

Abstract
Defects in the photoreceptor-specific gene encoding aryl hydrocarbon receptor interacting protein like-1 (AIPL1) are linked to blinding diseases, including Leber congenital amaurosis (LCA) and cone dystrophy. While it is apparent that AIPL1 is needed for rod and cone function, the role of AIPL1 in cones is not clear. In this study, using an all-cone animal model lacking Aipl1, we show a light-independent degeneration of M- and S-opsin containing cones that proceeds in a ventral-to-dorsal gradient. Aipl1 is needed for stability, assembly and membrane association of cone PDE6, an enzyme crucial for photoreceptor function and survival. Furthermore, RetGC1, a protein linked to LCA that is needed for cGMP synthesis, was dramatically reduced in cones lacking Aipl1. A defect in RetGC1 is supported by our finding that cones lacking Aipl1 exhibited reduced levels of cGMP. These findings are in contrast to the role of Aipl1 in rods, where destabilization of rod PDE6 results in an increase in cGMP levels, which drives rapid rod degeneration. Our results illustrate mechanistic differences behind the death of rods and cones in retinal degenerative disease caused by deficiencies in AIPL1.
INTRODUCTION
Leber congenital amaurosis (LCA) is a severe disease that leads to blindness in humans. In children afflicted with LCA, both rod and cone light responses are extinguished at an early age (1). Mutations in multiple genes including AIPL1 are linked to LCA (2–7). Among these, changes in AIPL1 cause a severe form of LCA in humans (AIPL1-LCA) (4). In agreement with this observation, in mouse models lacking Aipl1, both rods and cones do not function and rapidly degenerate with complete loss of photoreceptor cells by 4 weeks of age (8,9). Mutations in AIPL1 are also linked to milder form of retinal dysfunction, such as dominant cone–rod dystrophy and juvenile retinitis pigmentosa (3).
Absence of Aipl1 in photoreceptor cells results in destabilization of rod phosphodiesterase-6 (PDE6), an enzyme crucial for phototransduction and survival of rod cells (9). Destabilization of PDE6 leads to an increase in cGMP levels and this event is thought to be the main driver behind rapid death of rod photoreceptor cells (9). In contrast, very little is known about the need for Aipl1 in cone function and subsequent loss of cone photoreceptor cells. In humans and primates, AIPL1 is expressed very early in retinal development. Both rods and cones show robust expression of AIPL1 (10). However, the expression of AIPL1 appears to be down-regulated in adult cones (11). Despite this finding, the importance of AIPL1 in human cones is apparent as mutations in AIPL1 lead to complete loss of vision or dominant cone–rod dystrophy, a disease characterized by initial loss of cone vision (3).
Our studies using a transgenic animal model expressing human AIPL1 exclusively in rod photoreceptor cells of the Aipl1−/− mouse (Tg hAIPL1; Aipl1−/−) unequivocally demonstrated the essential role played by Aipl1 in cone function in the murine retina (12). In comparison to the mouse model lacking Aipl1 in rod and cone photoreceptor cells, cone cells in this transgenic animal model survived longer, but ultimately degenerated, suggesting that rods play a protective role in the survival of cones (12). Similar to rod cells, we observed reduction in cone PDE6 levels (12). However, very little is known about the connection between Aipl1 and PDE6 in cones. In addition, the mechanism behind the loss of cone cells in the absence of Aipl1 is not clear (9). Our paucity of knowledge about the role of Aipl1 in cones is partly due to a low representation of cones in the murine retina and our inability to express functional PDE6 catalytic subunits in heterologous protein expression system. In this study, using two different animal models, the transgenic model where cones lack functional Aipl1 (Tg hAIPL1; Aipl1−/−) and an animal model with an all-cone retina lacking Aipl1 (Nrl−/− Aipl1−/−), we investigated the need for AIPL1 in cone function and the mechanism behind cone cell death in the absence of AIPL1.
RESULTS
Generation and characterization of all-cone animal model lacking Aipl1
To study the role of AIPL1 in cone function and stability, we used an animal model lacking the Nrl transcription factor. In Nrl−/− animals, all presumptive rod photoreceptor cells are converted to cones (13). Nrl−/− mutant mice were crossed with Aipl1−/− animals. The heterozygous mice obtained from this cross were further bred to generate Nrl−/− Aipl1+/− and Nrl−/−Aipl1−/− animals. Since no obvious differences in cone photoreceptor cell viability or light-dependent electrical responses were observed between Nrl−/−Aipl1+/+ or Nrl−/−Aipl1+/− mice (Supplementary Material, Fig. S1), we used Nrl−/− Aipl1+/− littermates as controls in this work. The genotypes of the animals used in this study were determined as described in the Methods section and verified by reverse transcriptase-polymerase chain reaction (RT-PCR, Fig. 1A). We measured photoreceptor cell function by recording electroretinograms (ERG) under both dark- (scotopic) and light-adapted (photopic) conditions. A normal photopic ERG response in Nrl−/− Aipl1+/− controls was observed at the earliest age tested (Fig. 1B). In contrast, no light-adapted electrical response could be elicited in Nrl−/− Aipl1−/− animals (Fig. 1B). As expected, both Nrl−/− Aipl1+/− and Nrl−/− Aipl1−/− animals lacked scotopic ERG response (Supplementary Material, Fig. S1). In sum, these results demonstrate the crucial role of Aipl1 in functioning of cone photoreceptor cells in the murine retina.
Figure 1.

Aipl1 is essential for cone photoreceptor function. (A) RT-PCR analysis using retinal RNA extracted from indicated animals at P12 showing expression of Aipl1 and Nrl. A house keeping gene, Hprt (hypoxanthine-guanine phosphoribosyltransferase), serves as a loading control. (B) Photopic ERG from all-cone mice lacking Aipl1 (Nrl−/− Aipl1−/−) and littermate controls recorded at P14. Photopic ERGs were measured at 0.4 log cd s/m2 xenon white flash with steady background light.
Differential degeneration of cones lacking Aipl1
We observed rapid degeneration of rods photoreceptor cells in Aipl1-deficient animals (9). To assess whether cones undergo similar rates of cell death in an all-cone mouse model, we stained retinal sections using 4-,6-diamidino-2-phenylindole (DAPI) to visualize nuclei (Fig. 2A). At post-natal day 12, we found laminated nuclear layers with no differences in the number of photoreceptor outer nuclei in both dorsal and ventral regions of retina from Nrl−/− Aipl1−/− and Nrl−/− Aipl1+/− littermate control animals (Fig. 2A). However, rapid degeneration of cone photoreceptor cells starting at P14 was observed in the ventral retina with complete degeneration of the outer nuclei by P100 (Supplementary Material, Fig. S2 and Fig. 2). In contrast, cone nuclei in the dorsal retina underwent slower degeneration starting at P30 with one to two layers of nuclei surviving until P200 (Fig. 2A and B). The degeneration is specific to cone photoreceptor cells, as no changes were observed in the inner nuclear layer (INL) or ganglion cell layer (GCL) (Fig. 2). Differential degeneration of cones, with rapid cell death in ventral areas compared with dorsal regions of retina, was light independent (Fig. 2C). Since cone opsins (red/green = M and Blue = S) are expressed in an opposing dorsal–ventral gradient in the murine retina, we investigated whether the uneven rate of cone degeneration was due to selective death of M- or S-opsin expressing cones by whole-mount immunocytochemistry. Although present at P12, both M- and S-opsin expression were both absent in the ventral region of retinas from Nrl−/− Aipl1−/− animals at P30 (Supplementary Material, Fig. S3). No appreciable changes in S or M opsin staining were observed in the dorsal region of retina from Nrl−/− Aipl1−/− and littermate controls at P12 or P30 (Supplementary Material, Fig. S3). We conclude that Aipl1 is essential for the survival of cones, and that there is a dramatic regional variance in the rate of light-independent cone degeneration.
Figure 2.

Loss of Aipl1 leads to rapid and differential degeneration of cones in retina. (A) Retinal sections from all-cone mice lacking Aipl1 and littermate control stained with DAPI showing laminated nuclear layers. Top panel represents sections from the dorsal region of retina at various ages from P12 to P200. Sections from the ventral region of retina are shown in the bottom panel. (B) Quantitation of rows of outer nuclei in the dorsal (D) and ventral (V) regions of Nrl−/− Aipl1+/−and Nrl−/− Aipl1−/− at various ages. Counts were performed as described previously (32). Counts are an average from retinal sections at four different locations obtained from at least three sections each from three independent animals. (C) Retinal sections from all-cone mice lacking Aipl1 and littermate control reared in complete darkness. All sections were stained with DAPI showing laminated nuclear layers. Top panel represents sections from the dorsal region of retina at P30. Sections from the ventral region of retina are shown in the bottom panel. RPE, retinal pigment epithelium; ONL, outer nuclear layer; INL, inner nuclear layer and GCL, ganglion cell layer. Scale bar = 10 μm applies to all panels.
Aipl1 is essential for stability of phosphodiesterase and retinal guanylate cyclase in cones
The lack of electrical activity in response to light suggested defects in proteins crucial for phototransduction. We used immunoblotting with specific antibodies to quantify relative levels of retinal proteins involved in phototransduction prior to significant photoreceptor cell degeneration. Among these, the levels of cone PDE6 catalytic subunit, an enzyme responsible for second-messenger cGMP hydrolysis and retinal guanylate cyclase-1 (RetGC1), an enzyme that mediates cGMP synthesis in response to changes in calcium, were dramatically reduced in Nrl−/− Aipl1−/− animals in comparison to littermate controls. We observed 10% of wild-type levels of both enzymes in Nrl−/− Aipl1−/− animals (Fig. 3A and B). In contrast, the levels of cone transducin α-subunit (GαT2) and cone arrestin (mCAR) were not significantly altered (Fig. 3A and B). Guanylate cyclase activating protein-1 (GCAP-1), the calcium-sensitive activator of RetGC1, was reduced by 30% in Nrl−/− Aipl1−/− animals. The observed reduction in RetGC1 and PDE6 was at the post-transcriptional level as the message levels remained unchanged between Nrl−/− Aipl1+/− and Nrl−/− Aipl1−/− animals (Fig. 3C). The results from our immunoblotting studies indicate that the lack of ERG response and cone degeneration is likely due to a reduction in both RetGC1 and PDE6 in cones. Additionally, our results show that Aipl1 acts to control the levels of RetGC1 and PDE6 by a post-transcriptional mechanism.
Figure 3.

Destabilization of cone PDE6 and RetGC1 in animals lacking Aipl1. (A) Immunoblotting of retinal extracts from Nrl−/− Aipl1−/− compared with Nrl−/− Aipl1+/− mice at P12 with antibodies against indicated proteins. Equal amounts (150 µg) of total proteins were loaded in each lane. As expected, Aipl1 was not observed in Nrl−/− Aipl1−/− double mutant mice. Tubulin serves as a loading control. Protein molecular weight marker is shown on the left. (B) Quantitation of cone PDE6, RetGC1 and GαT2 levels in retinal extracts from Nrl−/− Aipl1+/−and Nrl−/− Aipl1−/− double knockout animals. Band densities were measured in LI-COR odyssey and normalized to levels present in Nrl−/− Aipl1+/−animals. Each column shown is the mean of normalized values obtained from three independent samples. *P < 0.001, Nrl−/− Aipl1+/− versus Nrl−/− Aipl1−/− double knockout mice by Student's t-test. (C) RT-PCR results showing message levels of RetGC1 (Gucy2e) and cone PDE6α′ (Pde6c).
Aipl1 interacts with the catalytic subunit of cone PDE6
Our previous study showed that Aipl1 interacts with PDE6α, the rod PDE6 catalytic subunit (14). To investigate whether a similar interaction occurs between Aipl1 and cone PDE6α' subunit, we performed co-immunoprecipitation (co-IP) using a B-6 monoclonal antibody (14). B-6 antibody recognized native Aipl1 protein from retinal extracts of Nrl−/− animals and was efficient in depleting 100% of expressed Aipl1 (Fig. 4, top panel). In comparison, non-specific mouse IgG control did not recognize Aipl1. While we observed significant co-IP of PDE6α′ subunit (11%) along with Aipl1, PDE6γ′ was not found in co-IP (Fig. 4, middle and bottom panels). We did not observe RetGC1 subunit in our pull-downs suggesting a lack of interaction between Aipl1 and RetGC1 under our experimental conditions (data not shown). The co-IP results from immunoblotting were further confirmed by LC-MS/MS mass spectrometry. Both Aipl1 and cone PDE6α′ subunits were identified with 100% confidence, while no spectra corresponding to RetGC1 or PDE6γ′ were found in our mass-spec analysis (data not shown). Altogether, our results demonstrate an interaction between Aipl1 and cone PDE6α′ subunit.
Figure 4.
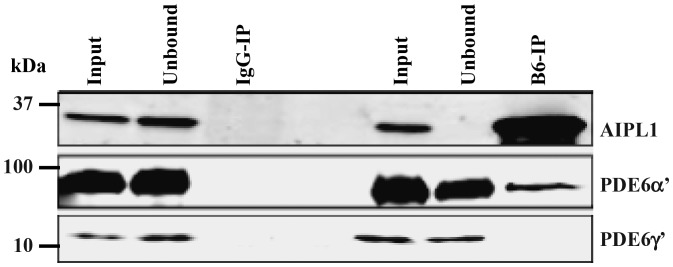
Aipl1 interacts with catalytic subunit of cone PDE6. IP of Aipl1 using a mouse monoclonal antibody (B6-IP) from retinal extracts of all-cone mice (Nrl−/−) (right panel). IP with non-specific mouse IgG serves as a control (left panel). Aipl1, PDE6α′ and PDE6γ′ subunits were analyzed by immunoblotting. IP samples are five times more concentrated than input or soluble fraction. Protein molecular weight marker is shown on the left.
Aipl1 is needed for proper assembly of cone PDE6
To understand the link between Aipl1 and the dramatic reduction in cone PDE6 levels observed in Nrl−/− Aipl1−/− animals, we investigated the synthesis, stability and assembly of the cone PDE6 catalytic subunits. As expression of functional PDE6 in tissue culture is not feasible, we cultured retinas from P12 Nrl−/− Aipl1+/− and Nrl−/− Aipl1−/− animals. Pulse-label using radioactive [35S] -methionine at the indicated times after depletion of endogenous methionine was performed as described in the methods. Immunoprecipitation (IP) with the indicated antibodies followed by fluorography was used to measure the level of protein synthesis in response to increasing amounts of labeling time. We found no differences in the rate of PDE6 (α′ and γ′) synthesis, suggesting that AIPL1 does not affect the translation of PDE6 protein from its mRNA (Fig. 5A). On the other hand, pulse-chase experiments showed dramatic change in the stability of synthesized PDE6 (α′ and γ′). In comparison to littermate Nrl−/− Aipl1+/− controls, the majority of synthesized PDE6 (α′ and γ′) in retinal cultures from Nrl−/− Aipl1−/− animals was not stable after 3 h of chase (Fig. 5B). As a control, we demonstrate that the synthesis and stability of the transducin subunit, GαT2, was not affected by the lack of Aipl1 (Fig. 5A and B).
Figure 5.

Assembly and stability of cone PDE6 is dependent on Aipl1. (A) Protein synthesis was monitored in retinas isolated from Nrl−/−Aipl1+/− or mice lacking Aipl1 (Nrl−/− Aipl1−/−). The retinas were labeled with radioactive methionine for the indicated amount of time. The amount of proteins synthesized was measured by IP using antibodies that recognize Aipl1, PDE6α′, PDE6γ′ or cone transducin (GαT2) followed by fluorography. (B) Stability of the synthesized protein was tested after 1.5 h of radioactive labeling followed by indicated 0–3 h of chase using non-radioactive methionine. GαT2 serves as a control. (C) PDE6 assembly verified after 1.5 h of radioactive labeling followed by IP with ROS-I antibody and fluorography. For all experiments in this panel, retinas from P12 animals were used.
In our effort to probe the cause behind the altered stability of PDE6 in the absence of Aipl1, we checked the assembly of cone PDE6 holoenzyme. Previous studies including ours have shown that ROS-I, a monoclonal antibody, exclusively recognizes functional and assembled rod and cone PDE6 subunits (12). We used IP with ROS-I to assay the assembly status of cone PDE6 in retinal extracts from P12 Nrl−/− Aipl1+/− and Nrl−/− Aipl1−/− animals. Retinas from animals of both genotypes were cultured in the presence of [35S] -methionine for 1.5 h followed by IP with ROS-I antibody and fluorography. Although there was no difference in the levels of synthesized PDE6 subunits in these extracts after 1.5 h of labeling (Fig. 5A, middle panels), assembled cone PDE6 was observed only in Nrl−/− Aipl1+/− retinal extracts, suggesting a lack of or defective PDE6 assembly in retinas from Nrl−/− Aipl1−/− animals (Fig. 5C). Altogether, these results show the importance of AIPL1 in stability and assembly of cone PDE6.
Absence of Aipl1 affects the membrane association of cone PDE6
Cone PDE6 is a peripheral membrane protein anchored in the membrane by a prenylated C-terminus. Based on the presence of leucine as the last amino acid residue of the C-terminal “CAAX” box motif (C = cysteine, A = Aliphatic amino acid residue, X=leucine), cone PDE6α′ is thought to be geranylgeranylated (15). But PrenBase, a software program, predicts PDE6α′ to be farnesylated (http://mendel.imp.ac.at/PrePS/PRENbase). This observation is interesting in light of our previous finding that Aipl1 interacts with farnesylated proteins and likely enhances the membrane association of prenylated proteins (16). To test whether cone PDE6 membrane association is affected in the absence of Aipl1, we performed isotonic extraction of PDE6 using retinal extracts from P12 Nrl−/− Aipl1+/− and Nrl−/− Aipl1−/− animals. While approximately half (50%) of cone PDE6 was associated with membrane in littermate controls, 97% of cone PDE6 was found in the cytosolic fraction in the absence of Aipl1 (Fig. 6). Interestingly, RetGC1, a transmembrane protein, although reduced, exhibited a localization pattern consistent with littermate controls and was exclusively present in the membrane fraction. Cone transducin (GαT2) used as a control localized similarly in both P12 Nrl−/− Aipl1+/− and Nrl−/− Aipl1−/− animals (Fig. 6). Our studies demonstrate that cone PDE6 requires Aipl1 for proper association with membranes.
Figure 6.

Aipl1 contributes to association of PDE6 with membranes. Isotonic protein extraction examining the association of proteins with membranes. (A) After sub-cellular fractionation of retinal extracts, total (T), soluble cytosolic fraction (C) and re-suspended membrane fraction (M) were analyzed by western blotting with indicated antibodies. (B) Protein density in each fraction was measured using Li-COR Odyssey and normalized to protein density found in total fraction. Cone PDE6 and RetGC1 values shown are the mean of normalized measurements obtained from three independent samples. *P < 0.01, Nrl−/− Aipl1+/− versus Nrl−/− Aipl1−/− double knockout mice by Student's t-test.
RetGC1 is mislocalized in cones lacking Aipl1
We next examined whether reduction in RetGC1 levels was due to defective trafficking to outer segments as previous studies indicate that RetGC1 and PDE6 may travel together from cone inner to outer segments (17). Since Nrl−/− animals possess shorter cone outer segments, for the following experiments, we used transgenic animals that express human AIPL1 (hAIPL1) in rods on an Aipl1−/− background (Tg hAIPL1; Aipl1−/−). Rod photoreceptor cells in these animals express hAIPL1 and are similar to rod cells from wild-type animals expressing murine Aipl1. In contrast, cone photoreceptor cells lack Aipl1, are non-functional and degenerate after P18 (12). In agreement with our earlier study (12), cone PDE6 levels were reduced and rarely detected (data not shown). Cone transducin (GαT2) localized normally to the outer segments of both Tg hAIPL1; Aipl1−/− and littermate Aipl1+/− controls (Fig. 7B and D). In contrast, RetGC1 was mislocalized or completely absent in many cone cells (Fig. 7C, see also magnified panels 1 and 2) when compared with littermate controls (Aipl1+/−) (Fig. 7A). Localization of cone arrestin, M- and S- opsin is unaffected in the absence of Aipl1 (data not shown and Supplementary Material, Fig. S4). We further confirmed the mislocalization of RetGC-1 by double-labeling with S-opsin (Supplementary Material, Fig. S5). These results concur with our earlier experiments showing a dramatic reduction in RetGC1 and cone PDE6 levels and in addition demonstrate defective trafficking of RetGC1 in cone photoreceptor cells lacking Aipl1.
Figure 7.

Absence of Aipl1 leads to mislocalization of RetGC1. Frozen retinal sections from P15 littermate control Aipl1+/− (A and B) and animals lacking Aipl1 in cones (Tg hAIPL1; Aipl1−/−) (C and D) (Scale bar = 10 μm). Peanut agglutinin (PNA) which stains cone sheath is shown in red. Panels A and C show sections stained with antibody against RetGC1 (Green). Panels below are magnified to clarify the localization pattern (Scale bar = 20 μm). In Tg hAIPL1; Aipl1−/− animals, RetGC1 was either mislocalized in cone inner segment (C, middle enlarged panel 1) or absent (C, bottom enlarged panel 2). Panels B and D showing similar localization of GαT2 (Green) to cone outer segment in littermate controls (B) and experimental animals (D).
Absence of Aipl1 leads to decreased levels of cGMP in retina
Increased cGMP levels due to non-functional or defective PDE6 has been implicated in rod photoreceptor cell death in rd1 and Aipl1−/− animals (9,18). In an effort to test whether a similar raise in the cGMP levels played a role in cone degeneration, we measured cGMP levels in Nrl−/−Aipl1−/− retina.
Retinal extracts were obtained from animals kept either in complete darkness or exposed to bright light (30 cd m−2) for 20 min to induce light-dependent activation of cone PDE6. In the dark, when PDE6 is not active, littermate controls had higher levels of cGMP (Fig. 8A). In response to light exposure, consistent with light activation of PDE6, cGMP levels decreased 5-fold (Fig. 8A). In contrast, animals lacking Aipl1 failed to exhibit significant light-dependent changes in retinal cGMP levels (Fig. 8A). Retina from Nrl−/−Aipl1−/− animals under both dark- and light-adapted conditions had similar levels of cGMP as light-adapted littermate controls (Fig. 8A). These results show the lack of light-dependent activation of PDE6 in the absence of Aipl1. Additionally, it shows that cones lacking Aipl1 experience low levels of cGMP irrespective of light conditions. Since we observed a difference in the rate of degeneration between dorsal and ventral regions of Nrl−/−Aipl1−/− animals, we examined cGMP levels in different retinal regions prior to the onset of cone photoreceptor cell degeneration. Both dorsal and ventral portions of retinas from animals lacking Aipl1 showed a similar reduction in cGMP levels in comparison to controls (Fig. 8B). Reduction in cGMP levels in the absence of Aipl1 is likely due to decrease in RetGC1 levels in both dorsal and ventral regions of retina lacking Aipl1. To test this hypothesis, we examined the protein levels in both dorsal and ventral regions of retina at P12. Both cone PDE6 and RetGC1 were equally reduced between ventral and dorsal regions of Nrl−/−Aipl1−/− retinas (Supplementary Material, Fig. S6).
Figure 8.

cGMP levels are reduced in retina lacking Aipl1. (A) cGMP levels in P15 retina obtained from animals adapted to dark (D) or light (L) conditions. Each value shown is the mean of cGMP levels (pmol/mg protein) obtained from three independent samples. *P < 0.005, Nrl−/−Aipl1+/− versus Nrl−/− Aipl1−/− double knockout mice by Student's t-test. The small reduction in cGMP levels between dark and light conditions in Nrl−/− Aipl1−/− was statistically not significant. (B) Dissected dorsal (D) and ventral (V) regions of retina from Nrl−/− Aipl1−/− and littermate control Nrl−/− Aipl1+/− at P12 were used to measure cGMP levels by an enzyme immunoassay kit. Each value shown is the mean of cGMP levels obtained from three independent samples. *P < 0.01, Nrl−/− Aipl1+/− versus Nrl−/− Aipl1−/− double knockout mice by Student's t-test.
DISCUSSION
The causes of cone cell death have been studied extensively with the focus on non-autonomous death of cones in humans and in animal models of retinitis pigmentosa (19). In this study, we explored the reason behind autonomous death of cones and the loss of cone function caused by deficiency in Aipl1. Both cone PDE6 and RetGC1, essential enzymes involved in cGMP metabolism are dramatically reduced in the absence of Aipl1. Consistent with the reduction in RetGC1 levels, cGMP levels are down-regulated in the entire retina prior to significant degeneration. The lack of Aipl1 in cones severely impaired the membrane association and assembly of cone PDE6, while RetGC1 was mislocalized in cone inner segments. In addition, we observed a differential rate of cone degeneration. Both M- and S-opsin expressing cones in the ventral region degenerate much faster than cones in the dorsal area of the retina. Based on our results, we propose that lowered cGMP accelerates the degeneration of cones in Nrl−/− animals.
In our previous study, we showed that Nrl−/− cones lacking PDE6α′ (Nrl−/− cpfl1) animals retain residual rod PDE6 expression and are able to evoke light-mediated visual response. In contrast, the animal model (Nrl−/− Aipl1−/−) described in this study demonstrated complete loss of vision likely due to the destabilization of cone PDE6 and residual rod PDE6 in the absence of Aipl1.
In the following discussion, we will compare and contrast the role for Aipl1 in rods and cone photoreceptor cells of the murine retina. Loss of Aipl1 leads to defective assembly of the rod PDE6 heteromer resulting in severe instability of rod PDE6 (14). Similarly, a cone PDE6 heteromeric complex is misassembled in the absence of Aipl1, affecting its stability. Interestingly, the membrane association of cone PDE6 in the absence of Aipl1 is abolished. Both rod and cone PDE6 remain in the cytosolic fraction (data not shown and Fig. 6). This finding is in contrast with results from animal models lacking post-prenylation processing (PPP) in the retina (20). In cone photoreceptor cells lacking PPP, cone PDE6 assembles properly, stability is dramatically affected, but their membrane association is never abolished (Kolandaivelu et al., manuscript in preparation). Altogether, these findings suggest that prenylation of PDE6, a key determinant for membrane association and/or assembly is affected by the lack of Aipl1. Efforts are underway to determine the status of PDE6 lipid modification in the Nrl−/− Aipl1−/− cones.
IP of native Aipl1 from mouse retinal extracts demonstrated an interaction between Aipl1 and the cone PDE6 catalytic subunit. A portion of cone PDE6 (11%) interacted with Aipl1 (Fig. 4). This finding is consistent with the role of Aipl1, an inner segment protein, acting as a chaperone for PDE6. Aipl1 interacts with rod PDE6α that is modified by a farnesyl lipid group, in agreement with our earlier study proposing that AIPL1 interacts with farnesylated proteins (14). A recent in vitro study confirmed the interaction of Aipl1 with the farnesyl lipid moiety (21). The identity of the prenyl group attached to the cone PDE6α′ subunit is not known. Based on the last amino acid residue, where X = leucine, cone PDE6α′, is thought to be geranylgeranylated. However, online prenbase program predicts cone PDE6α′ to be farnesylated. Further studies are needed to identify the prenyl group attached to cone PDE6 which will help to clarify the mechanism behind AIPL1–PDE6 interaction.
The most dramatic difference between Aipl1 deficiency in rods and cones is the 90% reduction in the level of RetGC1 in cones prior to significant degeneration (Fig. 3). A minor reduction in GCAP-1 and RD3, a protein needed for stable expression of RetGC1, was also observed (Fig. 3 and Supplementary Material, Fig. S7). We do not believe that small changes in GCAP-1 or RD3 could lead to a dramatic reduction in RetGC1 levels in cones lacking Aip11. Reduction in GCAP-1 is consistent with previous studies demonstrating decrease in GCAP-1 in the absence of RetGC1 (22). Interestingly, RetGC1 trafficking to outer segments was severely affected in cones lacking Aipl1. Based on the earlier model proposed for trafficking of cone OS resident proteins (17,23), we speculate that transport vesicles containing defective PDE6 and RetGC1 are targeted for proteasomal degradation. Loss of RetGC1 is likely a collateral consequence due to defective cone PDE6 in the absence of Aipl1.
In agreement with the lack of detectable ERG response from AIPL1-LCA patients, both rods and cones degenerate in the absence of Aipl1 (8,9). In rods, the lack of Aipl1 is thought to raise calcium to pathological levels due to increased cGMP caused by the absence of functional PDE6 (9). In cone-only mice lacking Aipl1, both PDE6 and RetGC1 along with cGMP levels are down-regulated in the entire retina. Reduced cGMP levels, leading to closure of cGMP-gated channels, are likely to reduce calcium levels below a threshold needed for long-term cone photoreceptor survival (24). Lower levels of cGMP are a consequence of reduced levels of RetGC1 present in cones lacking Aipl1. It is also possible that reduced RetGC1 activity along with partially active PDE6 but uncoupled from cone transducin may account for diminished cGMP levels in Nrl−/− Aipl1−/− cones.
Degeneration of rods proceeds from the center to the periphery in murine retinas (8,9). In Nrl−/− Aipl1−/− cones, irrespective of cone opsin expression, the ventral retina shows faster degeneration than the dorsal retina. Differential or region-specific cone degeneration was noted in multiple animal models (22,25). Of interest, is the increased ventral cone degeneration observed in cones lacking RetGC1 (22). However, this difference is dramatic in all-cone animal model lacking Aipl1 and is opsin independent (Supplementary Material, Fig. S3). The precise reason for differential degeneration is not clear. Cone death in Nrl−/− animals occurs over a span of several months (Fig. 2). However, microglial activation, a sign of retinal degeneration, was noted as early as 2 weeks in Nrl−/− animals prior to cone loss (26). Ventral region of Nrl−/− retina exhibited both early and significantly higher levels of microglial activation (26). These findings indicate an increased susceptibility of ventral cones to death in Nrl−/− animals. Factors that affect cone survival in Nrl−/− animals include increased oxidative stress due to hyperoxic conditions, retinal detachment and impaired phagocytosis (26,27). In Nrl−/− Aipl1−/− animals, a reduced cGMP level exacerbates existing susceptibility of ventral cones to death leading to rapid degeneration. Alternatively, dorsally enriched cone genes such as Smug1, encoding for a DNA repair enzyme and complement factor I (Cfi), encoding a modulator of complement pathway may play a role in protecting dorsal cones from degeneration (28). Interestingly, reduced CFI activity is associated with higher risk for age-related macular degeneration in humans suggesting a functional role for Cfi in protection of dorsal cones in Nrl−/− Aipl1−/− animals (29). Further studies are warranted to test this hypothesis.
Our studies have uncovered a novel role for AIPL1 in stability of RetGC1 apart from cone PDE6. Defects in RetGC1 and AIPL1 are linked to LCA and cone–rod dystrophy (3,30). In some AIPL1-LCA patients, selective foveal cone photoreceptor loss with some preservation of peripheral photoreceptor cells was observed (5). Taken together, we propose that a subset of AIPL1-linked blinding conditions may be due to loss of RetGC1 or a combination of RetGC1 and PDE6.
MATERIALS AND METHODS
Animals
Nrl−/− Aipl1−/− mutant mice were generated by crossing heterozygous Nrl+/− Aipl1+/− mutant animals. Nrl mutant mice were a generous gift from Dr Anand Swaroop and Aipl1 mutant mice were generated as described earlier (9). Animals were identified by a PCR using primers as described previously (9,31). The conditions used for the PCR amplification of Nrl and Aipl1 mutant alleles were 95 °C for 2 min followed by 30 cycles of (95°C, 30 s; 56°C, 30 s; 72°C, 30 s). Animals were maintained in complete darkness, or cyclic light conditions, and physiological experiments were performed under dim red illumination using a Kodak number 1 Safelight filter (transmittance > 560 nm). All experimental procedures involving animals in this study were approved by Institutional Animal Care and Use Committee of the West Virginia University.
Antibodies
Polyclonal (4365P) and monoclonal (B-6) antibodies to the AIPL1 protein were raised as described previously (14). Polyclonal antibodies to cone PDE6 catalytic subunit (3184P) were raised in rabbit by immunization with purified fragment of mouse cone PDE6 α′ subunit (12). Commercial antibodies used in this study included the following: Rod transducin α and cone transducin α antibodies (Santa Cruz), Tubulin antibody (Sigma), and PKCα (B.D. Biosciences). Antibodies to PDE6 subunits (ROS-1) and PDE6 γ inhibitory subunits (R4842) were generously provided by Drs. Ted Wensel (Baylor College) and Rick Cote (University of New Hampshire). IS4 and Cone arrestin antibody was a gift from Dr Wolfgang Baehr at University of Utah. RetGC1 antibody used in this study was obtained from Dr David Garbers (Deceased; University of Texas Southwestern Medical School, Dallas, TX).
RT-PCR
Retinae from enucleated mouse eyes (2) were flash-frozen on dry ice in 200 μl of TRIzol reagent (Invitrogen). Total RNA was isolated as per manufacturer's instructions. Oligo (dT)-primed reverse transcription reactions were performed with 2.5 μg of total RNA by using SuperScript III (Invitrogen) to obtain cDNA, which was then used as a template in PCR (31). The conditions used for PCR were 95°C for 2 min followed by 95°C for 30 s, 56°C for 30 s, and 72°C for 45 s, for 33 cycles. The quantitative RT-PCR analysis were performed using 200 ng of cDNA from Nrl−/− and Nrl−/− Aipl1−/− littermates in triplicate using MyiQ PCR cycler (Bio-Rad) and MyiQ SYBR Green Supermix (Bio-Rad). The primers used in this analysis as described previously (31). Threshold values were normalized to hypoxanthine phosphoribosyltransferase (Hprt), a housekeeping gene
Western blotting
Retinae from enucleated eyes (2) were homogenized by sonication (Microson Ultrasonic cell disruptor, five pulses 10 s at power setting 6) in 150 μl of 1× Urea-SDS buffer (6 m Urea, 125 mm Tris–HCl, pH 6.8, 4% SDS, 0.2% bromphenol blue, 10 mm dithiothreitol) in a 1.5 ml microcentrifuge tube on ice. After homogenization, protein concentration was measured with a NanoDrop spectrophotometer (ND-1000, Thermo Scientific). Equal concentrations (150 μg) of the total protein sample were separated by SDS–PAGE gel electrophoresis, transferred to Immobilon-FL membrane (Millipore) and probed with indicated antibodies. Primary antibodies listed in the previous section were diluted in a 1:1 ratio of blocking buffer (Rockland) and 1× PBST [1× phosphate buffered saline (PBS)/0.1% Tween-20] and used at 1:1000. After overnight incubation of primary antibodies, immunoblots were washed three times with 1× PBST for 10 min each. Secondary antibodies, odyssey goat anti-rabbit Alexa 680 and/or odyssey goat anti-mouse Alexa 680 (LI-COR Biosciences) were then added to blots at 1:50 000 (diluted with 1X PBST) for 30 min. Immunoblots were washed three times with 1× PBST for 10 min each and then scanned using an Odyssey Infrared Imaging System (LI-COR Biosciences). The protein density was measured using an Odyssey Infrared Imaging System according to manufacturer's instructions.
Electroretinography
Electroretinography (ERGs) were performed as described earlier (31). Briefly, mice were anesthetized with isofluorane (5% in 2.5% oxygen) and placed on a heating stage at 37°C with a nose cone supplying isofluorane (1.0% in 2.5% oxygen) during the testing period. Eyes were dilated for 10 min prior to recording and lubricated with methylcellulose (1/5 dilution with 1× PBS or H2O). A silver wire electrode rested on the methylcellulose above the cornea with a needle reference electrode inserted on top of the head, between the ears. Mice were placed into a Ganzfield chamber and light flashes were given at varying intensities. Dark-adapted ERGs using flashes of intensity −3.6 log cd s/m2 were performed after mice were kept in the dark for 12 h. Light-adapted ERGs were performed after mice were equilibrated to background light (30 cd m−2) for 10 min and flashes at 0.4 log cd s/m2 were administered in the presence of constant background lighting.
Immunocytochemistry
Mouse eyes were enucleated, punctured with a fine needle in the dorsal region and incubated for 10 min in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) in PBS at room temperature. Both cornea and lens from partially fixed tissue were removed to prepare eyecups, which were further fixed in 4% paraformaldehyde for 2 h at room temperature and then cryoprotected in 20% sucrose in PBS overnight at 4°C. The tissue was then frozen in an optimal cutting temperature (OCT) compound on dry ice and stored at −80°C. Sections were cut at 16 μm and collected on Superfrost Plus slides (Fisher Scientific). For immunocytochemistry, slides were washed in PBST (1× PBS with 0.1% Triton X-100) and blocked with buffer containing 2% goat serum (Invitrogen) for 1 h at room temperature. Following removal of blocking buffer, primary antibodies were added at 1:1000 dilution and incubated overnight at 4°C. After primary antibody incubation, slides were washed in 1× PBST for three times and incubated with secondary antibody (Odyssey Alexa Fluor-488, or Alexa Fluor-568, LI-COR Biosciences) at 1:1000 dilutions for 2 h at room temperature. DAPI (1:5000 DAPI, Molecular Probes) or propidium iodide (Invitrogen, 2 µg/ml working concentration) were used as a nuclear stain. DAPI or PI was added prior to final washes with 1× PBST for three times. Slides were mounted with Fluromount-G (Southern Biotech) and cover slipped. Confocal imaging was performed at the WVU Microscope Imaging Facility with a Zeiss LSM 510 laser scanning confocal on a LSM Axioimager upright microscope using excitation wavelengths of 405, 488 and 543 nm. Rhodamine peanut agglutinin (PNA, Vector Laboratories, CA, USA) was used at a 1:500 dilution for 1 h during secondary antibody incubation.
Preparation and immunocytochemistry of whole-mount retina
Whole eyes were enucleated and the dorsal side was marked by puncturing the cornea with a 25G 5/8 precision glide needle for orientation purposes. The whole eye was fixed in 4% paraformaldehyde in 1× PBS for 30 min. The eye was then removed from fixative and the cornea and lens were dissected away carefully to preserving the known orientation. A relaxing incision (2–3 mm) was made starting from the needle marked dorsal retina toward the optic nerve. The retina was carefully isolated from the retinal pigment epithelium and free-floating retinas were returned to 4% paraformaldehyde for 6 h. For immunocytochemistry, the tissue was washed with 1× PBS (three times for 30 min each) and nonspecific binding sites were blocked by incubating with blocking buffer for 4 h. After blocking, retinas were incubated for 12 h with primary antibodies (diluted 1:500 in 1XPBST). Whole retinal tissues were washed in 1× PBST (two times for 30 min each) and 1× PBS (30 min) before incubating overnight in Odyssey goat anti-rabbit Alexa 488 IgG secondary antibody (diluted 1:1000, LI-COR Biosciences). After removal of secondary antibody, retinas were further washed as described earlier. Before imaging, the retina was placed on a Superfrost Plus slide (Fisher Scientific) with the outer segments of the photoreceptor cells facing down. Radial cuts 2–3 mm in length were made both to flatten the otherwise concave tissue and to divide the tissue into four quadrants: dorsal–rostral, dorsal–caudal, ventral–rostral and ventral–caudal. Finally, the whole retinal tissue was flat mounted, vitreal side up, on to the slide (lines of nail polish served as spacers to preserve the vertical structure of the outer retina) and were cover slipped for imaging.
Pulse labeling and IP
Three retinas from P12 animals were incubated in pre-warmed DMEM medium lacking dl-methionine (Invitrogen) for 30 min at 37°C in a 95% O2, 5% CO2 incubator. To reduce variability among various samples, we combined retinas from three different animals. After endogenous methionine depletion, retinas were transferred in DMEM media containing L-[35S]-methionine (1 mCi/mmol, Perkin Elmer) for indicated times. For the pulse-chase experiments, radioactive dl-methionine incorporation was performed for an hour and 30 min followed by two washes in DMEM media and further incubated for indicated times in fresh DMEM medium supplemented with cold l-methionine at 500 µm final concentration. After pulse label or chase, retinas were removed and immediately frozen at –80°C on dry ice to prevent further incorporation of label and protein degradation. Frozen retinas were homogenized with 1× PBS mix containing protease inhibitor cocktail (Roche), 0.5% Triton X-100 and 1 mm iodoacetamide. The homogenized retinal extracts were pre-cleared with 20 μl of immunopure immobilized protein A plus beads by incubating at 4°C for 1 h. Supernatants after centrifuging at 10 000 rpm (Eppendorf centrifuge 5424/5424R) for 5 min at 4°C were collected. IP was carried out using indicated antibodies using collected supernatants (input) as described previously (14). Proteins were separated by 4–20% SDS–polycarylamide gel (PAGE) (Bio-Rad, Hercules, CA, USA). For detection of radioactive samples, the gels were impregnated with a fluorography enhancer (En3Hance: Perkin Elmer) and were dried, exposed to autoradiography on Biomax MR Film (Kodak) for several days at −80°C.
Cellular fractionation
Four retinas from Nrl−/− Aipl1+/− and Nrl−/− Aipl1−/− animals were solubilized by homogenization using a pellet pestle (VWR) in a 1.5 ml microcentrifuge tube on ice with 200 µl of isotonic buffer (PBS) containing protease and phosphatase inhibitor cocktail (Roche). Solubilized samples were centrifuged at low-speed centrifugation at 5000g for 10 min at 4°C. The supernatant (total) was collected to another tube and unbroken cellular debris was discarded. The low speed supernatant was further centrifuged at 100 000g for 30 min at 4°C. The high-speed supernatant (cytosol) was removed. The pellet (membrane) was then homogenized in the same volume as supernatant fraction. All collected fractions were subjected to immunoblot analysis to check for the distribution of cytosolic and membrane levels of proteins.
cGMP assay
Dissected retinas (4) were homogenized in 0.1 N hydrochloric acid (HCl), an aliquot of sample was used to measure protein concentration after neutralization with 0.5 m Tris, pH 8.0 by using a NanoDrop spectrophotometer. Rest of retinal homogenates was boiled for 5 min and clarified by centrifugation at 6000 × g for 5 min at 4°C. The acidic supernatant was neutralized in 0.5 m Tris, pH 8.0. Equal amounts of samples (1 mg of total protein) were used to measure cGMP levels using a Direct cGMP enzyme immunoassay kit (Assay Designs) as described by the manufacturer. Average of three independent experiments performed in duplicate was used in the calculation of cGMP levels. To minimize the individual variations between mice, retinas were pooled from different mice for each time point assayed.
FUNDING
This work was supported by National Institutes of Health grant RO1EY017035 (VR), West Virginia Lions, Lions Club International Foundation and an Unrestricted challenge grant from Research to Prevent Blindness (RPB) to West Virginia University.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Wolfgang Baehr, Rick Cote, David Garbers, Robert Molday and Ted Wensel for sharing antibodies generated in their laboratories. The Nrl mutant mice were generous gift from Dr Anand Swaroop. We are indebted to Drs Peter Mathers, Maxim Sokolov and Jeffrey Christiansen for suggestions and the members of Dr Ramamurthy laboratory for their help and support throughout this study.
Conflict of Interest statement. None declared.
REFERENCES
- 1.den Hollander A.I., Roepman R., Koenekoop R.K., Cremers F.P. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Sohocki M.M., Bowne S.J., Sullivan L.S., Blackshaw S., Cepko C.L., Payne A.M., Bhattacharya S.S., Khaliq S., Qasim Mehdi S., Birch D.G., et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat. Genet. 2000;24:79–83. doi: 10.1038/71732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sohocki M.M., Perrault I., Leroy B.P., Payne A.M., Dharmaraj S., Bhattacharya S.S., Kaplan J., Maumenee I.H., Koenekoop R., Meire F.M., et al. Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Mol. Genet. Metab. 2000;70:142–150. doi: 10.1006/mgme.2000.3001. [DOI] [PubMed] [Google Scholar]
- 4.Dharmaraj S., Leroy B.P., Sohocki M.M., Koenekoop R.K., Perrault I., Anwar K., Khaliq S., Devi R.S., Birch D.G., De Pool E., et al. The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Arch. Ophthalmol. 2004;122:1029–1037. doi: 10.1001/archopht.122.7.1029. [DOI] [PubMed] [Google Scholar]
- 5.Jacobson S.G., Cideciyan A.V., Aleman T.S., Sumaroka A., Roman A.J., Swider M., Schwartz S.B., Banin E., Stone E.M. Human retinal disease from AIPL1 gene mutations: foveal cone loss with minimal macular photoreceptors and rod function remaining. Invest. Ophthalmol. Vis. Sci. 2011;52:70–79. doi: 10.1167/iovs.10-6127. [DOI] [PubMed] [Google Scholar]
- 6.Pennesi M.E., Stover N.B., Stone E.M., Chiang P.W., Weleber R.G. Residual electroretinograms in young Leber congenital amaurosis patients with mutations of AIPL1. Invest. Ophthalmol. Vis. Sci. 2011;52:8166–8173. doi: 10.1167/iovs.11-8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Testa F., Surace E.M., Rossi S., Marrocco E., Gargiulo A., Di Iorio V., Ziviello C., Nesti A., Fecarotta S., Bacci M.L., et al. Evaluation of Italian patients with leber congenital amaurosis due to AIPL1 mutations highlights the potential applicability of gene therapy. Invest. Ophthalmol. Vis. Sci. 2011;52:5618–5624. doi: 10.1167/iovs.10-6543. [DOI] [PubMed] [Google Scholar]
- 8.Dyer M.A., Donovan S.L., Zhang J., Gray J., Ortiz A., Tenney R., Kong J., Allikmets R., Sohocki M.M. Retinal degeneration in Aipl1-deficient mice: a new genetic model of Leber congenital amaurosis. Brain Res. Mol. Brain Res. 2004;132:208–220. doi: 10.1016/j.molbrainres.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 9.Ramamurthy V., Niemi G.A., Reh T.A., Hurley J.B. Leber congenital amaurosis linked to AIPL1: a mouse model reveals destabilization of cGMP phosphodiesterase. Proc. Natl Acad. Sci. USA. 2004;101:13897–13902. doi: 10.1073/pnas.0404197101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Spuy J., Kim J.H., Yu Y.S., Szel A., Luthert P.J., Clark B.J., Cheetham M.E. The expression of the Leber congenital amaurosis protein AIPL1 coincides with rod and cone photoreceptor development. Invest. Ophthalmol. Vis. Sci. 2003;44:5396–5403. doi: 10.1167/iovs.03-0686. [DOI] [PubMed] [Google Scholar]
- 11.van der Spuy J., Chapple J.P., Clark B.J., Luthert P.J., Sethi C.S., Cheetham M.E. The Leber congenital amaurosis gene product AIPL1 is localized exclusively in rod photoreceptors of the adult human retina. Hum. Mol. Genet. 2002;11:823–831. doi: 10.1093/hmg/11.7.823. [DOI] [PubMed] [Google Scholar]
- 12.Kirschman L.T., Kolandaivelu S., Frederick J.M., Dang L., Goldberg A.F., Baehr W., Ramamurthy V. The Leber congenital amaurosis protein, AIPL1, is needed for the viability and functioning of cone photoreceptor cells. Hum. Mol. Genet. 2010;19:1076–1087. doi: 10.1093/hmg/ddp571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mears A.J., Kondo M., Swain P.K., Takada Y., Bush R.A., Saunders T.L., Sieving P.A., Swaroop A. Nrl is required for rod photoreceptor development. Nat. Genet. 2001;29:447–452. doi: 10.1038/ng774. [DOI] [PubMed] [Google Scholar]
- 14.Kolandaivelu S., Huang J., Hurley J.B., Ramamurthy V. AIPL1, A protein associated with childhood blindness, interacts with alpha-subunit of rod phosphodiesterase (PDE6) and is essential for its proper assembly. J. Biol. Chem. 2009;284:30853–30861. doi: 10.1074/jbc.M109.036780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anant J.S., Ong O.C., Xie H.Y., Clarke S., O'Brien P.J., Fung B.K. In vivo differential prenylation of retinal cyclic GMP phosphodiesterase catalytic subunits. J. Biol. Chem. 1992;267:687–690. [PubMed] [Google Scholar]
- 16.Ramamurthy V., Roberts M., van den Akker F., Niemi G., Reh T.A., Hurley J.B. AIPL1, A protein implicated in Leber's congenital amaurosis, interacts with and aids in processing of farnesylated proteins. Proc. Natl Acad. Sci. USA. 2003;100:12630–12635. doi: 10.1073/pnas.2134194100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baehr W., Karan S., Maeda T., Luo D.G., Li S., Bronson J.D., Watt C.B., Yau K.W., Frederick J.M., Palczewski K. The function of guanylate cyclase 1 and guanylate cyclase 2 in rod and cone photoreceptors. J. Biol. Chem. 2007;282:8837–8847. doi: 10.1074/jbc.M610369200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lolley R.N., Farber D.B., Rayborn M.E., Hollyfield J.G. Cyclic GMP accumulation causes degeneration of photoreceptor cells: simulation of an inherited disease. Science. 1977;196:664–666. doi: 10.1126/science.193183. [DOI] [PubMed] [Google Scholar]
- 19.Punzo C., Xiong W., Cepko C.L. Loss of daylight vision in retinal degeneration: are oxidative stress and metabolic dysregulation to blame? J. Biol. Chem. 2012;287:1642–1648. doi: 10.1074/jbc.R111.304428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christiansen J.R., Kolandaivelu S., Bergo M.O., Ramamurthy V. RAS-converting enzyme 1-mediated endoproteolysis is required for trafficking of rod phosphodiesterase 6 to photoreceptor outer segments. Proc. Natl Acad. Sci. USA. 2011;108:8862–8866. doi: 10.1073/pnas.1103627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majumder A., Gopalakrishna K.N., Cheguru P., Gakhar L., Artemyev N.O. Interaction of aryl hydrocarbon receptor-interacting protein-like 1 with the farnesyl moiety. J. Biol. Chem. 2013 doi: 10.1074/jbc.M113.476242. 10.1074/jbc.M1113.476242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coleman J.E., Zhang Y., Brown G.A., Semple-Rowland S.L. Cone cell survival and downregulation of GCAP1 protein in the retinas of GC1 knockout mice. Invest. Ophthalmol. Vis. Sci. 2004;45:3397–3403. doi: 10.1167/iovs.04-0392. [DOI] [PubMed] [Google Scholar]
- 23.Karan S., Frederick J.M., Baehr W. Involvement of guanylate cyclases in transport of photoreceptor peripheral membrane proteins. Adv. Exp. Med. Biol. 2008;613:351–359. doi: 10.1007/978-0-387-74904-4_41. [DOI] [PubMed] [Google Scholar]
- 24.Fain G.L. Why photoreceptors die (and why they don't) Bioessays. 2006;28:344–354. doi: 10.1002/bies.20382. [DOI] [PubMed] [Google Scholar]
- 25.Michalakis S., Geiger H., Haverkamp S., Hofmann F., Gerstner A., Biel M. Impaired opsin targeting and cone photoreceptor migration in the retina of mice lacking the cyclic nucleotide-gated channel CNGA3. Invest. Ophthalmol. Vis. Sci. 2005;46:1516–1524. doi: 10.1167/iovs.04-1503. [DOI] [PubMed] [Google Scholar]
- 26.Roger J.E., Ranganath K., Zhao L., Cojocaru R.I., Brooks M., Gotoh N., Veleri S., Hiriyanna A., Rachel R.A., Campos M.M., et al. Preservation of cone photoreceptors after a rapid yet transient degeneration and remodeling in cone-only Nrl−/− mouse retina. J. Neurosci. 2012;32:528–541. doi: 10.1523/JNEUROSCI.3591-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mustafi D., Kevany B.M., Genoud C., Okano K., Cideciyan A.V., Sumaroka A., Roman A.J., Jacobson S.G., Engel A., Adams M.D., et al. Defective photoreceptor phagocytosis in a mouse model of enhanced S-cone syndrome causes progressive retinal degeneration. FASEB J. 2011;25:3157–3176. doi: 10.1096/fj.11-186767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corbo J.C., Myers C.A., Lawrence K.A., Jadhav A.P., Cepko C.L. A typology of photoreceptor gene expression patterns in the mouse. Proc. Natl Acad. Sci. USA. 2007;104:12069–12074. doi: 10.1073/pnas.0705465104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van de Ven J.P., Nilsson S.C., Tan P.L., Buitendijk G.H., Ristau T., Mohlin F.C., Nabuurs S.B., Schoenmaker-Koller F.E., Smailhodzic D., Campochiaro P.A., et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat. Genet. 2013;45:813–817. doi: 10.1038/ng.2640. [DOI] [PubMed] [Google Scholar]
- 30.Olshevskaya E.V., Ermilov A.N., Dizhoor A.M. Factors that affect regulation of cGMP synthesis in vertebrate photoreceptors and their genetic link to human retinal degeneration. Mol. Cell Biochem. 2002;230:139–147. [PubMed] [Google Scholar]
- 31.Kolandaivelu S., Chang B., Ramamurthy V. Rod phosphodiesterase-6 (PDE6) catalytic subunits restore cone function in a mouse model lacking cone PDE6 catalytic subunit. J. Biol. Chem. 2011;286:33252–33259. doi: 10.1074/jbc.M111.259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott P.A., Kaplan H.J., Sandell J.H. Anatomical evidence of photoreceptor degeneration induced by iodoacetic acid in the porcine eye. Exp. Eye Res. 2011;93:513–527. doi: 10.1016/j.exer.2011.06.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.