

Abstract
Transcriptional dysregulation has been proposed to play a major role in the pathology of Huntington's disease (HD). However, the mechanisms that cause selective downregulation of target genes remain unknown. Previous studies have shown that mutant huntingtin (Htt) protein interacts with a number of transcription factors thereby altering transcription. Here we report that Htt directly interacts with methyl-CpG binding protein 2 (MeCP2) in mouse and cellular models of HD using complimentary biochemical and Fluorescent Lifetime Imaging to measure Förster Resonance Energy Transfer approaches. Htt–MeCP2 interactions are enhanced in the presence of the expanded polyglutamine (polyQ) tract and are stronger in the nucleus compared with the cytoplasm. Furthermore, we find increased binding of MeCP2 to the promoter of brain-derived neurotrophic factor (BDNF), a gene that is downregulated in HD, in the presence of mutant Htt. Finally, decreasing MeCP2 levels in mutant Htt-expressing cells using siRNA increases BDNF levels, suggesting that MeCP2 downregulates BDNF expression in HD. Taken together, these findings suggest that aberrant interactions between Htt and MeCP2 contribute to transcriptional dysregulation in HD.
INTRODUCTION
Transcriptional dysregulation is an early event in Huntington's disease (HD) pathology and is present across multiple HD models (1). Human, cellular and transgenic mouse models of HD demonstrate alterations of specific genes at the level of mRNA expression (1–6). Given that mRNA up- and down-regulation of key molecules may underlie the neuronal dysfunction in HD, correction of these transcriptional abnormalities has great potential as a novel therapeutic approach. However, the mechanisms that cause selective alterations in the expression of target genes remain unknown.
Mutant Htt interacts with numerous transcription factors (1) such as CREB-binding protein (CBP) (7–9), TATA-binding protein (TBP) (10), p53 (8), Sp1 and its coactivator TAFII 130 (11,12), mSin3a (8,13), nuclear co-repressor (NCoR) (14), NF-Y (15), CtBP (16), REST/NRSF (17,18) and the transcriptional activator CA150 (19), raising the possibility that the polyglutamine (polyQ) repeat expansion alters the interaction of Htt with these proteins thereby disrupting their normal function. Thus, one mechanism whereby mutant Htt alters transcription is through abnormal interactions with transcription factors.
Before it was discovered that mutations in methyl-CpG binding protein 2 (MeCP2) gene cause Rett syndrome (20–22), MeCP2 was known to bind to methylated DNA (23,24), thereby repressing gene expression (25,26). DNA methylation has been shown to induce long-term changes in gene expression through direct interference with transcription factor binding and recruitment of chromatin remodeling enzymes regulated through the actions of MeCP2. Thus, MeCP2 recruits transcriptional co-repressors to silence transcription through modifying surrounding chromatin structure and providing a link between DNA methylation, chromatin remodeling and gene expression (27,28).
Previous studies from our laboratory have demonstrated that Htt is present in the nucleus and that it binds DNA (29). However, it is yet unclear whether the interaction between Htt and DNA is direct and what the effect of this interaction is within the nucleus. Therefore, identifying other Htt binding partners or interactors is a logical step in addressing these issues. In addition, we have shown that mutant Htt alters histone modifications such as acetylation (5) and ubiquitylation (30). These findings suggest that there may be more players involved in creating this transcriptional profile in HD. Given that MeCP2 plays a pivotal role in regulating gene expression through its interactions with histone deacetylases, we propose that increases in MeCP2 association with gene promoters may be an underlying mechanism of altered gene expression in HD. Here, we demonstrate that Htt interacts with MeCP2 in a mouse and cellular model of HD using traditional colocalization studies. Moreover, in order to gain insight into the localization of the protein–protein and protein–DNA interactions within intact cells, we complemented traditional colocalization studies with Fluorescent Lifetime Imaging (FLIM), a powerful approach to assess Förster Resonance Energy Transfer (FRET) between interacting molecules in the cell lines (31). Furthermore, we demonstrate that alterations in MeCP2 binding at the brain-derived neurotrophic factor (BDNF; gene name: Bdnf) gene promoter is sufficient to decrease BDNF expression in HD.
RESULTS
Increased interactions between Htt and MeCP2 in mutant Hdh mouse striatum and STHdh cell lines
In order to determine whether Htt interacts with MeCP2, we used the striatum from 10-month-old wild-type (Hdh7/7) and mutant (Hdh111/111) Hdh mouse model of HD for co-immunoprecipitation studies. For the first experiment, we used anti-MeCP2 antibody for the immunoprecipitation and anti-Htt antibody 2166 (anti-Htt-2166) for the immunoblots. The results demonstrate that there is a significant increase in MeCP2 binding to Htt in Hdh111/111 striatum compared with wild-type (Fig. 1A). The data were analyzed with a two-way ANOVA, which revealed a significant main effect of genotype × Htt fragment [F(9,48) = 4.66, P < 0.0002; n = 3–5], main effect of Htt fragment [F(3,48) = 12.99, P < 0.0001; n = 3–5], as well as a significant main effect of genotype [F(3,48) = 4.46, P < 0.0076, n = 3–5] (Fig. 1B). Next, the reverse immunoprecipitation was carried out using anti-Htt-2166 for the immunoprecipitation and anti-MeCP2 antibody for the immunoblots. These results confirmed the first experiment and demonstrated an increase between MeCP2 and Htt interaction in the Hdh111/111 striatum compared with wild-type (Fig. 1C and D). The two-way ANOVA revealed a significant genotype x binding interaction [F(1,12) = 8.1, P < 0.015; n = 3–5], and a trends towards a main effect of genotype [F(1,12) = 4.34, P < 0.059, n = 3–5] (Fig. 1D).
Figure 1.

Co-immunoprecipitation assay demonstrating MeCP2–Htt interactions in Hdh mouse striatum. (A) Co-immunoprecipitation assay in Hdh mouse striatum with anti-MeCP2 antibody followed by immunoblot with MAb2166 antibody against Htt protein demonstrates that a significant increase in Htt immunoreactivity for the 83, 47 and 25 kDa Htt fragments in the Hdh111/111 striatum compared with Hdh7/7. (B) Integrated density values for Htt fragment immunoreactivity from a total of five immunoblots. (C) Co-immunoprecipitation assay in mouse striatum with MAb2166 antibody against Htt protein followed by immunoblot with an anti-MeCP2 antibody demonstrates a significant increase in MeCP2 immunoreactivity in immunoprecipitated Hdh111/111 striatum compared with Hdh7/7. Control IgG co-immunoprecipitation followed by immunoblot with an anti-MeCP2 antibody in Hdh striatum. (D) Integrated density values for MeCP2 immunoreactivity from a total of four immunoblots; ***P < 0.001, *P < 0.05. IP, immunoprecipitation.
Similar co-immunoprecipitation studies were next carried out in the STHdh cells lines, a cellular model of HD derived from the striatal neurons of the Hdh mouse. These results demonstrated that there was a significant increase in MeCP2 binding to Htt in STHdh111/111 cells (Fig. 2). The data were analyzed with a two-way ANOVA, which revealed a significant main effect of genotype x binding [F(1,12) = 5.21, P < 0.04; n = 4] as well as a significant main effect of genotype [F(1,12) = 15.71, P < 0.002] (Fig. 3B).
Figure 2.

Co-immunoprecipitation assay demonstrating MeCP2-Htt interactions. (A) Western blot in STHdh cell lines with MeCP2 antibody demonstrates that there is no significant difference in MeCP2 protein levels between the mutant and WT cells. (B) Co-immunoprecipitation assay in STHdh cell lines with MAb2166 antibody against Htt protein followed by immunoblot with an anti-MeCP2 antibody. There is a significant increase in MeCP2 immunoreactivity in immunoprecipitated STHdh111/111 samples compared with STHdh7/7 cells. (C) Integrated density values for MeCP2 immunoreactivity from a total of four immunoblots, *P < 0.05. IP, immunoprecipitation.
Figure 3.
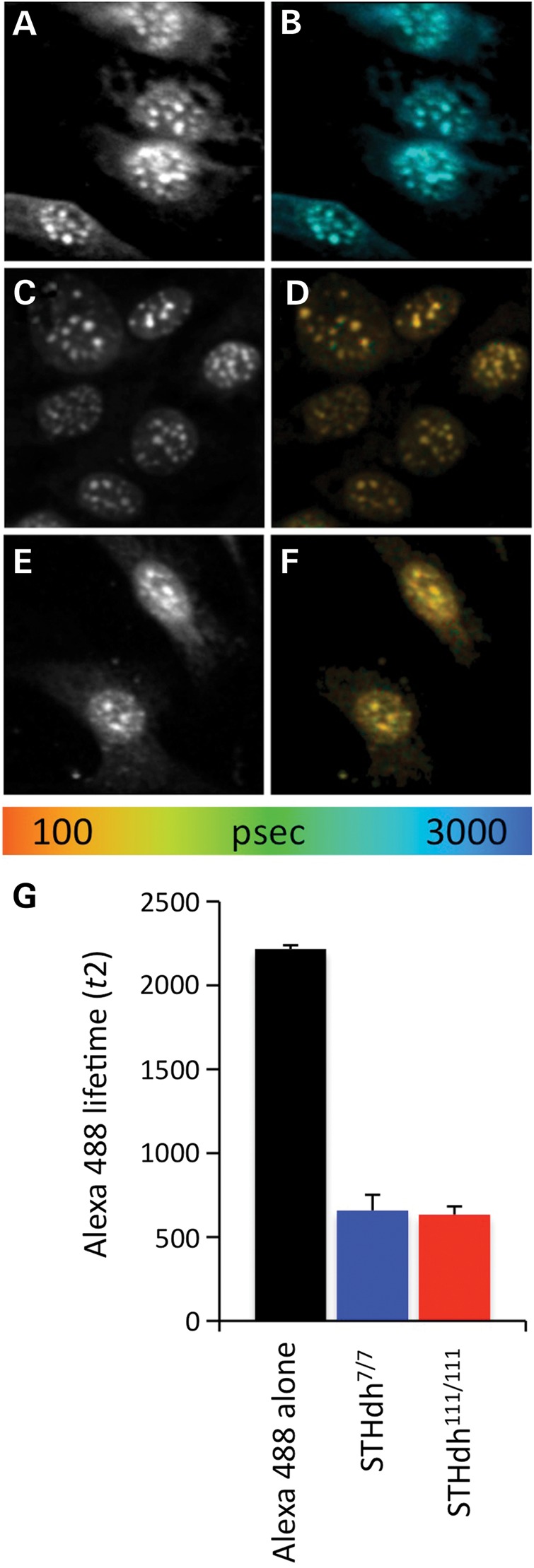
FLIM analysis of MeCP2 interaction with HP1α in STHdh cell lines. Fluorescent imaging of the donor fluorophore (Alexa488) in HP1α/Alexa488 and MeCP2/Cy3 immunostaining (A, C and E). Fluorescent imaging of the donor fluorophore with fluorescent lifetimes pseudocolored (see color scale) and mapped on the image pixel-by-pixel (B, D and F). (A and B) FLIM analysis of the donor fluorophore (HP1α/Alexa488) in the no acceptor condition, STHdh7/7 cells. (C and D) FLIM analysis of HP1α/Alexa488 immunostaining of STHdh7/7 cells in the presence of MeCP2/Cy3, acceptor fluorophore. (E and F) FLIM analysis of HP1α/Alexa488 immunostaining of STHdh111/111cells in the presence of MeCP2/Cy3, acceptor fluorophore. (G) Average fluorescent lifetime of the HP1α/Alexa488 donor fluorophore in STHdh7/7 cells in the absence of acceptor fluorophore (Alexa488 alone, black bars) and in the presence of MeCP2/Cy3 acceptor fluorophore in STHdh7/7 (blue bars) and STHdh111/111cells (red bars).
Increased interactions between Htt and MeCP2 in mutant huntingtin-expressing cells
To confirm our co-immunoprecipitation findings, suggestive of Htt–MeCP2 interactions in both mouse striatum as well as cell lines, we turned to FLIM studies using the same antibodies for each protein. Using FLIM analysis, the proximity of two fluorophores can be measured relative to one another. The baseline lifetime of the donor fluorophore is measured in the absence of the acceptor fluorophore, thus representing the no-FRET state. When the acceptor fluorophore is added, the lifetime of the donor fluorophore decreases from the baseline. This shortened lifetime indicates physical proximity between the acceptor and donor fluorophores. Any change in the donor fluorophore lifetimes in two experimental states indicates differences in physical proximity between the donor and acceptor fluorophores. Here, we are using antibody-linked donor and acceptor fluorophores or an antibody-linked donor fluorophore and a nucleic acid dye for the acceptor fluorophore as a proxy for measuring physical interactions between their antigens.
To demonstrate interactions between MeCP2 and its binding partners, we chose to first demonstrate interactions between MeCP2 and HP1α as a positive control (Fig. 3) as HP1α is a well-known MeCP2 interactor (32). AlexaFluor 488 labeled HP1α was used as the donor fluorophore with Cy-3 labeled MeCP2 as the acceptor. Donor fluorophore lifetimes in the absence of the acceptor resulted in a baseline lifetime of 2217 ± 23 ps. In the presence of Cy-3 labeled MeCP2, the lifetime decreased in both STHdh7/7 and STHdh111/111 cells indicating interactions between MeCP2 and HP1α [t(38) = −4.4788; P < 0.0001; 16 (STHdh7/7), 24 (STHdh111/111)]. Comparing the change in lifetime between STHdh7/7 and STHdh111/111 cells revealed a small, non-significant difference (658 ± 94 ps in STHdh7/7 cells versus 633 ± 50 ps in STHdh111/111 cells) in interaction between MeCP2 and HP1α [t(38) = −4.4788; P < 0.0001; 16 (STHdh7/7), 24 (STHdh111/111)]. Thus, FRET–FLIM can detect interactions between MeCP2 and its known interacting protein HP1α.
In order to determine whether MeCP2 interacts with Htt, cells were immunostained for both Htt and MeCP2. We labeled Htt with the donor fluorophore, AlexaFluor488, and MeCP2 with the acceptor fluorophore, Cy3. Prior to FLIM analysis, the cells were imaged using conventional fluorescence microscopy to ensure correct staining patterns (data not shown). During FLIM analysis, we noticed an obvious difference between Htt–MeCP2 interactions in the nuclear and cytoplasmic compartments (Fig. 4). Therefore, we performed separate analyses on the nucleus and cytoplasm by drawing our region of interest on these separate compartments during the analysis.
Figure 4.
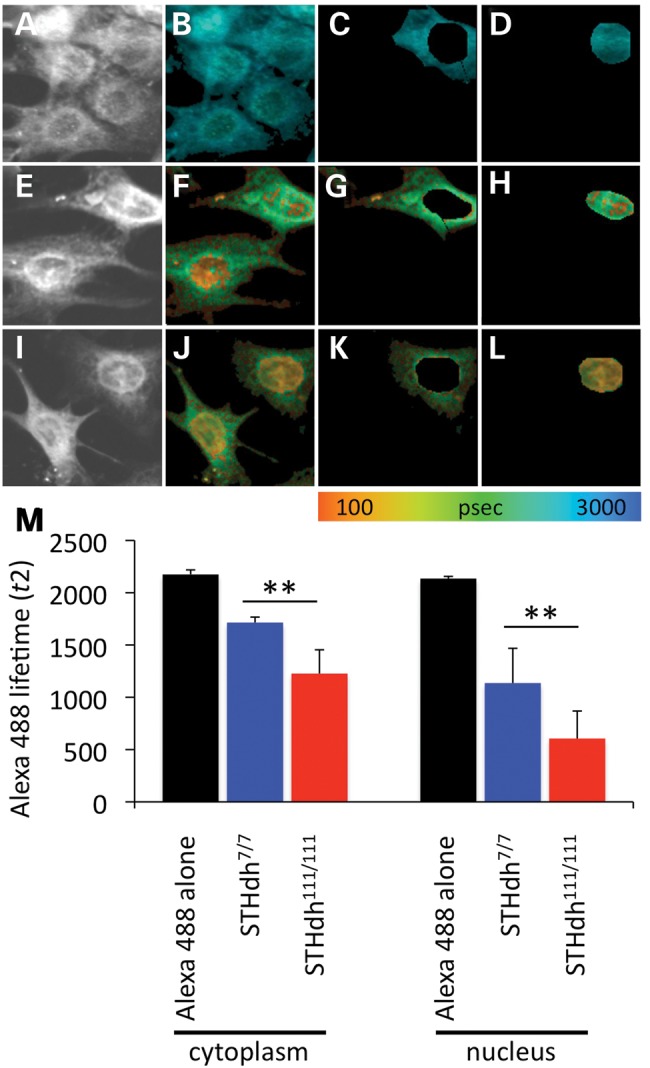
FLIM analysis of MeCP2 interaction with Htt in STHdh cell lines. Fluorescent imaging of the donor fluorophore in Htt/Alexa488 and MeCP2/Cy3 immunostaining (A, E and I). Fluorescent imaging of the donor fluorophore with fluorescent lifetimes pseudocolored (see color scale) and mapped on the image pixel-by-pixel (B–D, F–H, J–L). (A–D) FLIM analysis of the donor fluorophore (Htt/Alexa488) in the no acceptor condition, STHdh7/7 cells. (E–H) FLIM analysis of Htt immunostaining of STHdh7/7 cells in presence of MeCP2/Cy3, acceptor fluorophore. (I–L) FLIM analysis of Htt/Alexa488 immunostaining of STHdh111/111 cells in the presence of MeCP2/Cy3, acceptor fluorophore. Cytoplasmic (C, G and K) and nuclear (D, H and L) compartments were analyzed separately. (M) Average fluorescent lifetime of Htt/Alexa488 donor fluorophore in STHdh7/7 cells in the absence of acceptor fluorophore (Alexa488 alone, black bars) and in the presence of MeCP2/Cy3 acceptor fluorophore in STHdh7/7 (blue bars) and STHdh111/111 cells (red bars).
Within the nucleus, FLIM analysis of AlexaFluor 488-labeled Htt in the absence of the acceptor fluorophore resulted in an average donor lifetime of 2136 ± 21 ps (n = 23 cells). When MeCP2 was labeled with the acceptor fluorophore, Cy3, the lifetime decreased to 1135 ± 333 ps in STHdh7/7 cells and to 606 ± 263 ps in STHdh111/111 cells [t(31) = 5.0837; P < 0.0001; 16 (STHdh7/7), 17 (STHdh111/111)] (Fig. 4). These results suggest that MeCP2–Htt interactions in the nucleus are increased when mutant Htt is present.
Comparatively, Htt–MeCP2 interactions were weaker in the cytoplasmic compartment for both STHdh7/7 and STHdh111/111 cells as revealed by a one-way ANOVA followed by Bonferroni's test [F(3, 62) = 57.9; P < 0.0001]. In the absence of acceptor fluorophore, the baseline lifetime of AlexaFluor-labeled Htt was 2175 ± 43 ps (n = 46 cells). In the presence of Cy3-labeled MeCP2, the lifetime decreased to 1714 ± 53 ps in STHdh7/7 cells and to 1227 ± 227 ps in STHdh111/111 cells [t(31) = 8.3531; P < 0.0001; 16 (STHdh7/7), 17 (STHdh111/111)] (Fig. 4). As was observed in the nuclear compartment, cytoplasmic MeCP2–Htt interactions were significantly increased when Htt is in its mutant form. Taken together, the FLIM data indicate that Htt and MeCP2 interact in both the cytoplasm and nucleus of STHdh cells. However, in the presence of the polyQ expansion in mutant Htt, MeCP2–Htt interactions are stronger.
Increased interactions between Htt and MeCP2 may underlie alterations in BDNF expression in mutant huntingtin-expressing cells
Our results indicate that MeCP2 interacts with Htt and that this interaction is increased in the presence of the expanded polyQ tract. Therefore, we hypothesized that increased MeCP2 interaction with mutant Htt could contribute to gene expression changes in HD. We chose to examine the expression of BDNF in our experimental system as it is decreased in HD (4,33). Moreover, MeCP2 regulation of BDNF gene is well characterized (34,35). Specifically, the removal of MeCP2 from the CpG-island within BDNF promoter IV leads to enhanced expression of BDNF gene (34).
We measured Bdnf transcript levels using reverse transcription quantitative real-time PCR (RT–qPCR) in addition to MeCP2 binding at Bdnf promoters using chromatin immunoprecipitation (ChIP). As a negative control, we also measured microtubule associated protein-2 (Map2) transcript levels and binding of MeCP2 to its promoter. While there was no change in Map2 levels, there was a significant decrease in Bdnf exon IV-containing transcript in the STHdh111/111 compared with STHdh7/7 cells in agreement with results from other HD models. The data were analyzed with a two-way ANOVA, which revealed a significant main effect of gene [F(1,12) = 7.19, P < 0.02; n = 4] (Fig. 4A) (Fig. 5A). Next, we measured the binding of MeCP2 at Bdnf promoter IV and the Map2 promoter in the cell lines using ChIP. The two-way ANOVA revealed a significant effect of genotype [F(1,11) = 5.35, P < 0.003; n = 4], gene [F(1,11) = 14.01, P < 0.04; n = 4], as well as genotype x gene interaction [F(1,11) = 7.47, P < 0.019; n = 4], which demonstrates a significant increase in MeCP2 association with Bdnf promoter IV in the mutant cells compared with WT (Fig. 5B). Together, these results suggest that increased MeCP2 binding at Bdnf exon IV promoter can underlie decreases in Bdnf expression in HD.
Figure 5.

MeCP2 binding at BDNF promoter IV decreases BDNF expression in STHdh111/111 cells as measured by ChIP. (A) There is a significant decrease in Bdnf exon IV-containing transcripts in STHdh111/111 cells compared with STHdh7/7 (n = 4; *P < 0.05). There is no change in Map2 transcript levels between the cell lines. (B) There is a significant increase in MeCP2 binding to Bdnf promoter IV in STHdh111/111 compared with STHdh7/7 as measured by ChIP (n = 4; **P < 0.01). There is no change in MeCP2 binding at the Map2 gene promoter.
In order to determine whether increased MeCP2 association at the Bdnf promoter is necessary to decrease Bdnf transcript levels, we decreased MeCP2 levels in STHdh111/111 cells. Transfection of MeCP2 siRNA decreased MeCP2 protein expression compared with mock and non-targeting control (NTC) siRNA by 48 and 72 h (Fig. 6A). Two-way ANOVA revealed a significant main effect of treatment [F(2,18) = 22.79, P < 0.0001; n = 3] as well as a significant treatment × time interaction [F(4,18) = 3.48, P < 0.028] (Fig. 6B). If increased MeCP2 binding at gene promoters results in decreased transcript levels in STHdh111/111 cells, then lowering cellular MeCP2 levels should decrease MeCP2 binding at gene promoters correcting mRNA abnormalities. Thus, we measured Bdnf mRNA levels in STHdh111/111 cells following treatment with MeCP2 siRNA for 72 h. There was a significant increase in Bdnf mRNA expression in the STHdh111/111 cells following MeCP2 knockdown when compared with non-transfected cells (Fig. 6C). Analysis of these data with two-way ANOVA revealed a significant main effect of treatment [F(1,18) = 4.79, P < 0.04; n = 4].
Figure 6.

Decreases in MeCP2 levels using siRNA in STHdh111/111 cells normalizes BDNF expression. (A) Western blot analysis demonstrates that there is a significant decrease in MeCP2 protein levels following MeCP2 siRNA (100 nM) treatment of cells at 48 and 72 h Mock or 100 nM non-targeting control (NTC) siRNA transfected cells do not show a significant change in MeCP2 levels compared with non-transfected cells. There is no change in GAPDH levels in response to MeCP2 siRNA treatment. (B) Integrated density values for MeCP2 immunoreactivity from a total of three immunoblots (*P < 0.05; **P < 0.01). (C) There is a significant increase in Bdnf exon IV-containing transcript levels in STHdh111/111 cells following MeCP2 siRNA treatment compared with mock and NTC treatment (n = 4; *P < 0.05).
DISCUSSION
In the present report, we provide the first evidence indicating that Htt protein interacts directly with MeCP2 in established HD mouse and cellular models. Furthermore, this interaction is enhanced in the presence of the expanded polyQ tract and is stronger in the nucleus compared with cytoplasm as measured by FLIM. Furthermore, our findings demonstrate that MeCP2 binding to gene promoters in mutant cells may underlie alterations in gene expression in HD. There was an increase in MeCP2 binding at the Bdnf gene promoter, a gene with downregulated expression in HD, in the mutant cells compared with wild-type. These findings suggest that the increased interaction between mutant Htt and MeCP2 is a novel underlying mechanism of transcriptional dysregulation in HD.
Htt and MeCP2 interact in both the cytoplasm and nucleus of STHdh cells. However, in the presence of the polyQ expansion, the MeCP2–Htt interaction is stronger, indicating that either there is an increased number of interacting Htt and MeCP2 molecules or stronger interactions. These alterations cannot be simply attributed to changes in MeCP2 levels in the STHdh111/111 cells, as there was no change in MeCP2 levels between the cell lines (Fig. 2B). Moreover, donor lifetime is an intrinsic property of the fluorophore and is not concentration dependent (36,37). As we have demonstrated, increased interaction between MeCP2 and mutant Htt within the nucleus is sufficient to alter transcript levels of Bdnf. Additionally, alterations in MeCP2–mutant Htt within the cytoplasm may contribute to the neurological phenotype in HD. Indeed, in a recent study by Roux et al. (38), it was demonstrated that Htt alters BDNF trafficking in a MeCP2-deficient mouse model of Rett Syndrome. These authors found decreased Htt and huntingtin-associated protein 1 (HAP1) as well as abnormal BDNF distribution, suggesting that MeCP2 in conjunction with Htt regulates axonal trafficking. Thus, these findings together suggest that while Htt–MeCP2 interactions in the nucleus may alter transcription, this interaction in the cytoplasm can alter other cellular functions such as protein trafficking.
The complex mechanisms regulating eukaryotic gene expression involve DNA methylation, chromatin remodeling through histone modifications that can in turn affect transcription factor activity and binding to gene promoters. Thus far, studies have focused on histone modifications and transcription factor activity as potential mechanisms that underlie changes in gene expression in HD. However, more recently, the mechanisms by which DNA methylation is targeted to specific regions of genome and interpreted by MeCP2 has become clear (25,26). DNA methylation has been shown to induce long-term changes in gene expression through direct interference with transcription factor binding and recruitment of chromatin remodeling enzymes regulated through the actions of MeCP2. Given that MeCP2 plays a pivotal role in regulating gene expression, increases in MeCP2 association at gene promoters may contribute to altered gene expression in HD and other disorders. Multiple genes, in addition to Bdnf, are under the direct transcriptional control of MeCP2. Thus the downstream effect of alterations of MeCP2 at their gene promoters can provide the underlying molecular mechanisms that are manifested in severe neurological deficits seen in HD and Rett Syndrome. Although there was no change in the overall levels of MeCP2 protein or mRNA levels between the cell lines, there was an increase in MeCP2 association with BDNF promoter IV in the mutant cells compared with wild-type. BDNF promotes the development, differentiation, maintenance, and survival of neurons in the CNS (39–41) and previous studies have shown that BDNF mRNA, protein, and trafficking are reduced in neurons and glia in HD (33,42,43). MeCP2 has been shown to repress BDNF promoter IV expression in the absence of neuronal activity (34,35,44). Thus, a prerequisite step in BDNF promoter IV-containing gene expression is de-repression caused by the dissociation of MeCP2 from promoter IV (34). Our data indicate that perhaps polyQ expansion hinders the dissociation of MeCP2 from BDNF promoter IV thereby decreasing BDNF expression.
MeCP2 was recently shown to bind at numerous sites throughout the genome (45) thus it may be involved in the regulation of transcripts that demonstrate altered expression in HD. In this brief report, we have focused on one gene, BDNF; determining whether MeCP2 binding at other gene promoters is altered in the presence of expanded polyQ warrants further study. MeCP2 binding at gene promoters may act by facilitating or recruiting other regulatory factors such as co-repressors or HDACs to these DNA regions in order to alter gene expression. Previous studies have demonstrated that the binding of MeCP2 to methyl-CpG sites translates into a gene-silencing signal via the recruitment of histone deacetylases (HDAC), histone methyltransferases, and other co-repressors (27,28). Specifically, MeCP2 has been shown to specifically interact with and recruit histone deacetylase 1 (HDAC 1) and histone deacetylase 2 (HDAC2) to gene promoters. Previous findings from our lab indicate that downregulated genes are associated with hypoacetylated histones in HD cellular and animal models (5). Therefore, given the known interaction between MeCP2 and HDACs 1 and 2, it is possible that MeCP2 recruits HDACs to gene promoters abnormally in the presence of mutant Htt leading to hypoacetylation of histones and in turn alterations in gene expression.
The findings here together with our previous work suggest that Htt has a role in the nucleus. Once mutated, the expanded polyQ Htt orchestrates a more repressive transcriptional environment by decreasing histone acetylation (5), increasing histone ubiquitylation (30), and perhaps recruiting MeCP2 to gene promoters. In summary, we demonstrated for the first time a direct interaction between Htt and MeCP2. As reported previously, we find a decrease in BDNF expression in HD cells that is caused by an increase in MeCP2 binding to BDNF promoter IV in the presence of expanded polyQ Htt. Our studies provide a novel mechanism underlying transcriptional dysregulation in HD. Additional investigations into the role of MeCP2 on alterations in gene expression in HD may provide further insight into the pathophysiology of HD as well as Rett Syndrome given the overlap in the motor symptoms in both disorders.
MATERIALS AND METHODS
Animals
The striatum of the knock-in Hdh mouse model (46) was a generous gift from Drs Lianna Orlando and Kim Kegel (Massachusetts General Hospital). Hdh mice contain chimeric human and mouse HD/Hdh exon 1 with a sequence encoding for the human polyproline tract together with either a full-length version of either wild-type (containing seven glutamines) or mutant (111 glutamines) Htt. We used the striatum from the wild-type (Hdh7/7) and homozygous (Hdh111/111) genotypes.
Cell culture
Striatal cell lines were established from wild-type and Hdh (Q111) knock-in embryonic mice (47) and were a generous gift from Dr Marcy MacDonald (Massachusetts General Hospital). STHdh cell lines express full-length versions of either wild-type (containing seven glutamines) or mutant (111 glutamines) Htt (47). Two different cell lines were used, corresponding to wild-type (STHdh7/7) and homozygous (STHdh111/111) genotypes. STHdh cells were used in passages 5–14. Cells were kept in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies, Gaithersburg, MD, USA) plus 10% fetal bovine serum at 33°C for propagation, and were placed at 39°C for 48 h to stop their division.
Immunostaining: Sytox Orange counterstain
STHdh7/7and STHdh111/111cells expressing wild-type or mutant huntingtin, respectively, were plated 24 h prior to fixing. Ice-cold methanol was used to fix the cells for 10 min. Cells were permeabilized with ice-cold acetone for 1 min. After gradual rehydration with PBS, cells were washed three times with PBS. Cellular RNA was digested with DNase-free RNase A (1 mg/ml) for 30 min at room temperature. Cells were blocked with 2% normal goat serum (Jackson Labs) and 0.1% Triton X-100 in PBS at room temperature for at least 1 h. Primary antibodies were diluted in blocking solution and incubated with the cells overnight at 4°C and then washed three times in PBS/0.1% Triton X-100. AlexaFluor 488-labeled secondary antibodies (goat anti-mouse (HP1α) or goat anti-rabbit (MeCP2) were incubated for 1 h at room temperature (Invitrogen; 1:200 dilution) followed by washes in PBS/0.1% Triton X-100. The nucleic acid stain Sytox Orange (Invitrogen) was added to a final concentration of 1 mm in PBS/0.1% Triton and incubated for 30 min followed by three washes of PBS. Negative controls (no acceptor staining) for the FLIM experiments omitted Sytox Orange. Cells were coverslipped using GVA Mount (Zymed Laboratories). Prior to FRET–FLIM experiments, staining was confirmed using a conventional epifluorescence microscope to ensure proper staining.
Immunostaining: double staining of interacting proteins
Antibody staining was similar to that discussed earlier with the elimination of RNase A treatment and Sytox Orange staining. The second protein stained in these reactions was counterstained with goat anti-mouse or goat anti-rabbit antibody conjugated to Cy3 (1:200 dilution; Jackson ImmunoResearch, West Grove, PA, USA). Cells were coverslipped in GVA Mount and imaged on a conventional epifluorescence microscope prior to FRET–FLIM imaging to ensure proper staining.
Antibodies for FLIM
Primary antibodies used for these experiments were: mouse anti-HP1α (Upstate 05–689; 5 mg/ml), mouse anti-huntingtin (Chemicon MAb2166; 1:50), rabbit anti-MeCP2 (AbCam ab2828; 1:200).
FLIM assays and analysis
Cells were imaged using a ×60 objective on an Olympus microscope equipped with a Ti-sapphire pulsed laser as the light source. A mode-locked Ti-sapphire laser (Spectra-Physics, Fremont, CA, USA) sent a femtosecond pulse every 12 ns to excite Alexa488 donor fluorophore. Images were acquired using a Bio-Rad (Hercules, CA, USA) Radiance 2000 multiphoton microscope. A high-speed Hamamatsu (Bridgewater, NJ, USA) detector and hardware and software from Becker and Hickl (Berlin, Germany) were used to measure donor fluorescence lifetimes on a pixel-by-pixel basis (48).
SPCImage software (Becker and Hickl) was used to analyze FLIM data as previously described (49). Briefly, the baseline fluorescence lifetime of the AlexaFluor 488 donor (t1) was calculated in the absence of FRET acceptor by fitting AlexaFluor488 lifetime to a single exponential decay curve (no acceptor negative control, FRET absent). In experimental settings where both donor and acceptor fluorophores are present, AlexaFluor 488 donor lifetime was fitted to two-exponential decay curves. The longer lifetime (t1, established in the no acceptor negative control) was fixed during the analysis, and the second, shorter lifetime (indicating FRET, t2) was calculated by the software and was used for all comparisons. Thus, changes in the t2 lifetime are a direct indication of changes in the physical proximity between the donor and acceptor fluorophores. Average per cell/nucleus lifetime was recorded; shortened lifetimes indicate closer proximity and/or increased numbers of interactions. t2 lifetimes were compared between STHdh7/7 and STHdh111/111cells by a two-tailed Student's t-test. In this analysis, the t1 lifetime, the non-FRETing portion, was excluded from the analysis. The t2 lifetimes were then mapped by pseudocolor over the image on a pixel-by-pixel basis. For the analysis of protein–DNA interaction, individual nuclei were defined as the region of interest. For the analysis of protein–protein interactions within individual cells, the region of interest was first defined as the nucleus and a separate analysis was performed using the cytoplasm as the region of interest.
Western blotting
Western blots were carried out as previously described (5,50). Briefly, 25 µg of protein was resuspended in sample buffer, boiled at 95°C for 5 min, and fractionated on a 4–20% glycine gels (Invitrogen, Carlsbad, CA, USA) for 90 min at 120 V. Proteins were transferred to PVDF membranes in transfer buffer (3% Tris base, 14.4% glycine, 20% MeOH) at 400 mA × 1 h, and the PVDF was then blocked with 5% milk in phosphate-buffered saline before immunodetection with antibodies. Primary antibody [MeCP2 (1:500; Abcam) and GAPDH (1:700; Abcam)] incubation overnight was followed by four washes (10 min, RT) in TBST before incubation with the secondary antibody for 1 h (HRP-conjugated goat anti-rabbit IgG; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). After four washes in PBS proteins were visualized using the ECL detection system (NEN, Boston, MA, USA). Coomassie gels were used to ensure equal protein loading for western blots.
Co-immunoprecipitation
The co-immunoprecipitations (co-IP) studies were carried out as previously described (30). Briefly, 50 µg of nuclear extracts from STHdh cells were incubated with 5 µg of anti-Htt (Chemicon MAb2166), or anti-MeCP2 (AbCam ab2828) antibody and 25 μl of protein A magnetic beads (Invitrogen) at 4°C for 8 h. The immune complexes were washed four times with Gal4 buffer (20 mm HEPES, 300 mm NaCl, 10% glycerol) and extracted by heating at 95°C for 5 min in 25 ml of 2X Tris–glycine sample buffer. Immune complexes were subjected to western blot analysis for analysis of MeCP2 and Htt proteins.
ChIP assay
A modified ChIP technique was adapted from previously published studies from our laboratory in order to analyze DNA/protein complexes in dissected brain tissue (5,51) anti-MeCP2 antibody (Abcam) was added to isolated protein-DNA complexes containing MeCP2. BDNF promoter DNA was detected in the resulting ChIP-DNA by qPCR using specific primers for the multiple BDNF promoters. Exon-specific BDNF primers were designed based on previously published sequences (34,35,52). The following primer sequences were used for qPCR: BDNF exon IV forward, 5-AACAAGAGGCTGTGACACTATGCTC-3; reverse, 5-CAGTAAGTAAAGGCTAGGGCAGGC-3 (34). Threshold amplification cycle numbers (Tc) using iCycler software were used to calculate IP DNA quantities as percentage of corresponding inputs.
siRNA transfection
siRNA transfections were performed as previously described (30). STHdh111/111 cells were transfected with siRNA to mouse MeCP2 (Dharmacon, Lafayette, CO, USA) or non-targeting control siRNA (Dharmacon) using Lipofectamine 2000 (Invitrogen) in OPTI-MEM I (Invitrogen) for 24–72 h. Final concentration of the siRNAs in cell culture media was 100 nm.
RNA extraction and reverse transcription
RNA was extracted from cell lines using a RNeasy kit (Qiagen, Valencia, CA, USA) according to manufacturer's instructions and as previously described (50,53). Reverse transcription reactions were performed using specific primers in an iCycler (Bio-Rad) (25°C for 10 min, 42°C for 50 min, 70°C for 15 min) to quantify the amount of gene expression when compared with a standard curve (Superscript First Strand Synthesis System; Invitrogen, Carlsbad, CA, USA). The following primers were used: BDNF exon IV: forward 5′-TTCCACTATCAATAATTTAACTTCTTTGC-3′ and reverse 5′-CTCTTACTATATATTTCCCCTTCTCTTCAGT-3′; GAPDH: forward 5′-AACAGCAACTCCCATTC TTC-3′ and reverse 5′-TGGTCCAGGGTTTCTTACTC-3′. BDNF primers were based on previously published sequences (54). Quantitative real time-PCR was performed using 50 PCR cycles (95°C for 30 s, 57°C for 60 s, 72°C for 90 s) in an iCycler (Bio-Rad) with the use of SYBR-green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The threshold cycle for each sample was chosen from the linear range and converted to a starting quantity by interpolation from a standard curve run on the same plate for each set of primers. For each replicate, mRNA levels were normalized to their respective GAPDH mRNA levels.
FUNDING
This work was supported by the Hereditary Disease Foundation (G.S.V.) and HDSA Coalition for the Cure (J.-H.J.C.) and National Institutes of Health AG15379 (O.B.).
ACKNOWLEDGEMENTS
The authors would like to thank Dr Marcy E. MacDonald (Massachusetts General Hospital, Boston, MA, USA) for providing us with STHdh cell lines. Drs Anne Young, Kimberly Kegel and Lianna Orlando (Massachusetts General Hospital, Boston, MA, USA) for providing us with Hdh mouse brain samples.
REFERENCES
- 1.Cha J.H. Transcriptional signatures in Huntington's disease. Prog. Neurobiol. 2007;83:228–248. doi: 10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Augood S.J., Faull R.L., Emson P.C. Dopamine D1 and D2 receptor gene expression in the striatum in Huntington's disease. Ann. Neurol. 1997;42:215–221. doi: 10.1002/ana.410420213. [DOI] [PubMed] [Google Scholar]
- 3.Cha J.H., Kosinski C.M., Kerner J.A., Alsdorf S.A., Mangiarini L., Davies S.W., Penney J.B., Bates G.P., Young A.B. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc. Natl Acad. Sci. USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luthi-Carter R., Strand A., Peters N.L., Solano S.M., Hollingsworth Z.R., Menon A.S., Frey A.S., Spektor B.S., Penney E.B., Schilling G., et al. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum. Mol. Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 5.Sadri-Vakili G., Bouzou B., Benn C.L., Kim M.O., Chawla P., Overland R.P., Glajch K.E., Xia E., Qiu Z., Hersch S.M., et al. Histones associated with downregulated genes are hypo-acetylated in Huntington's disease models. Hum. Mol. Genet. 2007;16:1293–1306. doi: 10.1093/hmg/ddm078. [DOI] [PubMed] [Google Scholar]
- 6.Kuhn A., Goldstein D.R., Hodges A., Strand A.D., Sengstag T., Kooperberg C., Becanovic K., Pouladi M.A., Sathasivam K., Cha J.H., et al. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- 7.Kazantsev A., Preisinger E., Dranovsky A., Goldgaber D., Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl Acad. Sci. USA. 1999;96:11404–11409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steffan J.S., Kazantsev A., Spasic-Boskovic O., Greenwald M., Zhu Y.Z., Gohler H., Wanker E.E., Bates G.P., Housman D.E., Thompson L.M. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl Acad. Sci. USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nucifora F.C., Jr, Sasaki M., Peters M.F., Huang H., Cooper J.K., Yamada M., Takahashi H., Tsuji S., Troncoso J., Dawson V.L., et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 10.Huang C.C., Faber P.W., Persichetti F., Mittal V., Vonsattel J.P., MacDonald M.E., Gusella J.F. Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somatic Cell Mol. Genet. 1998;24:217–233. doi: 10.1023/b:scam.0000007124.19463.e5. [DOI] [PubMed] [Google Scholar]
- 11.Dunah A.W., Jeong H., Griffin A., Kim Y.M., Standaert D.G., Hersch S.M., Mouradian M.M., Young A.B., Tanese N., Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 12.Li S.H., Cheng A.L., Zhou H., Lam S., Rao M., Li H., Li X.J. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol. Cell. Biol. 2002;22:1277–1287. doi: 10.1128/mcb.22.5.1277-1287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boutell J.M., Thomas P., Neal J.W., Weston V.J., Duce J., Harper P.S., Jones A.L. Aberrant interactions of transcriptional repressor proteins with the Huntington's disease gene product, huntingtin. Hum. Mol. Genet. 1999;8:1647–1655. doi: 10.1093/hmg/8.9.1647. [DOI] [PubMed] [Google Scholar]
- 14.Yohrling G.J.T., Jiang G.C., DeJohn M.M., Miller D.W., Young A.B., Vrana K.E., Cha J.H. Analysis of cellular, transgenic and human models of Huntington's disease reveals tyrosine hydroxylase alterations and substantia nigra neuropathology. Brain Res. Mol. Brain Res. 2003;119:28–36. doi: 10.1016/j.molbrainres.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 15.Yamanaka T., Miyazaki H., Oyama F., Kurosawa M., Washizu C., Doi H., Nukina N. Mutant Huntingtin reduces HSP70 expression through the sequestration of NF-Y transcription factor. EMBO J. 2008;27:827–839. doi: 10.1038/emboj.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kegel K.B., Meloni A.R., Yi Y., Kim Y.J., Doyle E., Cuiffo B.G., Sapp E., Wang Y., Qin Z.H., Chen J.D., et al. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J. Biol. Chem. 2002;277:7466–7476. doi: 10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- 17.Zuccato C., Tartari M., Crotti A., Goffredo D., Valenza M., Conti L., Cataudella T., Leavitt B.R., Hayden M.R., Timmusk T., et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 18.Packer A.N., Xing Y., Harper S.Q., Jones L., Davidson B.L. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington's disease. J. Neurosci. 2008;28:14341–14346. doi: 10.1523/JNEUROSCI.2390-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holbert S., Denghien I., Kiechle T., Rosenblatt A., Wellington C., Hayden M.R., Margolis R.L., Ross C.A., Dausset J., Ferrante R.J., et al. The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in huntington's disease pathogenesis. Proc. Natl Acad. Sci. USA. 2001;98:1811–1816. doi: 10.1073/pnas.041566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 21.Liu J., Francke U. Identification of cis-regulatory elements for MECP2 expression. Hum. Mol. Genet. 2006;15:1769–1782. doi: 10.1093/hmg/ddl099. [DOI] [PubMed] [Google Scholar]
- 22.Moretti P., Zoghbi H.Y. MeCP2 dysfunction in Rett syndrome and related disorders. Curr. Opin. Genet. Dev., 2006;16:276–281. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Lewis J.D., Meehan R.R., Henzel W.J., Maurer-Fogy I., Jeppesen P., Klein F., Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 24.Meehan R.R., Lewis J.D., Bird A.P. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic. Acids. Res. 1992;20:5085–5092. doi: 10.1093/nar/20.19.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bienvenu T., Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- 26.Klose R., Bird A. Molecular biology. MeCP2 repression goes nonglobal. Science. 2003;302:793–795. doi: 10.1126/science.1091762. [DOI] [PubMed] [Google Scholar]
- 27.Nan X., Ng H.H., Johnson C.A., Laherty C.D., Turner B.M., Eisenman R.N., Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 28.Fuks F., Hurd P.J., Wolf D., Nan X., Bird A.P., Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 29.Benn C.L., Sun T., Sadri-Vakili G., McFarland K.N., DiRocco D.P., Yohrling G.J., Clark T.W., Bouzou B., Cha J.H. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly=to DNA in a polyglutamine-dependent manner. J. Neurosci. 2008;28:10720–10733. doi: 10.1523/JNEUROSCI.2126-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim M.O., Chawla P., Overland R.P., Xia E., Sadri-Vakili G., Cha J.H. Altered histone monoubiquitylation mediated by mutant huntingtin induces transcriptional dysregulation. J. Neurosci. 2008;28:3947–3957. doi: 10.1523/JNEUROSCI.5667-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas A.V., Berezovska O., Hyman B.T., von Arnim C.A. Visualizing interaction of proteins relevant to Alzheimer's disease in intact cells. Methods. 2008;44:299–303. doi: 10.1016/j.ymeth.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agarwal N., Hardt T., Brero A., Nowak D., Rothbauer U., Becker A., Leonhardt H., Cardoso M.C. MeCP2 interacts with HP1 and modulates its heterochromatin association during myogenic differentiation. Nucleic. Acids. Res. 2007;35:5402–5408. doi: 10.1093/nar/gkm599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zuccato C., Ciammola A., Rigamonti D., Leavitt B.R., Goffredo D., Conti L., MacDonald M.E., Friedlander R.M., Silani V., Hayden M.R., et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 34.Chen W.G., Chang Q., Lin Y., Meissner A., West A.E., Griffith E.C., Jaenisch R., Greenberg M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 35.Martinowich K., Hattori D., Wu H., Fouse S., He F., Hu Y., Fan G., Sun Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 36.Lakowicz J.R., Szmacinski H., Nowaczyk K., Johnson M.L. Fluorescence lifetime imaging of free and protein-bound NADH. Proc. Natl Acad. Sci. USA. 1992;89:1271–1275. doi: 10.1073/pnas.89.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elangovan M., Day R.N., Periasamy A. Nanosecond fluorescence resonance energy transfer-fluorescence lifetime imaging microscopy to localize the protein interactions in a single living cell. J. Microsc. 2002;205:3–14. doi: 10.1046/j.0022-2720.2001.00984.x. [DOI] [PubMed] [Google Scholar]
- 38.Roux J.C., Zala D., Panayotis N., Borges-Correia A., Saudou F., Villard L. Modification of Mecp2 dosage alters axonal transport through the Huntingtin/Hap1 pathway. Neurobiol. Dis. 2012;45:786–795. doi: 10.1016/j.nbd.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Huang E.J., Reichardt L.F. Neurotrophins: roles in neuronal development and function. Ann. Rev. Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poo M.M. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 41.Zweifel L.S., Kuruvilla R., Ginty D.D. Functions and mechanisms of retrograde neurotrophin signalling. Nat. Rev. Neurosci. 2005;6:615–625. doi: 10.1038/nrn1727. [DOI] [PubMed] [Google Scholar]
- 42.Gauthier L.R., Charrin B.C., Borrell-Pages M., Dompierre J.P., Rangone H., Cordelieres F.P., De Mey J., MacDonald M.E., Lessmann V., Humbert S., et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 43.Wang L., Lin F., Wang J., Wu J., Han R., Zhu L., Zhang G., Difiglia M., Qin Z. Truncated N-terminal huntingtin fragment with expanded-polyglutamine (htt552–100Q) suppresses brain-derived neurotrophic factor transcription in astrocytes. Acta. Biochim. Biophys. Sin. 2012;44:249–258. doi: 10.1093/abbs/gmr125. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Z., Hong E.J., Cohen S., Zhao W.N., Ho H.Y., Schmidt L., Chen W.G., Lin Y., Savner E., Griffith E.C., et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skene P.J., Illingworth R.S., Webb S., Kerr A.R., James K.D., Turner D.J., Andrews R., Bird A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wheeler V.C., White J.K., Gutekunst C.A., Vrbanac V., Weaver M., Li X.J., Li S.H., Yi H., Vonsattel J.P., Gusella J.F., et al. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- 47.Trettel F., Rigamonti D., Hilditch-Maguire P., Wheeler V.C., Sharp A.H., Persichetti F., Cattaneo E., MacDonald M.E. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 48.Bacskai B.J., Skoch J., Hickey G.A., Allen R., Hyman B.T. Fluorescence resonance energy transfer determinations using multiphoton fluorescence lifetime imaging microscopy to characterize amyloid-beta plaques. J. Biomed. Opt. 2003;8:368–375. doi: 10.1117/1.1584442. [DOI] [PubMed] [Google Scholar]
- 49.Berezovska O., Lleo A., Herl L.D., Frosch M.P., Stern E.A., Bacskai B.J., Hyman B.T. Familial Alzheimer's disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J. Neurosci. 2005;25:3009–3017. doi: 10.1523/JNEUROSCI.0364-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sadri-Vakili G., Kumaresan V., Schmidt H.D., Famous K.R., Chawla P., Vassoler F.M., Overland R.P., Xia E., Bass C.E., Terwilliger E.F., et al. Cocaine-induced chromatin remodeling increases brain-derived neurotrophic factor transcription in the rat medial prefrontal cortex, which alters the reinforcing efficacy of cocaine. J. Neurosci. 2010;30:11735–11744. doi: 10.1523/JNEUROSCI.2328-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen-Plotkin A.S., Sadri-Vakili G., Yohrling G.J., Braveman M.W., Benn C.L., Glajch K.E., DiRocco D.P., Farrell L.A., Krainc D., Gines S., et al. Decreased association of the transcription factor Sp1 with genes downregulated in Huntington's disease. Neurobiol. Dis. 2006;22:233–241. doi: 10.1016/j.nbd.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 52.Jiang X., Tian F., Du Y., Copeland N.G., Jenkins N.A., Tessarollo L., Wu X., Pan H., Hu X.Z., Xu K., et al. BHLHB2 Controls Bdnf promoter 4 activity and neuronal excitability. J. Neurosci. 2008;28:1118–1130. doi: 10.1523/JNEUROSCI.2262-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmidt H.D., Sangrey G.R., Darnell S.B., Schassburger R.L., Cha J.H., Pierce R.C., Sadri-Vakili G. Increased brain-derived neurotrophic factor (BDNF) expression in the ventral tegmental area during cocaine abstinence is associated with increased histone acetylation at BDNF exon I-containing promoters. J. Neurochem. 2012;120:202–209. doi: 10.1111/j.1471-4159.2011.07571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aid T., Kazantseva A., Piirsoo M., Palm K., Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]