, especially when combined with other risk factors such as diabetes mellitus, environmental exposure factors, and habitual factors such as cigarette smoke.
It is well-known that genetic susceptibility contributes to the pathogenic pathways that lead to atherogenesis and other multifactorial disorders. The genetic polymorphisms of genes involved in PAH biotransformations evidently bring forth a risk of disease development. Thus, occupational exposure or conditions from one’s living environment and habits can contribute to the production of PAH metabolites with adverse effects on human health. Investigations aimed at the effect of the gene-smoking allow for a better understanding of gene-environment interactions. They should ultimately lead to the creation of a personalized medicine concept regarding cardiovascular diseases (58,66).
We may conclude that prevention and early intervention could be enhanced by data on genetically susceptible individuals. Also, the elucidation of genetic factors and their association with environmental influences might prove useful for treating high-risk patients with the aim to reduce their risks for atherosclerotic events.
Footnotes
Potential conflict of interest
None declared.
References
- 1.Brinkmann J, Stolpmann K, Trappe S, Otter T, Genkinger D, Bock U, et al. Metabolically Competent Human Skin Models: Activation and Genotoxicity of Benzo[a]pyrene. Toxicol Sci. 2013;131:351–9. doi: 10.1093/toxsci/kfs316. http://dx.doi.org/10.1093/toxsci/kfs316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cerniglia CE. Biodegradation of polycyclic aromatic hydrocarbons. Biodegradation. 1992;3:351–68. http://dx.doi.org/10.1007/BF00129093. [Google Scholar]
- 3.Pandey AK, Chaudhary P, Singh SB, Arora A, Kumar K, Chaudhry S, Nain L. Deciphering the traits associated with PAH degradation by a novel Serratia marcesencs L-11 strain. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2012;47:755–65. doi: 10.1080/10934529.2012.660108. [DOI] [PubMed] [Google Scholar]
- 4.Söderström H, Hajšlová J, Kocourek V, Siegmund B, Kocan A, Obiedzinski MW, et al. PAHs and nitrated PAHs in air of five European countries determined using SPMDs as passive samplers. Atmospheric Environment. 2005;39:1627–40. [Google Scholar]
- 5.Allan IJ, Ruus A, Schaanning MT, Macrae KJ, Næs K. Measuring nonpolar organic contaminant partitioning in three Norwegian sediments using polyethylene passive samplers. Sci Total Environ. 2012;423:125–31. doi: 10.1016/j.scitotenv.2012.02.027. http://dx.doi.org/10.1016/j.scitotenv.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 6.Jakovljević I, Zužul S. Polycyclic aromatic hydrocarbons in air. Arh Hig Rada Toksikol. 2011;62:357–70. doi: 10.2478/10004-1254-62-2011-2095. http://dx.doi.org/10.2478/10004-1254-62-2011-2095. [DOI] [PubMed] [Google Scholar]
- 7.St Helen G, Goniewicz M, Dempsey D, Wilson M, Jacob P, 3rd, Benowitz NL. Exposure and Kinetics of Polycyclic Aromatic Hydrocarbons (PAHs) in Cigarette Smokers. Chem Res Toxicol. 2012;25:952–64. doi: 10.1021/tx300043k. http://dx.doi.org/10.1021/tx300043k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maliszewska-Kordybach B. Sources, concentrations, fate and effects of polycyclic aromatic hydrocarbons (PAHs) in the Environment. Part A PAHs in air. Polish Journal of Environmental Studies. 1999;8:131–6. [Google Scholar]
- 9.Lacquaniti A, Fenga C, Venuti VA, Pernice L, Catanzariti S, Sirna G, et al. Hydrocarbons and Kidney Damage: Potential Use of Neutrophil Gelatinase-Associated Lipocalin and Sister Chromatide Exchange. Am J Nephrol. 2012;35:271–8. doi: 10.1159/000336310. http://dx.doi.org/10.1159/000336310. [DOI] [PubMed] [Google Scholar]
- 10.Henkler F, Stolpmann K, Luch A. Exposure to polycyclic aromatic hydrocarbons: bulky DNA adducts and cellular responses. EXS. 2012;101:107–31. doi: 10.1007/978-3-7643-8340-4_5. http://dx.doi.org/10.1007/978-3-7643-8340-4_5. [DOI] [PubMed] [Google Scholar]
- 11.Wormley DD, Ramesh A, Hood DB. Environmental contaminant-mixture effects on CNS development, plasticity, and behavior. Toxicol Appl Pharmacol. 2004;197:49–65. doi: 10.1016/j.taap.2004.01.016. http://dx.doi.org/10.1016/j.taap.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 12.den Hartigh LJ, Lamé MW, Ham W, Kleeman MJ, Tablin F, Wilson DW. Endotoxin and polycyclic aromatic hydrocarbons in ambient fine particulate matter from Fresno, California initiate human monocyte inflammatory responses mediated by reactive oxygen species. Toxicol In Vitro. 2010;24:1993–2002. doi: 10.1016/j.tiv.2010.08.017. http://dx.doi.org/10.1016/j.tiv.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 13.Riccioni G, Sblendorio V. Atherosclerosis: from biology to pharmacological treatment. J Geriatr Cardiol. 2012;9:305–17. doi: 10.3724/SP.J.1263.2012.02132. http://dx.doi.org/10.3724/SP.J.1263.2012.02132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ross JS, Stagliano NE, Donovan MJ, Breitbart RE, Ginsburg GS. Atherosclerosis and cancer: common molecular pathways of disease development and progression. Ann N Y Acad Sci. 2001;947:271–92. http://dx.doi.org/10.1111/j.1749-6632.2001.tb03949.x. [PubMed] [Google Scholar]
- 15.Curfs DM, Knaapen AM, Pachen DM, Gijbels MJ, Lutgens E, Smook ML, et al. Polycyclic aromatic hydrocarbons induce an inflammatory atherosclerotic plaque phenotype irrespective of their DNA binding properties. FASEB J. 2005;19:1290–2. doi: 10.1096/fj.04-2269fje. [DOI] [PubMed] [Google Scholar]
- 16.Mayati A, Levoin N, Paris H, N’Diaye M, Courtois A, Uriac P, et al. Induction of intracellular calcium concentration by environmental benzo(a)pyrene involves a β2-adrenergic receptor/adenylyl cyclase/Epac-1/inositol 1,4,5-trisphosphate pathway in endothelial cells. J Biol Chem. 2012;287:4041–52. doi: 10.1074/jbc.M111.319970. http://dx.doi.org/10.1074/jbc.M111.319970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barouki R, Aggerbeck M, Aggerbeck L, Coumoul X. The aryl hydrocarbon receptor system. Drug Metabol Drug Interact. 2012;27:3–8. doi: 10.1515/dmdi-2011-0035. http://dx.doi.org/10.1515/dmdi-2011-0035. [DOI] [PubMed] [Google Scholar]
- 18.Huang MC, Chen FY, Chou MT, Su JG. Fluoranthene enhances p53 expression and decreases mutagenesis induced by benzo[a]pyrene. Toxicol Lett. 2012;208:214–24. doi: 10.1016/j.toxlet.2011.11.011. http://dx.doi.org/10.1016/j.toxlet.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 19.Mandal PK. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol B. 2005;175:221–30. doi: 10.1007/s00360-005-0483-3. http://dx.doi.org/10.1007/s00360-005-0483-3. [DOI] [PubMed] [Google Scholar]
- 20.Mimura J, Fujii-Kuriyama Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim Biophys Acta. 2003;1619:263–8. doi: 10.1016/s0304-4165(02)00485-3. http://dx.doi.org/10.1016/S0304-4165-(02)00485-3. [DOI] [PubMed] [Google Scholar]
- 21.Shimada T. Xenobiotic-metabolizing enzymes involved in activation and detoxification of carcinogenic polycyclic aromatic hydrocarbons. Drug Metab Pharmacokinet. 2006;21:257–76. doi: 10.2133/dmpk.21.257. http://dx.doi.org/10.2133/dmpk.21.257. [DOI] [PubMed] [Google Scholar]
- 22.Nims RW, Lubet RA. Induction of cytochrome P-450 in the Norway rat, Rattus norvegicus, following exposure to potential environmental contaminants. J Toxicol Environ Health. 1995;46:271–92. doi: 10.1080/15287399509532035. http://dx.doi.org/10.1080/15287399509532035. [DOI] [PubMed] [Google Scholar]
- 23.Herbstman JB, Tang D, Zhu D, Qu L, Sjödin A, Li Z, et al. Prenatal exposure to polycyclic aromatic hydrocarbons, benzo[a]pyrene-DNA adducts, and genomic DNA methylation in cord blood. Environ Health Perspect. 2012;120:733–8. doi: 10.1289/ehp.1104056. http://dx.doi.org/10.1289/ehp.1104056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Croom E. Metabolism of xenobiotics of human environments. Prog Mol Biol Transl Sci. 2012;112:31–88. doi: 10.1016/B978-0-12-415813-9.00003-9. http://dx.doi.org/10.1016/B978-0-12-415813-9.00003-9. [DOI] [PubMed] [Google Scholar]
- 25.McCarver DG, Hines RN. The ontogeny of human drug-metabolizing enzymes: phase II conjugation enzymes and regulatory mechanisms. J Pharmacol Exp Ther. 2002;300:361–6. doi: 10.1124/jpet.300.2.361. http://dx.doi.org/10.1124/jpet.300.2.361. [DOI] [PubMed] [Google Scholar]
- 26.Xu J, Jaiswal AK. NAD(P)H:quinone oxidoreductase 1 (NQO1) competes with 20S proteasome for binding with C/EBPalpha leading to its stabilization and protection against radiation-induced myeloproliferative disease. J Biol Chem. 2012;287:41608–18. doi: 10.1074/jbc.M112.387738. http://dx.doi.org/10.1074/jbc.M112.387738. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Hinson JA, Forkert PG. Phase II enzymes and bioactivation. Can J Physiol Pharmacol. 1995;73:1407–13. doi: 10.1139/y95-196. http://dx.doi.org/10.1139/y95-196. [DOI] [PubMed] [Google Scholar]
- 28.Chen C, Yu R, Owuor ED, Kong AN. Activation of antioxidant-response element (ARE), mitogen-activated protein kinases (MAPKs) and caspases by major green tea polyphenol components during cell survival and death. Arch Pharm Res. 2000;23:605–12. doi: 10.1007/BF02975249. http://dx.doi.org/10.1007/BF02975249. [DOI] [PubMed] [Google Scholar]
- 29.Rushmore TH, Kong AN. Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr Drug Metab. 2002;3:481–90. doi: 10.2174/1389200023337171. http://dx.doi.org/10.2174/1389200023337171. [DOI] [PubMed] [Google Scholar]
- 30.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. http://dx.doi.org/10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 31.Pushparajah DS, Umachandran M, Plant KE, Plant N, Ioannides C. Up-regulation of the glutathione S-transferase system in human liver by polycyclic aromatic hydrocarbons; comparison with rat liver and lung. Mutagenesis. 2008;23:299–308. doi: 10.1093/mutage/gen012. http://dx.doi.org/10.1093/mutage/gen012. [DOI] [PubMed] [Google Scholar]
- 32.da Fonseca RR, Johnson WE, O’Brien SJ, Vasconcelos V, Antunes A. Molecular evolution and the role of oxidative stress in the expansion and functional diversification of cytosolic glutathione transferases. BMC Evol Biol. 2010;10:281. doi: 10.1186/1471-2148-10-281. http://dx.doi.org/10.1186/1471-2148-10-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.St Helen G, Goniewicz ML, Dempsey D, Wilson M, Jacob P, 3rd, Benowitz NL. Exposure and kinetics of polycyclic aromatic hydrocarbons (PAHs) in cigarette smokers. Chem Res Toxicol. 2012;25:952–64. doi: 10.1021/tx300043k. http://dx.doi.org/10.1021/tx300043k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwano S, Nukaya M, Saito T, Asanuma F, Kamataki T. A possible mechanism for atherosclerosis induced by polycyclic aromatic hydrocarbons. Biochem Biophys Res Commun. 2005;335:220–6. doi: 10.1016/j.bbrc.2005.07.062. http://dx.doi.org/10.1016/j.bbrc.2005.07.062. [DOI] [PubMed] [Google Scholar]
- 35.Iwano S, Asanuma F, Nukaya M, Saito T, Kamataki T. CYP1A1-mediated mechanism for atherosclerosis induced by polycyclic aromatic hydrocarbons. Biochem Biophys Res Commun. 2005;337:708–12. doi: 10.1016/j.bbrc.2005.09.109. http://dx.doi.org/10.1016/j.bbrc.2005.09.109. [DOI] [PubMed] [Google Scholar]
- 36.Scholz H, Lund T, Dahle MK, Collins JL, Korsgren O, Wang JE, Foss A. The synthetic liver X receptor agonist GW3965 reduces tissue factor production and inflammatory responses in human islets in vitro. Diabetologia. 2009;52:1352–62. doi: 10.1007/s00125-009-1366-z. http://dx.doi.org/10.1007/s00125-009-1366-z. [DOI] [PubMed] [Google Scholar]
- 37.Stein O, Thiery J, Stein Y. Is there a genetic basis for resistance to atherosclerosis? Atherosclerosis. 2002;160:1–10. doi: 10.1016/s0021-9150(01)00664-5. http://dx.doi.org/10.1016/S0021-9150(01)00664-5. [DOI] [PubMed] [Google Scholar]
- 38.Knaapen AM, Curfs DM, Pachen DM, Gottschalk RW, de Winther MP, Daemen MJ, Van Schooten FJ. The environmental carcinogen benzo[a]pyrene induces expression of monocyte-chemoattractant protein-1 in vascular tissue: a possible role in atherogenesis. Mutat Res. 2007;621:31–41. doi: 10.1016/j.mrfmmm.2006.12.010. http://dx.doi.org/10.1016/j.mrfmmm.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 39.Moon KS, Lee HJ, Hong SH, Kim HM, Um JY. CYP1A1 and GSTM1/T1 genetic variation in predicting risk for cerebral infarction. J Mol Neurosci. 2007;32:155–9. doi: 10.1007/s12031-007-0028-1. http://dx.doi.org/10.1007/s12031-007-0028-1. [DOI] [PubMed] [Google Scholar]
- 40.Lo HW, Ali-Osman F. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance. Curr Opin Pharmacol. 2007;7:367–4. doi: 10.1016/j.coph.2007.06.009. http://dx.doi.org/10.1016/j.coph.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 41.Abbas A, Delvinquiere K, Lechevrel M, Lebailly P, Gauduchon P, Launoy G, Sichel F. GSTM1, GSTT1, GSTP1 and CYP1A1 genetic polymorphisms and susceptibility to esophageal cancer in a French population: different pattern of squamous cell arcinoma and adenocarcinoma. World J Gastroenterol. 2004;10:3389–93. doi: 10.3748/wjg.v10.i23.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dusinska M, Staruchova M, Horska A, Smolkova B, Collins A, Bonassi S, Volkovova K. Are glutathione S transferases involved in DNA damage signalling? Interactions with DNA damage and repair revealed from molecular epidemiology studies. Mutat Res. 2012;736:130–7. doi: 10.1016/j.mrfmmm.2012.03.003. http://dx.doi.org/10.1016/j.mrfmmm.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Hirata H, Hinoda Y, Okayama N, Suehiro Y, Kawamoto K, Kikuno N, et al. CYP1A1, SULT1A1, and SULT1E1 polymorphisms are risk factors for endometrial cancer susceptibility. Cancer. 2008;112:1964–73. doi: 10.1002/cncr.23392. http://dx.doi.org/10.1002/cncr.23392. [DOI] [PubMed] [Google Scholar]
- 44.Zhan P, Wang Q, Qian Q, Wei SZ, Yu LK. CYP1A1 MspI and exon7 gene polymorphisms and lung cancer risk: an updated meta-analysis and review. J Exp Clin Cancer Res. 2011;30:99. doi: 10.1186/1756-9966-30-99. http://dx.doi.org/10.1186/1756-9966-30-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hayashi SI, Watanabe J, Nakachi K, Kawajiri K. PCR detection of an A/G polymorphism within exon 7 of the CYP1A1gene. Nucleic Acids Res. 1991;19:4797. doi: 10.1093/nar/19.17.4797. http://dx.doi.org/10.1093/nar/19.17.4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cascorbi I, Brockmoller J, Roots I. A C4887A polymorphism in exon 7 of human CYP1A1: population frequency, mutation linkages, and impact on lung cancer susceptibility. Cancer Res. 1996;56:4965–9. [PubMed] [Google Scholar]
- 47.Seidegård J, Vorachek WR, Pero RW, Pearson WR. Hereditary differences in the expression of the human glutathione transferase active on trans-stilbene oxide are due to a gene deletion. Proc Natl Acad Sci U S A. 1988;85:7293–7. doi: 10.1073/pnas.85.19.7293. http://dx.doi.org/10.1073/pnas.85.19.7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiencke JK, Pemble S, Ketterer B, Kelsey KT. Gene deletion of glutathione S-transferase theta: correlation with induced genetic damage and potential role in endogenous mutagenesis. Cancer Epidemiol Biomarkers Prev. 1995;4:253–9. [PubMed] [Google Scholar]
- 49.Zhong SL, Zhou SF, Chen X, Chan SY, Chan E, Ng KY, et al. Relationship between genotype and enzyme activity of glutathione S-transferases M1 and P1 in Chinese. Eur J Pharm Sci. 2006;28:77–85. doi: 10.1016/j.ejps.2006.01.002. http://dx.doi.org/10.1016/j.ejps.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Bolt HM, Thier R. Relevance of the deletion polymorphisms of the glutathione S-transferases GSTT1 and GSTM1 in pharmacology and toxicology. Curr Drug Metab. 2006;7:613–28. doi: 10.2174/138920006778017786. http://dx.doi.org/10.2174/138920006778017786. [DOI] [PubMed] [Google Scholar]
- 51.Taspinar M, Aydos S, Sakiragaoglu O, Duzen IV, Yalcinkaya A, Oztuna D. Impact of genetic variations of the CYP1A1, GSTT1, and GSTM1 genes on the risk of coronary artery disease. DNA Cell Biol. 2012;31:211–8. doi: 10.1089/dna.2011.1252. http://dx.doi.org/10.1089/dna.2011.1252. [DOI] [PubMed] [Google Scholar]
- 52.Wang XL, Greco M, Sim AS, Duarte N, Wang J, Wilcken DE. Effect of CYP1A1 MspI polymorphism on cigarette smoking related coronary artery disease and diabetes. Atherosclerosis. 2002;162:391–7. doi: 10.1016/s0021-9150(01)00723-7. http://dx.doi.org/10.1016/S0021-9150-(01)00723-7. [DOI] [PubMed] [Google Scholar]
- 53.Wang XL, Wang J. Smoking-gene interaction and disease development: relevance to pancreatic cancer and atherosclerosis. World J Surg. 2005;29:344–53. doi: 10.1007/s00268-004-7819-0. http://dx.doi.org/10.1007/s00268-004-7819-0. [DOI] [PubMed] [Google Scholar]
- 54.Nørskov MS, Frikke-Schmidt R, Loft S, Sillesen H, Grande P, Nordestgaard BG, Tybjaerg-Hansen A. Copy number variation in glutathione S-transferases M1 and T1 and ischemic vascular disease: four studies and meta-analyses. Circ Cardiovasc Genet. 2011;4:418–28. doi: 10.1161/CIRCGENETICS.111.959809. http://dx.doi.org/10.1161/CIRCGENETICS.111.959809. [DOI] [PubMed] [Google Scholar]
- 55.Bazo AP, Salvadori D, Jr, Salvadori RA, Sodré LP, da Silva GN, de Camargo EA, et al. DNA repair gene polymorphism is associated with the genetic basis of atherosclerotic coronary artery disease. Cardiovasc Pathol. 2011;20:e9–15. doi: 10.1016/j.carpath.2009.12.004. http://dx.doi.org/10.1016/j.carpath.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 56.Maciel SS, Pereira Ada C, Silva GJ, Rodrigues MV, Mill JG, Krieger JE. Association between glutathione S-transferase polymorphisms and triglycerides and HDL-cholesterol. Atherosclerosis. 2009;206:204–8. doi: 10.1016/j.atherosclerosis.2009.02.011. http://dx.doi.org/10.1016/j.atherosclerosis.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 57.Manfredi S, Calvi D, del Fiandra M, Botto N, Biagini A, Andreassi MG. Glutathione S-transferase T1- and M1-null genotypes and coronary artery disease risk in patients with Type 2 diabetes mellitus. Pharmacogenomics. 2009;10:29–34. doi: 10.2217/14622416.10.1.29. http://dx.doi.org/10.2217/14622416.10.1.29. [DOI] [PubMed] [Google Scholar]
- 58.Manfredi S, Federici C, Picano E, Botto N, Rizza A, Andreassi MG. GSTM1, GSTT1 and CYP1A1 detoxification gene polymorphisms and susceptibility to smoking-related coronary artery disease: a case-only study. Mutat Res. 2007;621:106–12. doi: 10.1016/j.mrfmmm.2007.02.014. http://dx.doi.org/10.1016/j.mrfmmm.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 59.Hayek T, Stephens JW, Hubbart CS, Acharya J, Caslake MJ, Hawe E, et al. A common variant in the glutathione S transferase gene is associated with elevated markers of inflammation and lipid peroxidation in subjects with diabetes mellitus. Atherosclerosis. 2006;184:404–12. doi: 10.1016/j.atherosclerosis.2005.05.017. http://dx.doi.org/10.1016/j.atherosclerosis.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 60.Miller EA, Pankow JS, Millikan RC, Bray MS, Ballantyne CM, Bell DA, et al. Glutathione-S-transferase genotypes, smoking, and their association with markers of inflammation, hemostasis, and endothelial function: the atherosclerosis risk in communities (ARIC) study. Atherosclerosis. 2003;171:265–72. doi: 10.1016/j.atherosclerosis.2003.07.007. http://dx.doi.org/10.1016/j.atherosclerosis.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 61.Huang JV, Greyson CR, Schwartz GG. PPAR-γ as a therapeutic target in cardiovascular disease: evidence and uncertainty. J Lipid Res. 2012;53:1738–54. doi: 10.1194/jlr.R024505. http://dx.doi.org/10.1194/jlr.R024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hung RJ, Boffetta P, Brockmöller J, Butkiewicz D, Cascorbi I, Clapper ML, et al. CYP1A1 and GSTM1 genetic polymorphisms and lung cancer risk in Caucasian non-smokers: a pooled analysis. Carcinogenesis. 2003;24:875–82. doi: 10.1093/carcin/bgg026. http://dx.doi.org/10.1093/carcin/bgg026. [DOI] [PubMed] [Google Scholar]
- 63.Hansen AM, Mathiesen L, Pedersen M, Knudsen LE. Urinary 1-hydroxypyrene (1-HP) in environmental and occupational studies--a review. Int J Hyg Environ Health. 2008;211:471–503. doi: 10.1016/j.ijheh.2007.09.012. http://dx.doi.org/10.1016/j.ijheh.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 64.Binkova B, Chvatalova I, Lnenickova Z, Milcova A, Tulupova E, Farmer PB, Sram RJ. PAH-DNA adducts in environmentally exposed population in relation to metabolic and DNA repair gene polymorphisms. Mutat Res. 2007;620:49–61. doi: 10.1016/j.mrfmmm.2007.02.022. http://dx.doi.org/10.1016/j.mrfmmm.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 65.Izzotti A, Piana A, Minniti G, Vercelli M, Perrone L, De Flora S. Survival of atherosclerotic patients as related to oxidative stress and gene polymorphisms. Mutat Res. 2007;621:119–28. doi: 10.1016/j.mrfmmm.2006.12.012. http://dx.doi.org/10.1016/j.mrfmmm.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 66.Bašić M, Butorac A, Landeka Jurčević I, Bačun-Družina V. Obesity: genome and environment interactions. Arh Hig Rada Toksikol. 2012;63:395–405. doi: 10.2478/10004-1254-63-2012-2244. http://dx.doi.org/10.2478/10004-1254-63-2012-2244. [DOI] [PubMed] [Google Scholar]