

Abstract
There are overwhelming data supporting the inflammatory origin of some epilepsies (e.g., Rasmussen's encephalitis and limbic encephalitis). Inflammatory epilepsies with an autoimmune component are characterized by autoantibodies against membrane-bound, intracellular or secreted proteins (e.g., voltage gated potassium channels). Comparably, little is known regarding autoantibodies targeting nuclear antigen. We tested the hypothesis that in addition to known epilepsy-related autoantigens, human brain tissue and serum from patients with epilepsy contain autoantibodies recognizing nuclear targets. We also determined the specific nuclear proteins acting as autoantigen in patients with epilepsy.
Brain tissue samples were obtained from patients undergoing brain resections to treat refractory seizures, from brain with arteriovenous malformations or from post-mortem multiple sclerosis brain. Patients with epilepsy had no known history of autoimmune disease and were not diagnosed with autoimmune epilepsy. Tissue was processed for immunohistochemical staining. We also obtained subcellular fractions to extract intracellular IgGs. After separating nuclear antibody-antigen complexes, the purified autoantigen was analyzed by mass spectrometry. Western blots using autoantigen or total histones were probed to detect the presence of antinuclear antibodies in the serum of patients with epilepsy. Additionally, HEp-2 assays and antinuclear antibody ELISA were used to detect the staining pattern and specific presence of antinuclear antibodies in serum of patients with epilepsy.
Brain regions from patients with epilepsy characterized by blood-brain barrier disruption (visualized by extravasated albumin) contained extravasated IgGs. Intracellular antibodies were found in epilepsy (n=13/13) but not in multiple sclerosis brain (n= 4/4). In brain from patients with epilepsy, neurons displayed higher levels of nuclear IgGs compared to glia. IgG colocalized with extravasated albumin. All subcellular fractions from brain resections of patients with epilepsy contained extravasated IgGs (n=10/10), but epileptogenic cortex, where seizures originated from, displayed the highest levels of chromatin-bound IgGs. In the nuclear IgG pool, anti-histone autoantibodies were identified by two independent immunodetection methods. HEp-2 assay and ELISA confirmed the presence of anti-histone (n= 5/8) and anti-chromatin antibodies in serum from patients with epilepsy
We developed a multi-step approach to unmask autoantigens in the brain and sera of patients with epilepsy. This approach revealed antigen-bound antinuclear antibodies in neurons and free antinuclear IgGs in serum of patients with epilepsy. Conditions with blood-brain barrier disruption but not seizures, were characterized by extravasated but not chromatin-bound IgGs. Our results show that the pool of intracellular IgG in brain of patients with epilepsy consists of nucleus-specific autoantibodies targeting chromatin and histones. Seizures may be the trigger of neuronal uptake of antinuclear antibodies.
Keywords: neuroimmunology, seizure disorders, autoantigen, autoantibody, anti-histone antibody, anti-chromatin antibody, autoimmune
Introduction
Studies on inflammatory mechanisms in epilepsy have been burgeoning, with a 300% increase in published articles on PubMed from 1993-2003 compared to the previous decade. It is thus not surprising that new models of seizures have emerged. These models take into account the knowledge gained from clinical studies, and are based on mechanisms, receptors, and pathways that were formerly reserved for the immunologist (Galanopoulou, 2013; Marchi et al., 2007b; Marchi et al., 2009; Vezzani et al., 2012). Evidence to support a role for inflammation and autoimmunity in epilepsy has come from indirect and direct sources. For example, the anticonvulsant activity of steroids in some epilepsies (indirect (Marchi et al., 2011b)), together with the presence of inflammatory signs and markers in serum or cerebrospinal fluid (CSF) of patients (direct (Nabbout, 2012)) have been interpreted as clues suggestive of a role for the immune response. In addition, well-established models of seizures which were developed to specifically target neurons have been re-examined to reveal an underlying inflammatory etiology. For example, research has shown that a putative muscarinic convulsant, pilocarpine, acts by immune activation and not as previously suspected by a CNS exclusive action on muscarinic receptors (Fabene et al., 2008; Marchi et al., 2007b; Marchi et al., 2009; Uva et al., 2007). The role of inflammation in seizure disorders has therefore been recognized as an etiologic reality and as an important target for therapy (Marchi et al., 2009; Marchi et al., 2010; Marchi et al., 2012b; Vezzani et al., 2011).
There are three groups of “inflammation-related seizures” (IRS): 1) Seizures caused by the presence of a pathogen. These are perhaps the least studied cluster of IRS and include seizures due to meningitis, neurotropic pathogens, etc. In developing countries, pathogens are considered the highest risk factor for acute seizure and increase the risk of epilepsy by eleven fold (Bharucha et al., 2008). 2) A large family of IRS encompasses autoimmune epilepsy syndromes, where one of the etiological mechanisms is believed to be the presence of anti-neuronal autoantibodies typically targeting either ion channels, intracellular epitopes or neurotransmitter receptors (Graus and Dalmau, 2012; Lancaster and Dalmau, 2012; McKnight et al., 2005). 3) A number of seizure disorders lacking either of these features (pathogen or autoantibodies) can be classified as IRS based on a therapeutic response to immunomodulators (Granata et al., 2008; Granata et al., 2011), vascular changes consistent with an ongoing inflammatory process (e.g., blood-brain barrier disruption; for a review see (Janigro, 2012)), or concomitant brain changes that mimic some, but not all, signs of inflammation (Granata et al., 2011; Nabbout et al., 2011; Nabbout, 2012).
As mentioned above, the third type of IRS may be linked to blood-brain barrier (BBB) disruption. The BBB is the gatekeeper of immune privilege in the CNS (Bechmann et al., 2007; Galea et al., 2007). The BBB maintains ionic homeostasis which, in turn, controls neuronal excitability (de Vries et al., 2012; Janigro, 2012; Marchi et al., 2012b; Seiffert et al., 2004). Thus, BBB disruption (BBBD) not only causes loss of immune privilege but may also directly result in seizures (Marchi et al., 2007a). A reporter of BBB failure, extravasated albumin levels in CSF, has also been used to detect focal BBBD by immunohistochemistry. Interestingly, after diffusion through the CNS extracellular space, albumin accumulates in neurons and glia (David et al., 2009a; David et al., 2009b; Seiffert et al., 2004). Regions of focal BBBD can also be measured by detection of extravasated IgGs. Whether extravasated IgGs also enter into brain cells has not been fully elucidated (Michalak et al., 2012).
The presence of IgGs in brain from patients with epilepsy, together with our understanding of the pathophysiology of multiple sclerosis (MS), has been used to propose autoimmunity as an etiologic factor in seizure disorders. Autoantibodies to the NMDA, GABAB and AMPA receptors, as well as the voltage-gated potassium channel and its components LGI1 and CASPR2 have been detected in CSF or serum of patients with seizures (Lancaster and Dalmau, 2012). In addition, autoimmune diseases such as systemic lupus erythematosus (SLE) greatly influence seizure susceptibility (Adelow et al., 2012) Thus, seizure threshold can be lowered by direct action on CNS targets (e.g., glutamate receptors), by exposure to endotoxin (Galic et al., 2008) or by autoimmune targeting of a specific antigen, such as nuclear components. A recent paper has shown that even in absence of autoimmune disease, IgG can be found in brain of mice after lithium/pilocarpine-induced seizures (Michalak et al., 2012). This is also consistent with previous work showing that BBBD, as seen in regions of seizure generation in human brain, is characterized by large deposits of extravasated IgG (Michalak et al., 2012). However, to date, the significance or consequences of IgG extravasation into the CNS has not been fully elucidated.
The CNS of patients with epilepsy provides a unique environment where the coupling of seizure with inflammation, loss of immune privilege and cell death may provide a mechanism for the generation and uptake of autoantibodies against intracellular proteins. Therefore, we examined whether or not autoantibodies against intracellular proteins existed in the CNS and serum of patients with epilepsy where an autoimmune or infectious etiology was ruled out. By using a number of techniques and an approach based on comparison of different pathologies all characterized by BBBD, we isolated autoantigens from subcellular fractions of brain from patients with epilepsy. MS was used as a comparative “neuro-autoimmune” disease, and brain resections derived from cerebrovascular malformations as a means to study BBBD independent of seizures. Our results demonstrate the presence of antinuclear antibodies in brain and serum from patients with epilepsy, and the accumulation of autoantibodies in neuronal nuclei.
Materials and methods
The multimodal approach used for the experiments detailed in this section is depicted graphically in Figure 1.
Figure 1. Schematic representation of the experimental design used for different aspects of the research presented herein. The figure is divided in sections to highlight the three different phases of this research effort.
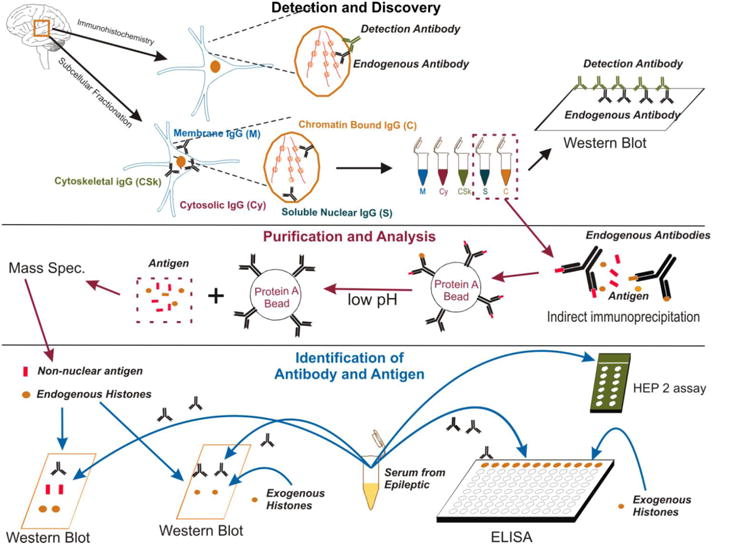
The phase labeled as Detection and Discovery describes the process of isolation of brain samples from surgically resected specimens and their use to unveil, by subcellular fractionation and immunohistochemical detection, antinuclear antibodies. The subcellular fractionation (see Figure 4) allows segregating antigen-antibody complexes from the membrane (M), cytoskeletal (CSk), cytosolic (Cy) and nuclear fractions. The nuclear fraction was further divided into chromatin-bound (C) and soluble (S). At this stage, these fractions were analyzed by Western blotting with use of specific anti-human IgG antibodies. In parallel experiments, formalin-fixed tissue was used to produce the results shown in Figures 2 and 3. Exogenous IgG antibodies (detection antibodies) were used to unveil endogenous IgG content in nuclear and subcellular fractions. After completion of these experiments a second phase (purification and analysis) was undertaken. The first step consisted of an indirect immunoprecipitation process where antigen-IgG complexes were dissociated to obtain free antigen fractions. These fractions were isolated from the brain; only soluble and chromatin-bound nuclear fractions were used. The antigens isolated by these means were further analyzed by mass spec. to unveil (identification of antibody and antigen) the nature of IgG targets. These putative autoantigens were divided into groups, endogenous histones or non-nuclear protein content. To confirm these findings, two separate means of investigations were used. In the first step, brain-derived putative autoantigens were probed by Western blotting to confirm or refute their suspected nature (histones, see Figure 6). In addition, serum samples from patients with epilepsy and controls were isolated to confirm the presence of antinuclear IgG in these samples. Again, the specificities for histones of these antibodies were tested by Western blotting and also by an ad hoc ELISA. An additional set of non-epilepsy patient controls consisted of using samples taken from brain from patients with multiple sclerosis (e.g., Figures 2 and 3) or brain samples isolated during surgery to relieve arteriovenous malformation (AVM) (Figure 4).
Patient Selection
Brain tissue specimens were obtained from patients conforming to the guidelines of the Declaration of Helsinki. All patients signed an informed consent according to institutional review protocols at the Cleveland Clinic Foundation. Patient information and experimental use of patient samples is summarized in Table 1. All brain tissue samples were obtained from surgical resections with the exception of post-mortem MS brain. Post-mortem samples were a generous gift of Dr. Bruce Trapp's laboratory at the Cleveland Clinic Foundation Lerner Research Institute. Inclusion criteria were willingness to participate to the study and lack of positive diagnosis for an autoimmune disease. One patient was identified as RA post-facto and is considered a positive control (Figure 6).
Table 1.
| 1 | 45 | F | Y | Y | Frontal lobe epilepsy | Encephalomalacia | Cortical Atrophy and WM degeneration | N | Meningioma | Carb, Lev | Brain | WB, IHC, IP, MS, |
| 2 | 18 | M | Y | Y | Temporal lobe epilepsy | Cortical malformation | Neuronomegaly and loss of cortical architecture | N | Lam, Lev, Pheno, Clor | Brain | WB | |
| 3 | 2 m | M | Y | Y | Temporal lobe epilepsy | Cortical malformation | Cortical Dysplasia, neuronal cytomegaly/dysmprphia, gliosis and microcalcifications | N | Lev, Lam, Oxcarb, Pheno | Brain | WB | |
| 4 | 23 | M | Y | Y | Temporal lobe epilepsy | Hippocampal sclerosis | neuron loss and gliosis | N | Lam, Lev | Brain | WB, IHC | |
| 5 | 19 | M | Y | Y | Occipital lobe epilepsy | Cortical malformation | Cortical dysplasia | N | Vegus nerve stimulator | Oxcarb, Clon | Brain | WB |
| 6 | 1 | M | Y | Y | Temporal lobe epilepsy | Hemimegalencephaly | Cortical dysplasia, WM atrophy and gliosis | N | Vegus nerve stimulator | Topi, Pheno | Brain | WB, IHC |
| 7 | 12 | F | Y | Y | Frontal Lobe epilepsy | Cortical malformation | Cortical Dysplasia and gliosis | N | Val, Zon, Lev | Brain | WB, IHC | |
| 8A | 14 | M | Y | Y | Temporal lobe epilepsy | Cortical malformation, encephalomalacia w/porencephaly | Cortical dysplasia, fibrotic leptomeninges | N | Topi, Lev, Clon | Brain | WB | |
| 8B | 14 | M | N | Y | Temporal lobe epilepsy | Cortical malformation, encephalomalacia w/porencephaly | Cortical dysplasia, fibrotic leptomeninges | N | Topi, Lev, Clon | Brain | WB | |
| 9 | 7 | M | Y | Y | Parieto-occipital lobe epilepsy | Ganglioglioma | Gangliogliomaw/cortical dysplasia | N | Carb, Lev | Brain | WB, IHC | |
| 10 | 27 | M | Y | Y | Temporal lobe epilepsy | Unknown | Unknown | N | Unknown | Unknown | Brain | IHC |
| 11 | 46 | F | Y | Y | Temporal lobe epilepsy | Unknown | Unknown | N | Unknown | Laco, Preg | Brain | IHC |
| 12 | 48 | M | Y | Y | Temporal lobe epilepsy | Unknown | Unknown | N | Unknown | Unknown | Brain | IHC |
| 13 | 14 | F | Y | Y | Temporal lobe epilepsy | Cortical malformation | Cortical malformation | N | Carb, Pheny, Val, Gabap, Lam, Pheno, Top, Oxcarb, Lev | Brain | IHC | |
| 14 | 36 | M | Y | Y | Temporal lobe epilepsy | Cavernous angioma | Cavernous angioma | N | Lev | Serum | WB-IP, WB-H, ELISA, HEp-2 | |
| 15 | 29 | F | Y | Y | Temporal lobe epilepsy | Unknown | Unknown | N | Anterior temporal lobectomy (hippocamp al sclerosis) | Carb | Serum | WB-IP, WB-H, ELISA, HEp-2 |
| 16 | 32 | M | Y | Y | Temporal lobe epilepsy | Unknown | Unknown | N | Lam, Lev | Serum | WB-IP, WB-H, ELISA, HEp-2 | |
| 17 | 20 | M | Y | Y | Epilepsy | Cortical malformation | Perisylvian polymicrogyria | N | Vegus Nerve stimulator | Laco, Val | Serum | WB-IP, WB-H, ELISA, HEp-2 |
| 18 | 21 | F | Y | Y | Temporal lobe epilepsy | Unknown | Unknown | N | Topi | Serum | ELISA, HEp-2 | |
| 19 | 33 | M | Y | Y | Temporal lobe epilepsy | Unknown | Unknown | N | Lev | Serum | ELISA, HEp-2 | |
| 20 | 30 | F | Y | Y | Temporal lobe epilepsy | Unknown | Unknown | N | Oxcarb, Clon | Serum | ELISA, HEp-2 | |
| 21 | 38 | M | Y | Y | Temporal lobe epilepsy | Unknown | Unknown | N | Val | Serum | ELISA, HEp-2 | |
| 22 | Unknown | Unknown | N | N | Multiple Sclerosis ** | Unknown | Unknown | Y | Unknown | Brain | IHC | |
| 23 | Unknown | Unknown | N | N | Multiple Sclerosis ** | Unknown | Unknown | Y | Unknown | Brain | IHC | |
| 24 | Unknown | Unknown | N | N | Multiple Sclerosis ** | Unknown | Unknown | Y | Unknown | Brain | IHC | |
| 25 | Unknown | Unknown | N | N | Multiple Sclerosis ** | Unknown | Unknown | Y | Unknown | Brain | IHC | |
| 26 | 48 | M | N | N | AVM w/med. controlled seizure | AVM | N | Embolization | Val, Zon, Lev | Brain | WB, IHC | |
| 27 | 23 | M | N | N | AVM w/no history of seizure | AVM | N | Embolization | Brain | WB, IHC | ||
| 28 | 37 | F | Y | N | AVM w/epilepsy | AVM | N | Embolization of left vertebral artery | Lev, Oxcarb, Pheny, Clon | Brain | WB, IHC | |
| 29 | 28 | F | N | N | AVM | N | Brain | MRI | ||||
| C1 | 26 | M | N | N | Healthy control | N | Serum | ELISA, WB-IP, WB-H, HEp-2 | ||||
| C2 | 33 | M | N | N | Healthy control | N | Serum | ELISA, WB-IP, WB-H, HEp-2 | ||||
| Pos. C 1 | 19 | F | Y | Y | Epilepsy | Unknown | Unknown | Y (Rheumatoid Arthritis) | Carb, Lam | Serum | WB-IP, ELISA, HEp-2 |
indicates post-mortem brain tissue samples. All other brain tissue specimens were obtained from surgical resections.
AED abbreviations: Carb = carbamazepine, Lev = levetiracetam, Lam = lamotrigine, Pheno = phenobarbital, Clor = clorazepate, Oxcarb = oxcarbazepine, Clon = clonazepam, Topi = topiramate, Val = valproate, Zon = zonisamide, Pheny = Phenytoin, Laco = lacosamide, Preg = pregabalin,
Experimental use abbreviations: WB = Western blot, IHC = immunohistochemistry, IP = immunoprecipitation, MS = mass spectrometry, WB-IP = Western blot using immunoprecipitation samples, WB-H = Western blot using purified histones, ELISA = enzyme linked immunosorbent assay, HEp-2 = human epithelial cell assay.
Figure 6. A multimodal analysis of serum from patients with epilepsy reveals the presence of specific anti-histone and anti-chromatin antibodies.
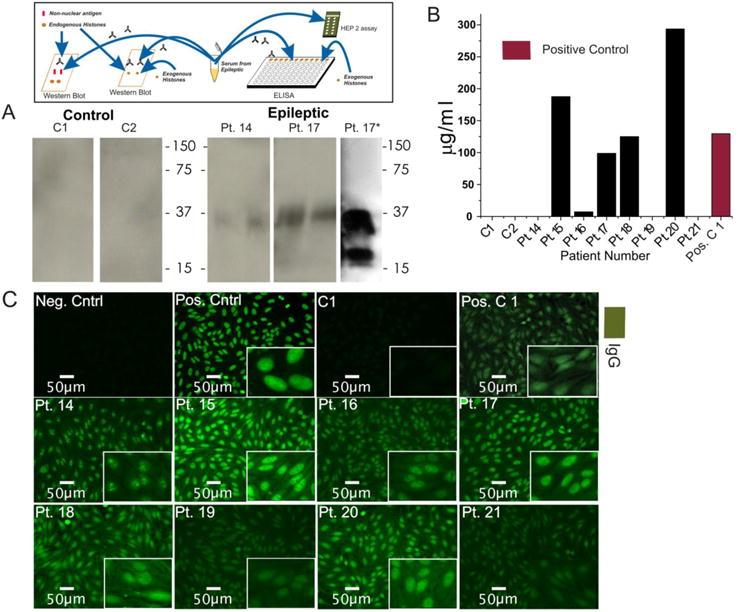
(A) Immunoprecipitation isolated nuclear antigen taken from the brain tissue of a patient with epilepsy was ran on a gel and probed with serum from patients with epilepsy and healthy controls. While control serum produces no signal, serum from patients with epilepsy produces a signal corresponding to histone complexes. Additionally, serum from Pt. 26 was used to probe a gel containing only purified histones (A Pt. 17*). Note the similar binding pattern between both gels using serum from patient 17. (B) ELISA quantifying anti-histone antibodies using the serum of patients with epilepsy and control patients. The ELISA did not detect anti-histone antibodies in control serum but did detect them in 5 of 8 patients with epilepsy. Going further, a HEp-2 assay using the same serum detected the presence of anti-chromatin antibodies (C). Patient 15 shows the speckled patter of general antinuclear staining, patient 14 and 16 show the specific binding pattern of anti-nucleolar and anti-centromere antibodies, respectively. The insert at the top of the figure refers to the phase of experimental design discussed in Figure 1.
Detection and Discovery
Immunohistochemical staining of brain tissue sections
Brain tissue was mounted using Tissue-Tek OCT compound (Sakura Finetek Europe B.V., The Netherlands) and sectioned at approximately 25 μm on a Leica CM3050 cryostat (Leica Microsystems Inc, Buffalo Grove, Il). Nine patients with epilepsy, 4 multiple sclerosis patients, 3 arteriovenous malformation (AVM) patients were included in these experiments.
Immunofluorescent detection of IgG and albumin in neurons, glia and brain parenchyma
Free-floating sections were stained for IgG and albumin. Non-specific binding was minimized by incubation in a 3% goat serum blocking solution at room temperature for 1 hour. Sections of brain tissue were incubated with monoclonal mouse anti-human albumin antibody (1:1000; Sigma-Aldrich, St. Louis, MO). Fluorescently-labeled secondary antibodies used were as follows: Alexa Fluor 594 polyclonal donkey anti-mouse IgG (1:100; Jackson Immunoresearch, West Grove, PA), and fluorescein conjugated polyclonal goat anti-human IgG (1:200, Vector Labs, Bulingame, CA).
3,3′-Diaminobenzidine staining of AVM patient brain tissue
3,3′-diaminobenzidine (DAB) staining of brain tissue sections was achieved using the method from Marchi, et. al., 2010.
Immunofluorescent detection of IgGs in neurons and astrocytes
Free-floating brain sections were stained for IgG and microtubule-associated protein 2 (MAP2). Adjacent sections were stained for IgG and glial fibrillary acidic protein (GFAP). Non-specific binding was minimized by incubation in a 3% goat serum blocking solution at room temperature for 1 hour. The following primary antibodies were used to stain the tissue sections: mouse monoclonal anti-human MAP2 (1:1000; Covance, Princeton, New Jersey), mouse monoclonal anti-human GFAP (1:500; Sigma-Aldrich, St. Louis, MO). The following secondary antibodies were used: goat anti-mouse polyclonal Alexa Fluor 594 (1:400, Jackson Immunoresearch, West Grove, PA), fluorescein goat polyclonal anti-human IgG (1: 200; Vector Labs, Burlingame, CA). Auto-fluorescence was minimized using Sudan Black B. Finally, tissue slices were placed on glass slides and mounted using a glass coverslip and Vectorshield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) to visualize nuclei (Vector Labs, Bulingame, CA). Images were obtained using a Leica Leitz fluorescent microscope and a Leica Microsystems upright confocal microscope with attached cameras (Leica Microsystems, Allendale, New Jersey). Fluorescence intensity and co-localization were measured using Q-Capture software (Q-Capture, Surrey, BC, Canada). Three-dimensional reconstruction of confocal images was performed using Velocity (PerkinElmer, Waltman Massachusetts).
Subcellular fractionation of brain tissue resections
Snap frozen tissue stored at −80°C was processed according to the protocol provided with the Thermo Scientific Subcellular Protein Fractionation Kit for Tissue (Thermo Fisher Scientific, Rockford Il). 200mg of tissue was used for each fractionation. Ten patients with epilepsy and 4 AVM patients were used for these experiments
Western blots for IgG from subcellular fractions
Proteins from subcellular fractions were separated via SDS-PAGE electrophoresis under non-denaturing conditions and transferred onto a polyvinylidene difluroride (PVDF) membrane (Millipore Corporation, Billerica, MA). Membranes were incubated overnight at 4°C with horseradish peroxidase (HRP) conjugated anti-human IgG (1:2,000; Calbiochem-Novabiochem Corporation, CA, U.S.A.). Proteins were visualized using Western Lightning Plus ECL (PerkinElmer, Waltman Massachusetts) and developed on Kodak Biomax MR Film (Eastman Kodak Company, Rochester, New York). IgG volume was quantified using Phoretix 2D software (Nonlinear USA Inc., Durham, North Carolina).
Purification and Analysis
Indirect Immunoprecipitation
Nuclear fractions were divided into soluble nuclear and chromatin-bound samples. Typically, indirect immunoprecipitation requires a step to allow the antibody-antigen complex to form. However, as antibody-antigen complexes were already present in cell nuclei, no antibody-antigen complex forming step was required. Nuclear fractions were incubated with protein-A coated agarose beads (Santa Cruz Biotechnology Inc., Dallas, Texas) for 3 hours at 4°C with gentle mixing. Beads were washed and centrifuged 4 times in 20% tween-PBS to remove as much unbound protein as possible. Samples were then placed in microcentrifuge tubes and exposed to a low pH (2.6) glycine-HCL solution for 3, 15 or 30 minutes. After centrifugation and removal of supernatant (containing antigen), the pH of the supernatant was neutralized with 1M Tris at pH 8.5.
Liquid Chromatography - Mass Spectrometry Analysis
Antigen samples, obtained as described above, were analyzed by SDS-Page electrophoresis. Bands were cut out of the gel, washed and dehydrated in acetonitrile. Bands were reduced using dithiothreitol and alkylated with iodoacetimide. Proteins were digested in gel overnight at room temperature using 10 ng/μl trypsin in 50 mM ammonium bicarbonate. Proteins were extracted from the polyacrylamide with acetonitrile (50%) and formic acid (5%). Extracts were evaporated in a Speedvac and resuspended in acetic acid (1%). Five μl volumes of extract were injected on a Dionex 15 cm × 75 μm id Acclain Pepmap C18, 2 μm, 100 angstrom, reversed-phase capillary chromatography column for liquid chromatography separation (Thermo Fisher Scientific Inc, Rockford, Il). Peptides were eluted from the column by acetonitrile/formic acid (0.1%) gradient at a flow rate of 0.25 μl/min and introduced to the mass spectrometry source on-line. For mass spectrometry analysis, a Finnigan LTQ-Orbitrap Elite hybrid mass spectrometer system (Thermo Fisher Scientific Inc, Rockford, Il) was used. The microelectrospray ion source was operated at 2.5 kV. The peptide digest was analyzed using the data-dependent multitask capability of the instrument acquiring full scan mass spectra to determine peptide molecular weights and ion spectra to determine the amino acid sequence.
Data from this experiment were analyzed using all collisionally-induced dissociation spectra collected. These were used to search the NCBI Mascot program with a human taxonomy filter. Manual interpretation, Sequest and Blast were used to verify Mascot matches.
Identification of potential antigens from mass spectrometry data
In our patient sample, time-dependent change in total antigen content was evaluated. The procedure (see above) was used to study both soluble nuclear and chromatin-bound fractions. Before collection, columns were washed 4× to remove unbound proteins. Thus, the remaining proteins were considered putative autoantigens; weakly-bound non-specific IgGs were also present in this sample. Qualitative analysis was achieved by examining the change in putative autoantigen count observed over time. A positive increase in a given autoantigen abundance (spectral count) was interpreted as elution of antigen tightly bound to IgGs. In contrast, a negligible increase or decrease was interpreted as non-specific binding of antigen to IgG. A transient increase or decrease at 15 min followed by a return to levels less than or equal to that observed at 3 min were not further analyzed. In short, putative autoantigen were further analyzed only if they were not obvious contaminants (e.g., keratins) and if their kinetic dissociation was indicative of specific antibody-antigen binding.
An additional validation step was used to unveil autoantigen. We compared the kinetics of soluble nuclear (presumably non-specific) vs. chromatin-bound (specific) complexes. This analysis revealed that, in the soluble nuclear fraction, the dissociation of protein from IgG was not time-dependent. In fact, increase, decrease or no change in total protein content were equally common. In contrast, the chromatin-bound IgG-antigen dissociation was observed by a definite time-dependent increase of dissociated autoantigen. The comparisons of these two behaviors for each putative autoantigen (see Figure 5) together with the observations above were merged into the following:
Figure 5. Mass spec. analysis revels histones are a likely target of autoantibodies in brain from patients with epilepsy.
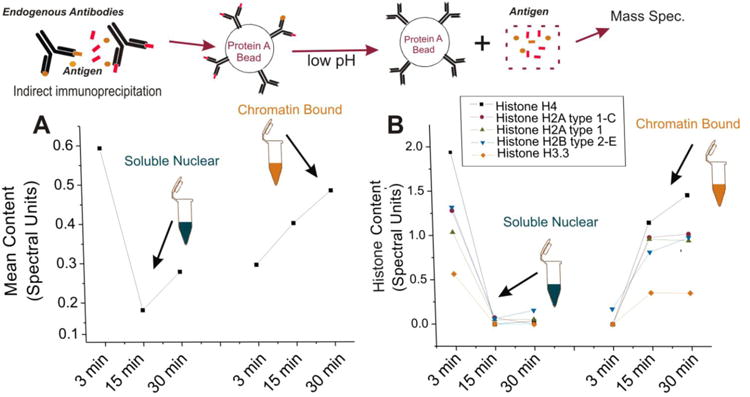
(A) The average change in antigen concentration in a sample obtained from brain of a patient with epilepsy. After exposure to low pH, there is a dramatic decrease in protein concentration between 3 and 15 minutes with a moderate increase after 30 minutes in the soluble nuclear fraction. In the chromatin-bound fraction, there is a constant increase in protein concentration over time. These kinetic differences suggest the presence of high affinity antibodies in the chromatin- bound fraction and low affinity antibodies (non-specific) in the soluble nuclear fraction. (B) When examining the dissociation kinetics for each protein found by mass spec., histones most closely matched the average dissociation profile seen in (A) suggesting that there was specific binding of antibodies to histones in the chromatin bound fraction. Refer to the legend for the dissociation kinetics of each histone observed. The insert at the top corresponds to the experimental steps in Figure 1.
where Q is a coefficient and where each value equals the normalized spectral count of a given autoantigen. CB = chromatin bound fraction, SN = soluble nuclear fraction. Q was used to discern between soluble unbound IgG, and chromatin-IgG or histone-IgG complexes.
All proteins associated with negative coefficients were considered to be likely autoantigen specifically bound to antibodies. Proteins were then ranked from most negative to least negative and any non-nuclear proteins were eliminated. The nuclear proteins with the most negative number were considered the primary targets of the autoantibodies (Table 2).
Table 2. Candidate autoantigens.
| Protein | CB-SN | Accession | # Mass (kDa) | Autoimmune Association | Support |
|---|---|---|---|---|---|
| Histone H4 | -2.83 | 4504301 | 11 | SLE | Burlingame, et al, 1994 |
| Histone H2A type 1-C | -2.07 | 4504245 | 14 | SLE | Imaoka, et al, 1990 |
| Histone H2A type 1 | -1.96 | 4504239 | 14 | SLE | Gu, et al, 2013 |
| Histone H2B type 2-E | -1.82 | 4504277 | 13 | ||
| Histone H3.3 | -0.92 | 4504279 | 15 | SLE | van Bavel, et al, 2011 |
| Histone H1.3 | -0.74 | 4885377 | 22 | ||
| Histone H2A.Z | -0.70 | 4504255 | 13 | ||
| Core histone macro-H2A.1 isoform 2 | -0/47 | 4758496 | 39 | ||
| Heterogeneous nuclear ribonucleoproteins C1/C2 isoform a | -0.18 | 117189975 | 33 | ||
| Heterogeneous nuclear ribonucleoprotein A3 | -0.16 | 34740329 | 39 | SLE, Scleroderma | Siapka, et. al 2007 |
| Histone H1.0 | -0.07 | 4885371 | 20 | ||
| Nucleoside diphosphate kinase B isoform a | -0.03 | 4505409 | 17 |
Autoimmune Association abbreviations: SLE = systemic lupus erythmatosus
Identification of Antibody and Antigen
Western Blots using serum to detect isolated nuclear antigen
Protein from 1 patient with epilepsy, serum from 8 patients with epilepsy, 2 control patients and 1 positive control patient with rheumatoid arthritis (RA) were used in this experiment. Isolated antigens from the immunoprecipitation experiment were separated via SDS-PAGE electrophoresis under non-denaturing conditions and transferred onto a PVDF membrane. Membranes were incubated at room temperature for 1 hour with serum from patients with epilepsy (1:1000). After repeated washing, membranes were incubated at room temperature for 2 hours with HRP-conjugated goat anti-human IgG (1:2,000; Calbiochem-Novabiochem Corporation, CA, U.S.A.). Proteins were visualized using Western Lightning Plus ECL and developed on Kodak Biomax MR Film.
Western blots using serum to detect purified histones
Total histones were separated by SDS-PAGE electrophoresis under non-denaturing conditions and transferred onto a PVDF membrane. Membranes were incubated at room temperature for 1 hour with serum from patients with epilepsy (1:500). After repeated washing, membranes were incubated at room temperature for 2 hours with HRP-conjugated goat anti-human IgG (1:2,000). Proteins were visualized using Western Lightning Plus ECL and developed on Kodak Biomax MR Film.
Anti-histone and anti-chromatin ELISA using serum
Serum obtained from human patients with epilepsy was diluted 1:300 in serum diluent (sterile filtered 0.5% bovine gamma-globulin, 5% gelatin, 0.05 mM Tween in 1× PBS) and analyzed for levels of anti-chromatin and anti-histone IgG autoantibodies. Microtiter plates (Immulon 2HB) were coated with purified chromatin or total histones over night at 4°C, blocked in 5% gelatin/PBS for 2hrs, and incubated with serum samples for 2 hrs. Secondary HRP-conjugated anti-human IgG antibodies were added for 1.5 hrs and plates were developed using 10mg/ml 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) in Mcllwain's buffer (0.09 M Na2HPO4, 0.06 M citric acid, pH 4.6). Samples were read on a spectrophotometer at 405 nm.
Human epithelial type 2 (HEp-2) cell assay
Slides provided by the manufacturer (Bio-Rad, Hercules, CA) were placed into a humidity chamber. Positive and negative controls provided with the manufacturers' kit were added (1:64 dilution) to two wells of the slides in addition to 3 human serum controls and serum from 8 patients with epilepsy (1:10 dilution). Slides were incubated for 20 minutes and then washed for 10 minutes in PBS. 25 μl of fluorescein conjugate was added to each well of the slide and incubated in a humidity chamber for 20 minutes. Images were obtained on a Leica Leitz fluorescent microscope with attached camera.
Results
For the results presented here we used a total of 32 subjects (including control). 21 brain samples and blood from 11 donors were analyzed. Of brain samples, 13 were obtained from patients affected by multiple drug resistant seizures, 4 were from patients treated by a neurosurgeon to repair AVMs and 4 were post-mortem brain samples from patients affected by MS. Brain samples were used for Western blotting, immunohistochemistry and general morphology. Eight patients with epilepsy, 2 healthy volunteers and 1 patient with epilepsy and RA were enrolled to donate blood samples used for ELISA, HEp-2 and Western blot data. The AVM samples were obtained from 1 patient with seizures responding to treatment, 1 patient with multiple drug resistant seizures and 2 with no seizure history. None of the MS patients had a history of epilepsy or seizures. Data obtained from the medical records of patients used, including volunteers, are shown in Table 1.
Our experimental design also encompassed another layer of distribution, namely presence or absence of recurring seizures. Thus, 1 AVM patient where seizures were present at time of surgery was grouped together with resected brain from patients with epilepsy where focal electrophysiological properties (“spiking cortex”, see (Najm et al., 2006)) were observed, while AVM patients with well-controlled or no history of seizures were grouped with resections from “non-spiking” areas of brain from patients with epilepsy. These criteria were only used for Figure 4.
Figure 4. Subcellular fractionation and comparison of AVM and brain tissue from patients with epilepsy reveal that BBBD alone is not sufficient to cause antibodies to bind to chromatin.
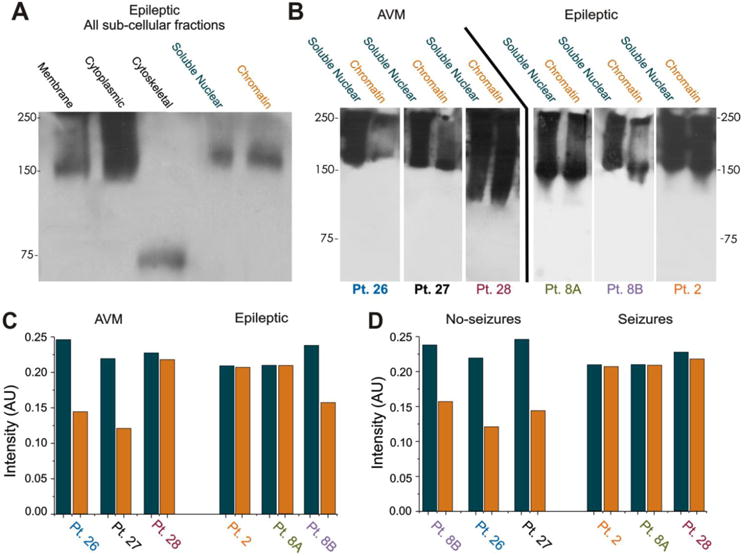
(A) Sub-fractioned brain of patients with epilepsy which was probed with HRP-conjugated anti-human IgG reveals abundant presence of immunoglobulin in all fractions tested. (B) Notably, chromatin-bound IgGs were quantitatively similar to levels in the soluble nuclear fraction of patients with epilepsy. Results were compared across several patients and against patients affected by AVM, a disease also characterized by widespread leakage of the BBB (Manninger et al., 2005; Tu et al., 2006), revealing that brain from patients with epilepsy, but not brain from AVM patients, accumulates significant amounts of IgGs in the chromatin-bound fraction. The bar graph in (C) shows the quantitative relationship between measured content in the 2 fractions based on pathology. The bar graph in (D) shows the same quantitative relationship between IgG in both fractions according to whether or not a patient had seizures. Note that regardless of pathology, BBBD with seizure produces larger amounts of IgG bound to chromatin than with BBBD alone.
BBB disruption in Multiple Sclerosis, Epilepsy, and AVM Brain
One of the goals of this study was to detect and localize immunoglobulins in the CNS. We first wished to understand the mechanisms by which these macromolecular complexes gain entry into the brain. There are two known mechanisms for IgG CNS ingress, namely passage across the BBB or synthesis by CNS B lymphocytes (Ankeny and Popovich, 2010). Our results suggest that the former was the predominant source of CNS IgGs in brain from patients with epilepsy. Figures 2A-B demonstrate the topographic overlap of extravasated immunoglobulins and albumin. Extravasated IgGs and albumin were found in the extracellular as well as the intracellular compartments (Figure 2 B1-B3). The arrows in B1-3 points to a neuron filled with IgGs and albumin.
Figure 2. Extravasation of IgGs in brain of patients with epilepsy coincides with regions of albumin extravasation.
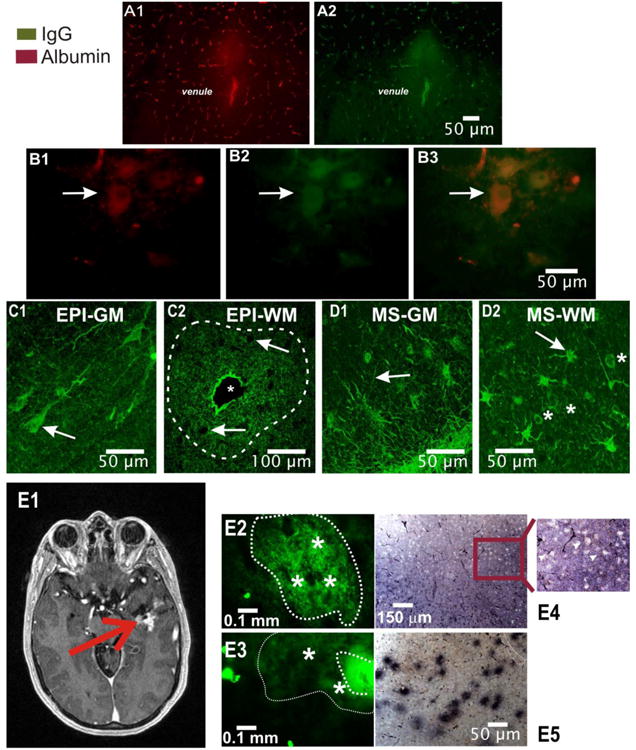
A–B) Similar pattern of IgG and albumin extravasation. The micrographs show the presence of extravasated albumin and IgGs in tissue samples resected from the brain of a patient with epilepsy. Note that patterns of albumin and IgG extravasation around a venule (A1 and A2) were virtually identical. Also note the intravascular presence of both albumin and IgG revealing the capillary network. B1-B3) Intracellular accumulation of albumin differs from intracellular localization of IgGs. Note that unlike in the case of albumin, IgGs were detected not only in the extracellular space and in the cytosol but also in the nucleus of neuronal cells (see also Figure 3; arrows point to neuronal cell bodies). C-D) Different patterns of IgG homing in brain from patients with epilepsy (C1-C2) compared to multiple sclerosis brain (D1-D2). In the case of brain from patients with epilepsy, as indicated by the arrow in C1, IgG accumulation was mostly restricted to neuronal cells. Large regions of extravasation were seen around blood vessels as indicated by the dotted line in C2. The asterisk in C2 indicates the lumen of a large venule. The arrows point to cell bodies which, in regions of white matter from the brain of patients with epilepsy, reveal cells with no intracellular accumulation of IgG in spite of the abundant presence of extracellular, extravasated immunoglobulin. In the case of multiple sclerosis, as shown in D1 and D2, regions of extravasated IgGs were inversely correlated to gray matter extravasation patterns (D1 vs. D2). In addition, note that in MS brain, cells identified as astrocytes revealed an uptake of IgGs in the cytosol but not in the nucleus (asterisk D2). E shows the same results in other tissue sections where DAB was used in lieu of immunofluorescence. Note the patchy profile of IgG extravasation (E5) and the lack of intracellular staining in E4. In summary, while both epilepsy and MS brain reveal regions of extravasated immunoglobulin, significant differences were found when analyzing gray matter and fiber tracts. EPI-GM = gray matter of patient with epilepsy, EPI-WM = white matter of patient with epilepsy, MS-GM = multiple sclerosis gray matter, MS-WM = multiple sclerosis gray matter
In brain tissue from patients with epilepsy, grey matter neurons were filled with IgGs (Figure 2 C1), while white matter extravasation of IgGs was restricted to the extracellular space (Figure 2 C2). Note the concentric feature of IgG extravasation; a large venule (indicated by an asterisk) gave rise to a circular pattern of leakage. Two types of control were used to confirm or refute the hypothesis that the fate of extravasated IgGs differs across pathologies. We used MS brain samples (Figure 2 D1 and D2) as well as cortical samples isolated during repair of AVM (Figure 2 E). In addition to differences between pathologies we also detected topographic segregation of intracellular uptake patterns in brain from patients with epilepsy vs. MS brain (compare Figure C and D). Unlike in grey matter from patients with epilepsy (Figure 2 C1), MS brain neuronal cell bodies were devoid of IgG content (Figure 2 D1). The arrows in C1 indicate parenchymal cells while in C2 they indicate ectopic neurons in white matter. In white matter of MS patients, extravasated IgGs were found in the extracellular compartment; in contrast to what was observed in neurons from patients with epilepsy, no nuclear IgGs were present (asterisk in D2). The arrow in D2 points to a glial cell.
An additional “control” for human brain with epilepsy and BBBD is the use of resections obtained during AVM repair. Figure 2 E1 shows the MRI of a typical AVM patient; the red arrow points to a region of extravasated contrast agent (gadolinium). The micrographs in Figure 2 E2 and Figure 2 E3 depict a pattern of IgG extravasation in a specimen obtained from another AVM patient. Note the lack of visible IgG accumulation in cell bodies (asterisks in E2), as well as the gradient of IgG extravasation (shown in E3). The dashed line indicates regions of maximum extravasation while the dotted line in E3 refers to a broader region of extravasation characterized by weak IgG signals. These results were confirmed by DAB-stained brain slices (Figure 2 E4 and E5) where variable extent of IgG extravasation as well as the absence of intracellular IgG was evident (enlargement in Figure E4).
Detection and Discovery
Figure 3 shows a typical outcome of experiments where frozen sections from human brain with epilepsy or MS brain were stained for the presence of neuronal or astrocytic markers as well as for DNA. In brain tissue resections from patients with epilepsy (Figure 3 A-B), intracellular and nuclear staining of IgG was readily revealed. However, intra-nuclear accumulation of immunoglobulin was restricted to neurons while astrocytes lacked intra-nuclear accumulation of IgGs despite robust uptake in the cytosol (arrows in A1 and B1). The right side panels (Figure 3 A3-B2) show a quantitative analysis of immunofluorescence. A computer-drawn line was used to detect the profile of nuclear (blue) or immunoglobulin-related (green) fluorescence in different cells. These results show the co-localization of IgG and DAPI in neurons (Figure 3 A3) but not in astrocytes (Figure 3 B2). To further demonstrate the co-localization of DAPI with IgGs, we used a three dimensional reconstruction of confocal images (Figure 3 A2). Regardless of the methods used, the results demonstrate the presence of fluorescently labeled immunoglobulin in neuronal nuclei. In comparison, although extravasation of IgG was widespread in MS brain, intracellular accumulation of IgG was absent (arrow in Figure 3 C1). The profile lines in Figure 3 C2 show two continuous neurons to demonstrate the lack of any IgG content in these cells.
Figure 3. Nuclear IgGs are exclusive to neurons in brain of patients with epilepsy.
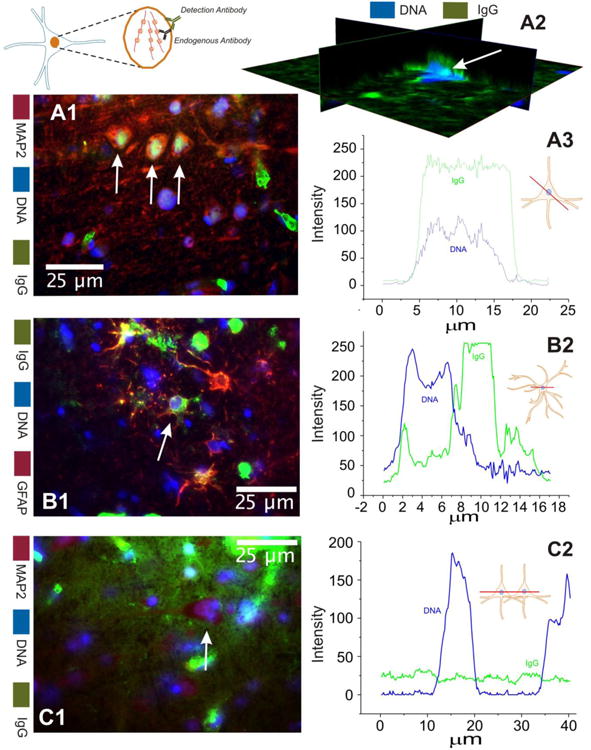
These figures compare results obtained in brain of patients with epilepsy (A-B) to multiple sclerosis tissue samples (C). The left panel shows the actual micrographs while the right panel shows a schematic representation of the values of IgGs (in green) or nuclear DNA (in blue). Note that in neurons, labeled by the neuronal marker MAP2, nuclear accumulation of IgGs was observed whereas in GFAP+ astrocytes from brain of patients with epilepsy (B) the content of intracellular IgG was limited to the cytosol and 2 processes. Nuclear co-localization is also observed in a confocal 3D reconstruction (A2) of brain tissue from a patient with epilepsy. In contrast, grey matter regions in MS brain (temporal lobe; C) revealed widespread extracellular leakage with neuronal cells (indicated by arrows) failed to reveal any significant intracellular accumulation of IgGs.
In order to determine the specific location of IgGs, subcellular fractionation was performed. The Western blot in Figure 4A shows the presence of intracellular IgGs in each subcellular fraction. The 75 kD band in the cytoskeletal fraction represents an IgG fragment corresponding to the heavy chain regions. Figure 4B, C and D show the difference between pathologies with BBB leakage with no seizures vs. BBB leakage in patients affected by seizures. AVM patients' brain displayed abundant IgGs in soluble nuclear fractions but a less remarkable level in the chromatin-bound fraction. In samples from patients with epilepsy, results demonstrated comparable amounts of IgGs in both soluble nuclear and chromatin-bound fractions. In addition, the AVM patient who also experienced seizures had similar levels of chromatin-bound IgGs as patients with epilepsy (Figure 4B, C and D patient #28). “Non-spiking” brain from patients with epilepsy displayed chromatin-bound IgG levels similar to those found in AVM patients without seizure (Figure 4B, C and D patient #8B).
Purification and analysis
A second major goal was to determine the molecular profiles of putative autoantigen in the nuclei of neurons from patients with epilepsy. The strategy used to unveil the nature of these autoantigens is described in detail in the methods section. Figure 5 summarizes the experimental design and results. After disassociation of antigen from the antibodies by low pH the soluble nuclear fraction displayed a dramatic drop in protein content over time (Figure 5A); this was the opposite of what was observed in the chromatin-bound fraction where a dramatic increase in protein content was observed (Figure 5A). This was interpreted as presence of specific binding of IgG to nuclear proteins and chromatin in comparison to free-floating or non-specifically bound protein-IgG complexes in the soluble fraction. When mass spectrometry analysis was performed, a number of non-nuclear proteins were found and discarded (see Supplemental Table 1). When narrowing down likely nuclear autoantigen candidates, the most prominent family of putative autoantigen was found in histones. The Legend in Figure 5B highlights the molecular descriptions of the histones found to be significantly associated with specific binding to IgG in the chromatin fraction. All histones tested behaved as predicted by the equation shown in the methods section and according to the time-dependent association of IgG and antigen shown in Figure 5A. This also implies that IgG-histone binding displayed high affinity owing to the fact that the kinetic profile was consistent with tight binding antibodies. This is in contrast to what was observed in the soluble fraction where histones were rapidly dissociated from antibodies. A summary of the molecular properties and quantitative analysis of histones and other putative autoantigens is shown in Table 2.
Identification of antigen and antibody
While the results in Figure 5 and Table 2 show an indirect approach to determine the molecular nature of putative autoantigens found in brain from patients with epilepsy, these results heavily rely on statistical analysis of mass spectrometry data. We performed three additional immunodetection experiments to confirm or disprove the presence of histone-specific IgGs in patients with epilepsy. We first used Western blotting to qualitatively analyze histones by molecular weight. To this end we prepared Western blots using protein extracts from the immunoprecipitation experiments shown in Figure 5. In other words, we used a mixture of nuclear protein that contained histones and other nuclear proteins obtained from brain tissue resections of patients with epilepsy as a target for patients' serum IgGs. In doing so we wished to test the hypothesis that the putative autoantigens isolated from the brain were also targets of IgGs present in serum of patients with epilepsy. The results are shown in Figure 6A. Note that, when control serum was used, no significant immunosignal was detected, whereas with serum from patients with epilepsy a distinct band was apparent. This band reflects the binding of IgGs isolated from serum of patients with epilepsy to autoantigen isolated from IgG present inside cells of human brain with epilepsy. This band also corresponds to the molecular weight of histones (kDa). To confirm that these bands indeed reflected the presence of histones, we ran a similar gel (Figure 6A; patient 17*) to demonstrate that total histones are recognized by the same serum used in the previous experiment. These results show that the molecular target of IgG extracted from neuronal nuclei of brain tissue resections of patients with epilepsy consists of histones.
While Western blots used to screen and confirm the presence of specific nuclear protein are useful for a few experiments, screening of a larger number of subjects is less convenient. To this end we used ELISA to quantitatively detect and quantify the presence of anti-histone IgG (Figure 6B). Note that in 5 of 8 patients with epilepsy a signal suggestive of autoimmunity against histones was present. Also note that in control subjects (labeled as C1) the signal was not present. These results confirm the Western blot findings shown in Figure 6A but also underscore the fact that these autoantibodies are not present in all patients with epilepsy. However, some patients with epilepsy have higher levels of antinuclear antibodies than the positive control rheumatoid arthritis patient (labeled Pos. C 1)
The presence of anti-histone antibodies can be indicative of a broader autoimmune response against multiple nuclear components. That is, many antinuclear antibodies recognize not just histones, but the histone-DNA complex (chromatin). Therefore, we tested the hypothesis that in addition to anti-histone antibodies, serum from patients with epilepsy also contains autoantibodies targeting chromatin (Figure 6C). Using the HEp-2 assay, a distinct pattern of staining was discovered. Serum from patients with epilepsy, but not serum from control subjects (Figure 6C C1) displayed various types of antinuclear staining. The insets in the figures show specific binding to nuclei or other sub-nuclear fractions (specifically nucleoli (Pt. 14) and centromeres (Pt. 16)). Note the similarity between the manufacturer's negative control and the serum control provided by the volunteer. The HEp-2 assay results demonstrate that a clinical tool for the diagnosis of autoimmune disease is able to detect the antinuclear antibodies in the serum of patients with epilepsy.
Discussion
The main finding of this research is the presence of antinuclear antibodies in brain and serum from patients with epilepsy. These findings were obtained in patients who did not present any signs, symptoms or clinical diagnoses of a typical autoimmune disease (such as SLE). Serum from a patient with epilepsy also diagnosed with RA was used as a positive control to underscore that the levels of autoimmune response in subjects with epilepsy were comparable to bona fide patients with autoimmune disease. Nuclear antibodies were predominately found in neurons and appeared to spare glia. Nuclear targets included histones and chromatin. Autoimmune IgGs are derived from the systemic circulation as their presence overlapped with areas of albumin leakage across the BBB. An alternative interpretation may support an indirect role of B lymphocytes in BBBD; this hypothesis needs to be tested by ad hoc experiments.
Significance
Many clinical studies suggest that aberrations of the immune system may be associated with seizures. Our results suggest the existence of a new category of autoimmune epilepsy. Additionally, while a number of studies have demonstrated the presence of antinuclear antibodies in sera of patients with epilepsy, we have for the first time shown that these antibodies are able to enter the nuclei of neurons. We focused on a population where antibodies against traditional “epilepsy related autoantigen” were not present and discovered that the misguided immune response in this population targeted the nuclear antigens histones and chromatin. A previous report by others concluded that subjects affected by epilepsy had levels of antinuclear antibodies similar to non-epilepsy controls (Ranua et al., 2004). In these studies no attempt was made to characterize these autoantigen as histones or chromatin. When the study was repeated in a controlled fashion, the authors reported a significant difference between prevalence of autoantibodies in patients with epilepsy compared to control (Eriksson et al., 2001; Peltola et al., 2000). Furthermore, these results were exclusively based on the HEp-2 assay, which is qualitative, and not with a clinical-grade ELISA test. In addition, the same group recently reported that antinuclear antibodies are associated with recurrent seizures in patients with refractory focal epilepsy (Peltola et al., 2000). Our results show a convergence of positive findings, since both qualitative (HEp-2) and quantitative tests (ELISA) demonstrated the presence of antinuclear antibodies in patients with epilepsy. We also used an extracted nuclear antigen fraction to confirm the presence of IgG-bound autoantigen which was found to be histones or chromatin.
Given the fact that areas of IgG leakage corresponded to areas of albumin extravasation, we concluded that ingress of IgGs in neurons occurred by a trans-endothelial route either across altered tight junctions or by other trans-cellular means. We found no evidence of local production of these immune molecules. Others also confirm the lack of significant B cell presence in brain tissue resections of patients with epilepsy where an infectious trigger is not suspected (Prayson and Frater, 2002).
In typical autoimmune epilepsy the presence of a pathogen is often suspected but not always demonstrated (Nabbout, 2012). Both bacterial and viral mechanisms have been proposed. The onset and development of the autoimmune response may be due to molecular mimicry or breaking of a barrier maintaining immunological privilege. The latter is typical for CNS disorders owing to the presence of a BBB. The question, however, remains of why an autoimmune disease should cause seizures which are characterized by neuronal hyperexcitability and synchronization, or alternatively do seizures trigger an autoimmune disease? There are several lines of evidence pointing to the devastating effect of antibodies targeting ion channels (Lang et al., 2003), glutamate receptors, voltage gated calcium and potassium channels, etc.(Graus and Dalmau, 2012; Hoftberger et al., 2012). In these pathologies the proposed and accepted cascade of evidence/events is as follows: 1) The immune system mounts a response against a pathogen or against a self-molecule perceived as such; 2) Antibodies are produced to target these proteins or a specific sequence; 3) Upon binding to its target the antibody promotes loss of function or excessive function of one of the several crucial components that regulate neuronal resting potential or synaptic function (see (Vincent et al., 2006) for details). We did not directly investigate whether autoantibodies may have an influence on and/or produce a downstream event that may impact comorbidities such as cognitive decline. Antinuclear IgGs may be yet another mechanism of seizure-induced neuronal cell loss originally described in hippocampal sclerosis but nowadays recognized also for neocortical seizure disorders. However further experiments are needed to determine the pathological mechanisms involves.
Consequences of intranuclear brain IgGs
How IgGs pass through a breached BBB is not known, nor is it understood how once they enter the cellular compartment IgG may act as “neurotoxins” (Levite and Ganor, 2008). There are many possible mechanisms for their “toxic” actions, including cell-induced death by an immune response, altered transcription owing to binding of IgGs to DNA, altered mechanisms of cell cycle or apoptosis, etc. Our results show no evidence of widespread neuronal cell death in the regions of albumin and IgG extravasation. In fact, the cells displaying the most intra-nuclear IgG content (for example Figure 3A) were characterized by healthy-appearing chromatin and nuclear content (Marchi et al., 2004). Others have suggested that IgGs are “toxic” and that they promote neuronal cell death in the lithium/pilocarpine model of status epilepticus or in human cortex (Levite and Ganor, 2008; Michalak et al., 2012).
Alternative hypotheses to speculate on the consequences of nuclear IgGs in human brain from patients with epilepsy may focus on comorbidities rather than seizures themselves. It is well known that a complex sequelae of pathologies follow the onset and progression of epilepsy. This is particularly true for pediatric populations where developmental delays, mental illness and other noxious consequences of prolong seizures are often encountered. It is possible that given a similar family history, seizure severity and age/gender patients deteriorate more rapidly when neuronal cells are exposed to nuclear IgGs. It is also possible, though it appears unlikely, that IgGs have a neuroprotective effect.
A role for the blood-brain barrier?
Seizures are characterized by widespread vascular changes that span from hyperemia to vascular leakage (Abe et al., 1997; Diehl et al., 1998; Ivens et al., 2010; Janigro, 2012; Marchi et al., 2011a; Marchi et al., 2011b; Marchi et al., 2011c; Marchi et al., 2012a; Weinand et al., 1997). We and others have shown that BBB disruption precedes seizures in patients with epilepsy and that BBBD causes seizures in humans and animals even in absence of a prior history of epilepsy (for review see (Marchi et al., 2012a)). There is therefore strong evidence that the BBB is impaired in patients with epilepsy, at least at time of seizure onset. BBB leakage results in extravasation of IgGs which is the first step towards their ingress into the nuclear compartment of neurons (Rigau et al., 2007). Thus, either BBBD, seizures, or both may be necessary for this to happen. To address this hypothesis, we performed experiments using two human pathologies also characterized by BBB leakage but not seizures; we used post-mortem samples of MS brain as well as brain samples isolated during surgeries to repair AVM. We also studied samples from patients with BBBD and seizures and compared those to BBBD but no ongoing seizures or negative seizure history. To this end, in addition to resections of brain from patients with epilepsy, we used a “non-spiking” region of resected temporal lobe (as a BBBD+ but no seizure) and AVM tissue from a patient presenting with multiple drug resistant seizures (as BBBD+ and seizure+) (see Table 1). The data in Figure 4 show that chromatin-bound autoantibodies were elevated in all samples from “spiking” cortex, regardless of its origin. This group contained all tissue isolated from foci of patients with epilepsy as well as AVM brain associated with seizures. The group with the least chromatin-bound IgGs encompassed brain samples from non-epilepsy patients and the brain tissue resected from “non-spiking” brain of patients with epilepsy. These preliminary results led us to conclude that leakage of the barrier is necessary for entry of IgG into the parenchyma but is not sufficient to allow binding of antibodies to chromatin.
If BBB dysfunction is not sufficient to cause accumulation of IgGs into the nucleus of neurons, what is the likely mechanism? We propose a scenario where seizures themselves are responsible for uptake of IgGs into the nuclear compartment of neurons. According to this scenario, prolonged excitation of neurons and other cellular elements occurring during a seizure, and the subsequent metabolic mismatch, act synergistically to decrease selective permeability of the cell membrane. According to this hypothesis, a specific receptor is not necessary, but rather, this uptake occurs due to a non-specific spreading depression-like episode. However, this scenario explains how IgGs enter into the cells but do not account for the presence in the nucleus. In fact, spreading depression affects all cells in a certain region but nuclear uptake was only present in neurons. How IgG may migrate from cytosol to nucleus remains unknown. A recent paper has shown that electrical stimulation characterized by low intensity (µA) and a frequency comparable to neuronal firing during an ictal event (50Hz) causes translocation of membrane-bound protein to the nucleus (Janigro et al., 2006). It is thus possible that field potential changes alone are sufficient to cause subcellular redistribution of macromolecules.
Potential pitfalls
One of the potential confounders in this study are the autoimmune side-effects of certain anticonvulsants. For example, carbamazepine is known to induce lupus-like symptoms or full-blown disease; this has been shown to occur primarily in female patients (Schoonen et al., 2010). This does not appear to be a significant factor in our study because of all the patients enrolled, only 5 were under carbamazepine therapy. In addition to carbamazepine, anecdotal reports have shown that lamotrigine and valproate can produce lupus-like symptoms (Bonnet et al., 2003; Chang and Cole, 2012). However, this was unlikely to be a factor in antinuclear antibody generation in our patients as none of them presented with symptoms of anti-convulsant drug-induced lupus. The probability of finding antinuclear antibodies was, in other words, unrelated to drug regimen. Others also found cohorts of patients with epilepsy undergoing carbamazepine therapy with no symptoms or signs suggestive of SLE (Ranua et al., 2004). It has to be noted, however, that in this study the presence of antinuclear antibodies was more common in female patients compared to male. Of the three patients with undetectable levels of auto-IgG, all were male while 60% of the patients with detectable levels of anti-histone antibodies were female. This finding is in agreement with numerous findings linking autoimmune disease to gender differences (Danchenko et al., 2006).
Another possible limitation of this study is the fact that we did not use CSF to test for autoantibodies. CSF analysis is a routine clinical approach to diagnosis infectious or autoimmune diseases. We did not have access to CSF samples for the patient population whose data is shown. Further experiments paired CSF and serum samples are being performed to confirm the diagnostic potential of antinuclear antibodies in epilepsy.
Conclusions
Our results point to a sterile inflammation mechanism (Rock et al., 2010) by which failure of the BBB promotes neuronal dysfunction. We focused on three neurological conditions (epilepsy, multiple sclerosis, AVM) all characterized by leakage of the BBB but found nucleus specific IgGs only in brain from patients with epilepsy and within these samples only in a population of neurons. IgGs directed towards histones and chromatin were subsequently found in sera from other patients with epilepsy suggesting that both compartments (circulatory and CNS) contain these antibodies and that antibodies found in serum are able to bind nuclear protein extracted from brain sample of patients with epilepsy. Thus, antinuclear autoantibodies may be a new mechanism of seizure-induced neuronal death.
Supplementary Material
{kind=link}
Highlights.
We examined non-autoimmune epileptic patients' brain and sera for autoantibodies
In areas of BBB leakage autoantibodies were found in neuronal nuclei of epileptics
Nuclear targets for these autoantibodies were found to be histones and chromatin
Autoantibodies in epileptic serum recognized nuclear antigen from epileptic brain.
BBBD and antinuclear antibody production may be a new mechanism of chronic epilepsy
Acknowledgments
This work was supported by: National Institutes of Health [R01NS078307, R01NS43284, R41MH093302, R21NS077236, R42MH093302, and R21HD057256 to DJ] and a Scientist Development Grant from the American Heart Association [13SDG13950015] and a Brain Behavior Research Foundation grant to CG.
Footnotes
Disclosure of conflicts of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Philip H. Iffland, II, Email: ifflanp@ccf.org.
Juliana Carvalho-Tavares, Email: carvalj@ccf.org.
Abhishek Trigunaite, Email: triguna@ccf.org.
Shumei Man, Email: mans@ccf.org.
Peter Rasmussen, Email: rasmusp@ccf.org.
Andreas Alexopoulos, Email: alexopa@ccf.org.
Chaitali Ghosh, Email: ghoshc@ccf.org.
Trine N. Jørgensen, Email: jorgent@ccf.org.
Damir Janigro, Email: janigrd@ccf.org.
References
- 1.Abe T, et al. Arterial vascular abnormality accompanying cerebral cortical dysplasia. AJNR Am J Neuroradiol. 1997;18:144–146. [PMC free article] [PubMed] [Google Scholar]
- 2.Adelow C, et al. Unprovoked seizures in multiple sclerosis and systemic lupus erythematosus: a population-based case-control study. Epilepsy Res. 2012;101:284–287. doi: 10.1016/j.eplepsyres.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Ankeny DP, Popovich PG. B cells and autoantibodies: complex roles in CNS injury. Trends in Immunology. 2010;31:332–338. doi: 10.1016/j.it.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bechmann I, et al. What is the blood-brain barrier (not)? Trends Immunol. 2007;28:5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Bharucha NE, et al. Epidemiology in developing countries. In: Engel G, Pedley TA, editors. Epilepsy: A comprehensive Textbook. Philadelphia, PA: Lippincott Williams and Wilkins; pp. 2008pp. 89–101. [Google Scholar]
- 6.Bonnet F, et al. Lupus-like syndrome and vasculitis induced by valpromide. J Rheumatol. 2003;30:208–209. [PubMed] [Google Scholar]
- 7.Chang RS, Cole AJ. Lamotrigine-Induced Lupus-Like Syndrome: A Case Report and Literature Review. Am J Ther. 2012 doi: 10.1097/MJT.0b013e3182491c31. [DOI] [PubMed] [Google Scholar]
- 8.Danchenko N, et al. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308–318. doi: 10.1191/0961203306lu2305xx. [DOI] [PubMed] [Google Scholar]
- 9.David Y, et al. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci. 2009a;29:10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.David Y, et al. Astrocytic Potassium and Glutamate Buffering Controls Synaptic Responses in A Frequency-Dependent Manner. Epilepsia. 2009b;50:86. [Google Scholar]
- 11.de Vries HE, et al. Inflammatory events at blood-brain barrier in neuroinflammatory and neurodegenerative disorders: Implications for clinical disease. Epilepsia. 2012;53:45–52. doi: 10.1111/j.1528-1167.2012.03702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diehl B, et al. The photic driving EEG response and photoreactive cerebral blood flow in the posterior cerebral artery in controls and in patients with epilepsy. Electroencephalogr Clin Neurophysiol. 1998;107:8–12. doi: 10.1016/s0013-4694(98)00036-4. [DOI] [PubMed] [Google Scholar]
- 13.Eriksson K, et al. High prevalence of antiphospholipid antibodies in children with epilepsy: a controlled study of 50 cases. Epilepsy Res. 2001;46:129–137. doi: 10.1016/s0920-1211(01)00273-x. [DOI] [PubMed] [Google Scholar]
- 14.Fabene PF, et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med. 2008;14:1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galanopoulou AS. Basic mechanisms of catastrophic epilepsy - Overview from animal models. Brain Dev. 2013 doi: 10.1016/j.braindev.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galea I, et al. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Galic MA, et al. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci. 2008;28:6904–6913. doi: 10.1523/JNEUROSCI.1901-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Granata T, et al. Immune-mediated epilepsies. Epilepsia. 2011;52:5–11. doi: 10.1111/j.1528-1167.2011.03029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Granata T, et al. Steroid treatment is effective in the treatement of status epilepticus in children. Epilepsia. 2008;1(0):276. [Google Scholar]
- 20.Graus F, Dalmau J. CNS autoimmunity: new findings and pending issues. Lancet Neurology. 2012;11:17–19. doi: 10.1016/S1474-4422(11)70280-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoftberger R, et al. Clinical neuropathology practice guide 5-2012: Updated guideline for the diagnosis of anti-neuronal antibodies. Clin Neuropathol. 2012;31:337–341. doi: 10.5414/NP300545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivens S, et al. Blood-brain barrier breakdown as a novel mechanism underlying cerebral hyperperfusion syndrome. J Neurol. 2010;257:615–620. doi: 10.1007/s00415-009-5384-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janigro D. Are you in or out? Leukocyte, ion, and neurotransmitter permeability across the epileptic blood-brain barrier. Epilepsia. 2012;53(1):26–34. doi: 10.1111/j.1528-1167.2012.03472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janigro D, et al. Alternating current electrical stimulation enhanced chemotherapy: a novel strategy to bypass multidrug resistance in tumor cells. BMC Cancer. 2006;6:72. doi: 10.1186/1471-2407-6-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lancaster E, Dalmau J. Neuronal autoantigens-pathogenesis, associated disorders and antibody testing. Nature Reviews Neurology. 2012;8:380–390. doi: 10.1038/nrneurol.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lang B, et al. New autoantibody mediated disorders of the central nervous system. Curr Opin Neurol. 2003;16:351–357. doi: 10.1097/01.wco.0000073937.19076.d5. [DOI] [PubMed] [Google Scholar]
- 27.Levite M, Ganor Y. Autoantibodies to glutamate receptors can damage the brain in epilepsy, systemic lupus erythematosus and encephalitis. Expert Rev Neurother. 2008;8:1141–1160. doi: 10.1586/14737175.8.7.1141. [DOI] [PubMed] [Google Scholar]
- 28.Manninger SP, et al. An exploratory study of ferumoxtran-10 nanoparticles as a blood-brain barrier imaging agent targeting phagocytic cells in CNS inflammatory lesions. AJNR Am J Neuroradiol. 2005;26:2290–2300. [PMC free article] [PubMed] [Google Scholar]
- 29.Marchi N, et al. Seizure-Promoting Effect of Blood-Brain Barrier Disruption. Epilepsia. 2007a;48(4):732–742. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marchi N, et al. The blood-brain barrier, inflammation and epilepsy. Trends in Immunology. 2011a invited review. [Google Scholar]
- 31.Marchi N, et al. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis. 2009;33:171–181. doi: 10.1016/j.nbd.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marchi N, et al. Transporters in Drug-Refractory Epilepsy: Clinical Significance. Clinical Pharmacology & Therapeutics. 2010;87:13–15. doi: 10.1038/clpt.2009.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchi N, et al. The blood-brain barrier hypothesis in drug resistant epilepsy. Brain. 2012a;135:e211. doi: 10.1093/brain/awr343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marchi N, et al. Efficacy of Anti-Inflammatory Therapy in a Model of Acute Seizures and in a Population of Pediatric Drug Resistant Epileptics. Plos One. 2011b;6 doi: 10.1371/journal.pone.0018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchi N, et al. Blood-brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia. 2012b;53:1877–1886. doi: 10.1111/j.1528-1167.2012.03637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchi N, et al. Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med. 2004;2:37. doi: 10.1186/1741-7015-2-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marchi N, et al. Modulation of peripheral cytotoxic cells and ictogenesis in a model of seizures. Epilepsia. 2011c doi: 10.1111/j.1528-1167.2011.03080.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchi N, et al. In vivo and in vitro effects of pilocarpine: relevance to epileptogenesis. Epilepsia. 2007b;48(10):1934–1946. doi: 10.1111/j.1528-1167.2007.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKnight K, et al. Serum antibodies in epilepsy and seizure-associated disorders. Neurology. 2005;65:1730–1736. doi: 10.1212/01.wnl.0000187129.66353.13. [DOI] [PubMed] [Google Scholar]
- 40.Michalak Z, et al. IgG Leakage May Contribute to Neuronal Dysfunction in Drug-Refractory Epilepsies With Blood-Brain Barrier Disruption. J Neuropathol Exp Neurol. 2012;71:826–838. doi: 10.1097/NEN.0b013e31826809a6. [DOI] [PubMed] [Google Scholar]
- 41.Nabbout R. Autoimmune and inflammatory epilepsies. Epilepsia. 2012;53(4):58–62. doi: 10.1111/j.1528-1167.2012.03614.x. [DOI] [PubMed] [Google Scholar]
- 42.Nabbout R, et al. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol. 2011;10:99–108. doi: 10.1016/S1474-4422(10)70214-3. [DOI] [PubMed] [Google Scholar]
- 43.Najm IM, et al. Definition of the epileptogenic zone in a patient with non-lesional temporal lobe epilepsy arising from the dominant hemisphere. Epileptic Disord. 2006;8(2):S27–S35. [PubMed] [Google Scholar]
- 44.Peltola JT, et al. Antiphospholipid and antinuclear antibodies in patients with epilepsy or new-onset seizure disorders. Am J Med. 2000;109:712–717. doi: 10.1016/s0002-9343(00)00617-3. [DOI] [PubMed] [Google Scholar]
- 45.Prayson RA, Frater JL. Rasmussen encephalitis: a clinicopathologic and immunohistochemical study of seven patients. Am J Clin Pathol. 2002;117:776–782. doi: 10.1309/AD8R-560C-4V11-C5E2. [DOI] [PubMed] [Google Scholar]
- 46.Ranua J, et al. Anticardiolipin and antinuclear antibodies in epilepsy--a population-based cross-sectional study. Epilepsy Res. 2004;58:13–18. doi: 10.1016/j.eplepsyres.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 47.Rigau V, et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain. 2007;130:1942–1956. doi: 10.1093/brain/awm118. [DOI] [PubMed] [Google Scholar]
- 48.Rock KL, et al. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–342. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schoonen WM, et al. Do selected drugs increase the risk of lupus? A matched case-control study. Br J Clin Pharmacol. 2010;70:588–596. doi: 10.1111/j.1365-2125.2010.03733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seiffert E, et al. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tu J, et al. Ultrastructure of perinidal capillaries in cerebral arteriovenous malformations. Neurosurgery. 2006;58:961–970. doi: 10.1227/01.NEU.0000210248.39504.B5. [DOI] [PubMed] [Google Scholar]
- 52.Uva L, et al. Acute induction of epileptiform discharges by pilocarpine in the in vitro isolated guinea-pig brain requires enhancement of blood-brain barrier permeability. Neuroscience. 2007 doi: 10.1016/j.neuroscience.2007.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vezzani A, et al. Inflammation and epilepsy. Handb Clin Neurol. 2012;107:163–175. doi: 10.1016/B978-0-444-52898-8.00010-0. [DOI] [PubMed] [Google Scholar]
- 54.Vezzani A, et al. Therapeutic potential of new antiinflammatory drugs. Epilepsia. 2011;52:67–69. doi: 10.1111/j.1528-1167.2011.03242.x. [DOI] [PubMed] [Google Scholar]
- 55.Vincent A, et al. Autoimmune channelopathies and related neurological disorders. Neuron. 2006;52:123–138. doi: 10.1016/j.neuron.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 56.Weinand ME, et al. Cerebral blood flow and temporal lobe epileptogenicity. J Neurosurg. 1997;86:226–232. doi: 10.3171/jns.1997.86.2.0226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.