

A vast number of biological processes are mediated by multivalent ligand-receptor interactions, including cell adhesion, host invasion by pathogens, pathogen neutralization by host and numerous cell regulatory signaling pathways.[1] Multivalency is especially important for carbohydrate-receptor interactions: whereas individual glycans[2] may bind with low affinity to a single binding site, the clustering of glycans creates a high-avidity interaction with clustered binding sites. This “carbohydrate cluster effect”[1b] has been demonstrated experimentally with synthetic multivalent carbohydrate ligands which bind well to protein targets. These ligands have included oligo- and polyvalent clusters of glycans on diverse scaffolds, including small molecules, dendrimers, polymers and even viral capsids.
To date, most glycocluster ligands have been designed for synthetic convenience rather than control of tertiary structure. However, the biological activity of the natural glycocluster may be influenced by tertiary structure and other elements which are not usually addressed in synthetic glycocluster designs, such as: 1) Glycan spacing and orientation – glycans are normally attached to synthetic scaffolds through long flexible linkers, and the scaffolds themselves are often flexible.[3] 2) Glycan internal flexibility – in a natural glycocluster, neighboring structures may restrict a glycan's conformational ensemble, but this fingerprint is lost when the glycan is placed on an artificial scaffold. 3) Non-carbohydrate recognition elements – some receptors may recognize a combination of glycans and peptide or lipid elements.[4] Few structures of glycocluster-receptor complexes have been solved, providing little data on which to base synthetic glycocluster designs. Even with unlimited structural data, this would be an exceedingly complex challenge for rational design.
As an alternative to rational design, we have been interested in directed evolution-based design of glycocluster ligands. Figure 1 outlines this concept: a library of scaffold molecules is glycosylated, generating a library of glycoclusters. The “best” glycoclusters are selected from the pool by binding to the target protein. These selection winners are then replicated to form a second-generation library and the process is repeated for several rounds until the pool is sufficiently enriched in high-affinity binders. We have chosen DNA as our glycocluster scaffolding material because DNA is easy to synthesize, easy to replicate by PCR, can fold into diverse sequence-dependent structures, and is amenable to sequence-specific “glycosylation” by glycan azides using CuAAC[5] (“click”) attachment to alkyne-modified nucleobases. Iterative selection/amplification of DNA structures (SELEX) is often performed to obtain DNAs which bind to a target.[6] Our method, by contrast, would yield DNA scaffolds whose major function would be to position and support glycans optimally for target binding. However, these DNAs might also contain elements which would interact directly with the target, mimicking any non-carbohydrate components necessary in the natural ligand.
Figure 1.

Directed evolution of glycosylated DNA scaffolds.
We decided to test this concept in the design of glycoclusters which mimic the epitope of 2G12, an antibody which protects against HIV infection and binds to a cluster of high-mannose glycans on the HIV envelope protein gp120.[7] Rationally-designed clusters of these glycans have been tested as vaccines to elicit 2G12-like antibodies, but without success.[8] Our evolution-based design would be the product of the procedure outlined in Figure 1, using a high-mannose glycan as the azide and 2G12 as the target protein. However, to enable PCR amplification of selection winners with such large modifications on the DNA bases, we have significantly redesigned the traditional SELEX protocol. Our method, which we term SELMA[9] (SELection with Modified Aptamers) is detailed in Scheme 1.
Scheme 1.

SELMA (SELection with Modified Aptamers)
The SELMA method (Scheme 1) begins with (a) a synthetic library of ssDNA hairpins containing a stem-loop, a (–)-sense random region (colored hollow bar) and primer sites 1 and 2. Polymerase extension with alkyne-substituted EdUTP instead of dTTP creates a dsDNA hairpin library (b), with alkyne-modified EdU bases only in the (+)-sense strand. CuAAC chemistry with a glycan azide transforms the alkynyl bases into “glyco-bases”, affording a glyco-dsDNA library (c). As before, the base modifications (now carbohydrates) are present only in the (+)-strand. Generation of the library is then completed by a strand displacement reaction: annealing of primer 2 inside the loop and polymerase extension with all-natural dNTPs creates an all-natural (+)-sense strand which displaces the glycoDNA strand, creating a library of glyco-ssDNA-dsDNA hybrids (d). The glyco-ssDNA (+)-sense strand now folds in a sequence dependent manner and exhibits a “phenotype”. The covalently-linked dsDNA region contains the same sequence with no unnatural bases and can be efficiently amplified by PCR, serving as the genetic “barcode”.[10] The best binders are then isolated from the library by capture on solid-phase-bound 2G12, and this small fraction of the library (e) is amplified by PCR (primers 1 and 2 + natural dNTP's) affording the (n+1)th-generation library without the hairpin portion (f). The (n+1)th-generation library is then restored to ssDNA hairpin form (i) by bidirectional polymerase extension with an overhanging biotinylated primer and removal of the biotinylated strand (g-i). A similar selection scheme has been proposed, but not reduced to practice, for TNA libraries.[11]
To facilitate rapid testing of our method, we chose to glycosylate our first library with easily-synthesized Man4 tetrasaccharide (Scheme 2), which comprises the majority of the carbohydrate recognized by 2G12. Crich-Kahne β-mannosylation[12] attached the core β-mannose 1 to cyclohexyl linker 2. Protection of the sulfonamide N-H was essential to ensure clean mono-coupling of 3 with trisaccharide donor 4. [13] Global deprotection of 5,[14] followed by azidation,[15] reliably afforded 100-mg quantities of the desired Man4-azide (6).
Scheme 2.
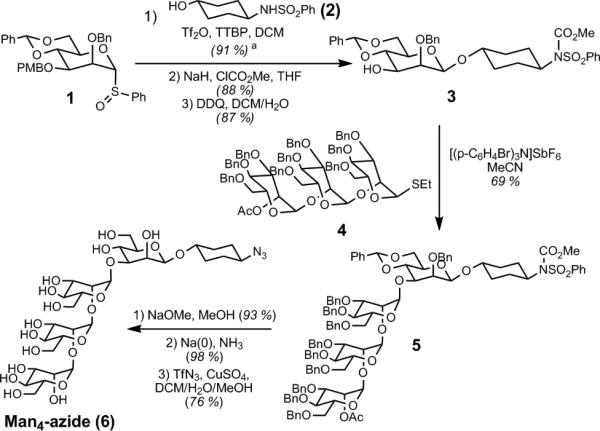
Synthesis of Man4-azide
With Man4-azide in hand, selection was then initiated with ~40 pmol of library (~2 × 1013 sequences). For the first cycle of SELMA, the state of the library at each selection stage was validated by observation in a PAGE gel (Figure 2). The original ssDNA hairpin library (Scheme 1a) ran as a poorly-staining smear (Figure 2, lane 1). After polymerase extension in the presence of dATP, dCTP, dGTP and EdUTP, the resulting duplex hairpin structure (Scheme 1b) ran on the gel as a narrow band (Figure 2, lane 2) with much less mobility than simple dsDNAs of similar length, due to the large molecular radius of the hairpin moiety. After CuAAC attachment of Man4 glycans, the glyco-dsDNA hairpins (Scheme 1c) ran as a diffuse band (Figure 2, lane 3) with still less mobility in the gel.[16] After strand displacement, the glyco-ssDNA-dsDNA hybrid structure of the library (see Scheme 1d) was confirmed by several observations and control experiments. First, it ran as a smear in the gel (Figure 2, lane 5). Additionally, treatment with exonuclease I (which digests the 3’-terminal ssDNA portion) resulted in the appearance of a sharp 80-bp band corresponding to the dsDNA portion (Figure 2, lane 6). By contrast, the glyco-dsDNA hairpins (Scheme 1c) showed no change upon exonuclease treatment (Figure 2, lanes 3 vs. 4). Heating the hybrids to 95 °C (but not 75 °C) destabilized the duplex portion of the hybrid structure, allowing the glycosylated strand to reinvade and expel the unglycosylated single strand. This results in a return to the glyco-dsDNA hairpin structure (Scheme 1c), which is impervious to the exonuclease (Figure 2, lane 8, same as lanes 3 & 4).
Figure 2.
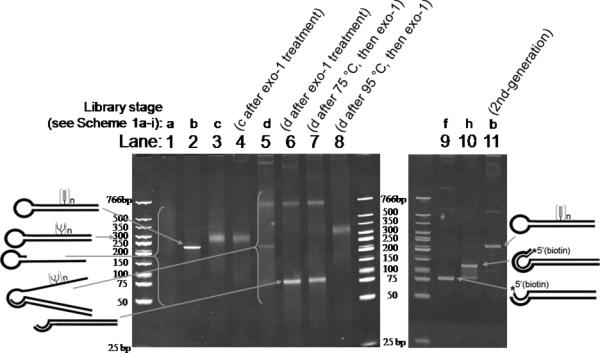
PAGE Analysis of Individual SELMA Steps.
After this confirmation of the desired dsDNA-ssDNA hybrid structure, we began selection: the library was incubated with 2G12 and the 2G12-bound fraction was captured with protein A beads. Bound glycoclusters were retrieved from the beads by thermal denaturation and subjected to PCR with biotinylated primer 2, giving the 2nd-generation library in dsDNA format (Scheme 1f), which ran as the expected sharp 80-bp band on the PAGE gel (Figure 2, lane 9). The library was then converted back to its ssDNA hairpin form (Scheme 1i) in three steps. Removal of primer-2-derived biotinylated strand with streptavidin beads and polymerase extension with an overhanging biotinylated primer afforded 120-bp dsDNA product (Scheme 1h and Figure 2, lane 10). Finally, removal of the biotinylated strand from the 120-bp duplex afforded the 2nd-generation library in ssDNA hairpin format (Scheme 1i). This ssDNA hairpin could now be extended with dATP, dCTP, dGTP and EdUTP, to produce the 2nd-generation library in dsDNA hairpin form (Scheme 1b) which again ran as a sharp band, identical to the first cycle (Figure 2, lane 11 vs. lane 2).
Now that all SELMA steps had been validated, the entire cycle was repeated through multiple rounds. Rounds 2, 4, and 6 included a negative selection to remove library members that bound to protein A beads. Enrichment of 2G12 binders in the population was assessed by monitoring the number of PCR cycles required to regenerate the library. Between rounds 5, 6 and 7, enrichment of the library leveled off, so the selection was terminated and the resulting PCR products were cloned. Sequencing of 20 randomly-selected clones yielded 19 full sequences, including 2 pairs of duplicates and 15 unique sequences with no apparent similarity (see Supporting Information).
Examination of these sequences showed that they contained 7-14 glycosylation (EdU) sites. Three glycoclusters (clones 4/5, 16/23, 18), each containing 10 glycosylation sites, were synthesized and their Kd's with 2G12 were measured in a filter binding assay. Glycoclusters 4/5, 16/23 and 18 displayed moderate affinity for 2G12, with values of Kd = 270 ± 40 nM, 220 ± 50 nM and 330 ± 30 nM, respectively (Figure 3a).[17] This moderate affinity, combined with the large number of glycosylation sites, suggested that high valency alone was responsible for the observed binding to 2G12. However, neither the starting library (of which > 75 % contained 7-15 glycosylations)[18] nor a random sequence containing 10 glycosylated positions showed detectable binding to 2G12. Therefore, the affinity of our selection winners is sequence dependent and not simply the result of high valency.
Figure 3.

Selection Results. a Preliminary 2G12- dependent filter binding data for clones 4/5,16/23 and 18, the starting library, and arbitrary sequence containing 10 glycosylation sites. b Effects of glycosylation, gp120 competition, and single/double-strandedness on 2G12 binding by glycocluster 16/23 determined by filter binding. c Mutagenesis study: Values of Kd and fraction bound (Fmax) for truncated and mutated glycocluster 16. Entry 1 is the parent sequence. Underlined sequence is the random region. S denotes Man4 -glycosylated EdU. † Kd and Fmax were calculated by fitting data points to y=(Fmax*x)/(Kd+x). Errors reported are the standard error of the curve fit in all cases except entry 1, for which the average of errors in entries 1-8 is reported. * The value of Kd reported in the text, in entries 1-8 and entries 9-21 were measured with different batches of 2G12, giving slightly different values of Kd for the parent clone 16 (text vs. entries 1 vs. 9). The Kd values in entries 10-21 should be compared only with entry 9. ** Kd was much greater than the maximum 2G12 concentration tested and Fmax was constrained to 1 to fit curve with finite Kd value.
We then performed several experiments with glycocluster 16/23 to clarify the elements necessary for binding to 2G12 (Figure 3b). When annealed to its complementary DNA strand, glycocluster 16/23 bound 2G12 significantly less efficiently, showing that binding is dependent on tertiary structure. Additionally, no binding was observed in the absence of glycosylation, strongly suggesting the binding contacts with 2G12 are mostly or exclusively made through glycans and not through DNA alone. Gratifyingly, binding was significantly diminished in the presence of gp120, showing that gp120 and glycocluster 16/23 compete for the same (or overlapping) site(s) on 2G12.
Next, we carefully dissected the binding determinants of glycocluster 16/23 through a series of mutagenesis experiments (Figure 3c), starting with truncation at both the 5’ and 3’ ends (entries 1-8). The extreme ends were not essential for binding to 2G12; however, truncations extending beyond the first and last glycosylation sites did result in total loss of binding. We then performed point mutagenesis, replacing each glycosylated EdU residue with cytosine (entries 9-21). Seven of these mutations produced little change in the value of Kd, but mutations in the 2nd, 4th, and 10th glycosylation positions (entries 11, 13 and 19) caused a drastic loss of binding (Kd >> 800 nM), suggesting that these glycans directly contact 2G12. However, glycoclusters containing only these three glycosylation sites (entries 20 and 21) failed to bind to 2G12, suggesting that the other glycans may be important for maintaining tertiary structure. We attempted to gain additional insight into this question by Mfold secondary structure prediction,[19] but the resulting structures did not provide an obvious explanation for the importance of the 2nd, 4th, and 10th glycosylation sites. Mfold calculation is probably of limited validity in this case, as it does not take into account the Man4-modification of ten bases.
In conclusion, this work is the first to demonstrate the feasibility of directed evolution for glycocluster design. We have shown that an “evolved” Man4 glycocluster binds to 2G12 and can compete with gp120. Future work will involve structural studies of these glycoclusters, refinement of the selection design, use of the full Man9 glycans and in vivo immunogenicity studies. In addition to the HIV vaccine problem, this method should be broadly applicable to the study of other multivalent interactions.
Supplementary Material
ACKNOWLEDGEMENTS
I. J. K. gratefully acknowledges Brandeis University and the NIH (R01 AI090745). L. H. gratefully acknowledges the NIH (U01 AI75466-01 and R01 GM054403).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Iain S. MacPherson, Department of Chemistry Brandeis University 415 South St. Waltham, MA 02454
J. Sebastian Temme, Department of Chemistry Brandeis University 415 South St. Waltham, MA 02454.
Sevan Habeshian, Department of Chemistry Brandeis University 415 South St. Waltham, MA 02454.
Krzysztof Felczak, Center for Drug Design University of Minnesota 516 Delaware St. SE MMC 204 Minneapolis, MN 55455.
Krzysztof Pankiewicz, Center for Drug Design University of Minnesota 516 Delaware St. SE MMC 204 Minneapolis, MN 55455.
Lizbeth Hedstrom, Departments of Biology and Chemistry Brandeis University 415 South St. Waltham, MA 02454.
Isaac J. Krauss, Department of Chemistry Brandeis University 415 South St. Waltham, MA 02454
References
- 1.a Mammen M, Choi S-K, Whitesides GM. Angew. Chem. Int. Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; b Lundquist JJ, Toone EJ. Chem. Rev. 2002;102:555–578. doi: 10.1021/cr000418f. [DOI] [PubMed] [Google Scholar]; c Kiessling LL, Gestwicki JE, Strong LE. Angew. Chem. Int. Ed. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.„Glycan“ is a generic term for a mono-, oligo-, or polysaccharide.
- 3.Examples of rigid scaffolds include cyclodextrins and calixarenes. For reviews, see Baldini L, Casnati A, Sansone F, Ungaro R. Chem. Soc. Rev. 2007;36:254–266. doi: 10.1039/b603082n.Ortiz Mellet C, Defaye J, García Fernández JM. Chem. Eur. J. 2002;8:1982–1990. doi: 10.1002/1521-3765(20020503)8:9<1982::AID-CHEM1982>3.0.CO;2-5.
- 4.a Ohyabu N, Hinou H, Matsushita T, Izumi R, Shimizu H, Kawamoto K, Numata Y, Togame H, Takemoto H, Kondo H, Nishimura S-I. J. Am. Chem. Soc. 2009;131:17102–17109. doi: 10.1021/ja903361f. [DOI] [PubMed] [Google Scholar]; b Brooks CL, Schietinger A, Borisova SN, Kufer P, Okon M, Hirama T, MacKenzie CR, Wang L-X, Schreiber H, Evans SV. Proc. Natl. Acad. Sci. U. S. A. 2010;107:10056–10061. doi: 10.1073/pnas.0915176107. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yang Q, Li CS, Wei YD, Huang W, Wang LX. Bioconjugate Chem. 2010;21:875–883. doi: 10.1021/bc9004238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Kolb HC, Finn MG, Sharpless KB. Angew. Chem.-Int. Edit. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; b Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem.-Int. Edit. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; c Gierlich J, Burley GA, Gramlich PM, Hammond DM, Carell T. Org. Lett. 2006;8:3639–3642. doi: 10.1021/ol0610946. [DOI] [PubMed] [Google Scholar]; d Gierlich J, Gutsmiedl K, Gramlich PM, Schmidt A, Burley GA, Carell T. Chem. Eur. J. 2007;13:9486–9494. doi: 10.1002/chem.200700502. [DOI] [PubMed] [Google Scholar]
- 6.Wilson DS, Szostak JW. Annu. Rev. Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- 7.a Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, Srinivasan K, Sodroski J, Moore JP, Katinger H. J. Virol. 1996;70:1100–1108. doi: 10.1128/jvi.70.2.1100-1108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sanders RW, Venturi M, Schiffner L, Kalyanaraman R, Katinger H, Lloyd KO, Kwong PD, Moore JP. J. Virol. 2002;76:7293–7305. doi: 10.1128/JVI.76.14.7293-7305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Scanlan CN, Pantophlet R, Wormald MR, Ollmann Saphire E, Stanfield R, Wilson IA, Katinger H, Dwek RA, Rudd PM, Burton DR. J. Virol. 2002;76:7306–7321. doi: 10.1128/JVI.76.14.7306-7321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, Kunert R, Zhu P, Wormald MR, Stanfield RL, Roux KH, Kelly JW, Rudd PM, Dwek RA, Katinger H, Burton DR, Wilson IA. Science. 2003;300:2065–2071. doi: 10.1126/science.1083182. [DOI] [PubMed] [Google Scholar]; e Hessell AJ, Rakasz EG, Poignard P, Hangartner L, Landucci G, Forthal DN, Koff WC, Watkins DI, Burton DR. PLoS Pathog. 2009;5:e1000433. doi: 10.1371/journal.ppat.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Wang J, Li H, Zou G, Wang LX. Org. Biomol. Chem. 2007;5:1529–1540. doi: 10.1039/b702961f. [DOI] [PubMed] [Google Scholar]; b Joyce JG, Krauss IJ, Song HC, Opalka DW, Grimm KM, Nahas DD, Esser MT, Hrin R, Feng MZ, Dudkin VY, Chastain M, Shiver JW, Danishefsky SJ. Proc. Natl. Acad. Sci. U. S. A. 2008;105:15684–15689. doi: 10.1073/pnas.0807837105. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Luallen RJ, Lin JQ, Fu H, Cai KK, Agrawal C, Mboudjeka I, Lee FH, Montefiori D, Smith DF, Doms RW, Geng Y. J. Virol. 2008;82:6447–6457. doi: 10.1128/JVI.00412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang SK, Liang PH, Astronomo RD, Hsu TL, Hsieh SL, Burton DR, Wong CH. Proc. Natl. Acad. Sci. U. S. A. 2008;105:3690–3695. doi: 10.1073/pnas.0712326105. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Astronomo RD, Kaltgrad E, Udit AK, Wang SK, Doores KJ, Huang CY, Pantophlet R, Paulson JC, Wong CH, Finn MG, Burton DR. Chem. Biol. 2010;17:357–370. doi: 10.1016/j.chembiol.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Our method is the first example of a selection in which dNTPs are modified after incorporation into the library. SELEX has previously been performed with dNTP's bearing modifications which are tolerated by polymerases. For selected examples, see: Keefe AD, Cload ST. Curr. Opin. Chem. Biol. 2008;12:448–456. doi: 10.1016/j.cbpa.2008.06.028.Li MY, Lin N, Huang Z, Du LP, Altier C, Fang H, Wang BH. J. Am. Chem. Soc. 2008;130:12636–12638. doi: 10.1021/ja801510d.Hollenstein M, Hipolito CJ, Lam CH, Perrin DM. ChemBioChem. 2009;10:1988–1992. doi: 10.1002/cbic.200900314.Vaught JD, Bock C, Carter J, Fitzwater T, Otis M, Schneider D, Rolando J, Waugh S, Wilcox SK, Eaton BE. J. Am. Chem. Soc. 2010;132:4141–4151. doi: 10.1021/ja908035g.
- 10.This covalent link between functional structure and its genetic barcode is similar to that used in mRNA display: Liu RH, Barrick JE, Szostak JW, Roberts RW. Rna-Ligand Interactions, Part B, Vol. 318. Academic Press Inc; San Diego: 2000. pp. 268–293.
- 11.Ichida JK, Zou K, Horhota A, Yu B, McLaughlin LW, Szostak JW. J. Am. Chem. Soc. 2005;127:2802–2803. doi: 10.1021/ja045364w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Kahne D, Walker S, Cheng Y, Van Engen D. J. Am. Chem. Soc. 1989;111:6881–6882. [Google Scholar]; b Crich D, Sun S. J. Org. Chem. 1996;61:4506–4507. doi: 10.1021/jo9606517. [DOI] [PubMed] [Google Scholar]
- 13.Geng X, Dudkin VY, Mandal M, Danishefsky SJ. Angew. Chem. Int. Ed. 2004;43:2562–2565. doi: 10.1002/anie.200353626. [DOI] [PubMed] [Google Scholar]
- 14.Iserloh U, Dudkin V, Wang Z-G, Danishefsky SJ. Tetrahedron Lett. 2002;43:7027–7030. [Google Scholar]
- 15.Nyffeler PT, Liang C-H, Koeller KM, Wong C-H. J. Am. Chem. Soc. 2002;124:10773–10778. doi: 10.1021/ja0264605. [DOI] [PubMed] [Google Scholar]
- 16.Completion of the CuAAC reaction was assessed by control reactions on an individual library clone containing 10 alkynes. PAGE and LC/MS analysis showed that ~40% of the sample was fully glycosylated. An additional ~40 and ~20 % were species lacking 1- and 2- glycosylations, respectively (see Supporting Information).
- 17.Wong and coworkers (see ref. 7d) have reported a Kd value of 210 nM for a 9-valent Man4 dendrimer binding to 2G12. However, their assay, based on competition binding to Man4-printed glass slides, was sensitive to glycan printing density. Given the large differences between our assay format and theirs, caution should be exercised in comparison of the two types of data.
- 18.The estimate of 7–15 glycosylations is calculated by adding 2 (the number of constant glycosylation sites in the primer 1 region) to the 5–13 glycosylations in the random region. The 75 % figure is calculated from the binomial distribution:
- 19.Zuker M. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.