

Abstract
RNA interference can be extremely useful in determining the function of an endogenously-expressed protein in its normal cellular environment. In this chapter, we describe a method that uses small interfering RNA (siRNA) to knock down mRNA and protein expression in cultured cells so that the effect of a putative regulatory protein on gene expression can be delineated. Methods of assessing the effectiveness of the siRNA procedure using real time quantitative PCR and Western analysis are also included.
Keywords: RNA interference, siRNA, Transcription regulation, Gene silencing
1. Introduction
Regulating gene expression in eukaryotic cells requires a complex array of proximal sequence-specific recognition sites and distal enhancer elements as well as the association of various populations of transcription factors and coregulatory proteins with these DNA regions. Together, these protein–DNA complexes modulate gene expression in response to various extracellular signals and changing cellular environments. The capacity of transcription factors and coregulatory proteins to influence gene expression has historically been defined by testing the effect of overexpressed proteins on expression of transiently-transfected heterologous promoters. In recent years, it has been possible to define the effects of endogenously-expressed proteins involved in regulating native gene expression by employing RNA interference (RNAi, refs. 1, 2). Using this method, an RNA transcript can be targeted for destruction by small interfering RNAs (siRNAs, refs. 3–5). An siRNA consists of 21 nucleotide coding and noncoding RNA strands with 3′overhangs (6–8) and has a sequence identical to a small portion of a target cellular RNA (9). When transfected into cells, the siRNA is recognized by RNA-induced silencing complex (RISC), which initiates transcript-specific destruction resulting in the subsequent decrease of the corresponding protein (6, 7, 10). Thus, RNAi can allow one to determine the effect of a protein in a cell where the RNA and protein are normally expressed and involved in cellular function. We have found that knocking down the expression of a single protein involved in influencing transcription oftentimes differentially alters expression of endogenous target genes (11–15). Importantly, these studies have shown that transient transfections using heterologous promoters are unable to fully recapitulate the gene-specific effects of regulatory proteins on native gene expression and that siRNA experiments provide more biologically-relevant evidence of a protein’s role in regulating gene expression than previous methods.
2. Materials
2.1. Preparation of Cultured Cells
Phenol red containing minimal essential medium, MEM (Gibco/Invitrogen, Carlsbad, CA): Supplement with 100 µM nonessential amino acids (NEAA), 10 mM HEPES, 2 mM l-glutamine, 100 U/mL Penicillin, 100 µg/mL Streptomycin, 25 µg/mL Gentamicin, and 5% calf serum (Atlanta Biologicals, Lawrenceville, GA).
Hanks buffered salt solution, HBSS (Cellgro, Herndon, VA).
Trypsin/EDTA: Dilute a 10× solution of 0.5% Trypsin and 7 mM EDTA (Gibco/Invitrogen, Carlsbad, CA) 1:10 in 1× HBSS.
Phenol red-free MEM (Cellgro, Herndon, VA) with antibiotics and serum (PRF): Supplement with 100 µM NEAA, 10 mM HEPES, 2 mM l-glutamine, 100 U/mL Penicillin, 100 µg/mL Streptomycin, 25 µg/mL Gentamicin and 5% charcoal dextran-treated calf serum. Calf serum (Atlanta Biologicals, Lawrenceville, GA) is treated with charcoal dextran to remove endogenous steroid hormones and growth factors.
Phenol red-free MEM without antibiotics (PRF-A): Supplement with 100 µM NEAA, 10 mM HEPES, 2 mM l-glutamine (see Note 1) and 5% charcoal dextran-treated calf serum. Calf serum (Atlanta Biologicals, Lawrenceville, GA) is treated with charcoal dextran to remove endogenous steroid hormones and growth factors.
Phenol red-free MEM without antibiotics or serum (PRFAS): Supplement with 100 µM NEAA, 10 mM HEPES and 2 mM l-glutamine.
2.2. siRNA Transfection
Control (see Note 2) and experimental (see Note 3) siRNA can be procured from a number of commercial vendors.
SiLentFect Lipid (BioRad, Hercules, CA), which we have found to be effective in transfecting siRNA into mammalian cell lines, is used in the protocol described. However, other transfection reagents can be utilized.
2.3. Preparation of Cell Lysate
TNE: 40 mM Tris-HCl pH 7.5, 140 mM NaCl, 1.5 mM EDTA. Store at 25°C.
Lysis Buffer: 20 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.2% (v/v) NP40. Make fresh from stock solutions.
2.4. Protein Assay
5× Protein Assay Solution (BioRad, Hercules, CA): Dilute 1:5 in dH2O and run through a Whatman filter (Fisher Scientific, Pittsburgh, PA). Store the 5× solution at 4°C. Make 1× solution fresh as needed.
Bovine Serum Albumin, BSA (BioRad, Hercules, CA): Dissolve to a concentration of 12.5 µg/mL in deionized water (dH2O) and store at 4°C. Standards are prepared by diluting the 12.5 µg/mL BSA to 10, 7.5, 5, and 2.5 µg/mL in dH2O.
2.5. SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE)
Mini-PROTEAN 3 gel system (BioRad, Hercules, CA).
Separating Gel Mix: 10% acrylamide prepared from a 40% 37.5:1 acrylamide/bisacrylamide, 750 mM Tris base pH 8.8, 0.1% SDS. Store at 4°C. Acrylamide is a neurotoxin when unpolymerized and should be handled with care.
Stacking Gel Mix: 4% acrylamide prepared from a 40% 37.5:1 acrylamide/bisacrylamide, 125 mM Tris base pH 6.8, 0.1% SDS. Store at 4°C. Acrylamide is a neurotoxin when unpolymerized and should be handled with care.
Ammonium persulfate (APS) (30% (w/v) ): Prepare in dH2O.
N,N,N,N′-tetramethyl-ethylenediamine (TEMED).
SDS Running Buffer: 25 mM Tris base, 192 mM glycine, 3.5 mM SDS. Store at 25°C.
SDS Sample Buffer (4×): 250 mM Tris-HCl pH 6.8, 300 mM SDS, 40% glycerin (v/v), 2% (v/v) β-mercaptoethanol, 0.02% (w/v) bromophenol blue. Store 1 mL aliquots at −20°C.
Prestained molecular weight markers, for example Kaleidoscope (BioRad, Hercules, CA).
2.6. Western Blot
Mini-Trans Blot Cell (BioRad, Hercules, CA).
Whatman paper, > 6 µm retention.
GE Nitrocellulose Hybridization and Transfer Membrane, 0.45 µm (Fisher Scientific, Pittsburgh, PA).
Transfer Buffer: 25 mM Tris base, 192 mM glycine, 3.5 mM SDS, 20% (v/v) methanol. Store at 4°C.
5% Milk Solution: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM thimerosal, 0.5% (v/v) Tween-20, 5% (w/v) instant dry milk.
Wash Buffer: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM thimerosal, 0.5% (v/v) Tween-20.
Supersignal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL).
Kodak BioMax XAR film (Fisher Scientific, Pittsburgh, PA).
2.7. Stripping and Reprobing Blots
Restore Western Blot Stripping Buffer (Pierce, Rockford, IL).
Wash Buffer: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM thimerosal, 0.5% (v/v) Tween-20.
5% Milk Solution: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM thimerosal, 0.5% (v/v) Tween-20, 5% (w/v) instant dry milk.
2.8. RNA Harvest and Isolation
Trizol (Invitrogen, Carlsbad, CA). Trizol is toxic and should be used in a ventilation hood. Store at 4°C.
Chloroform. Chloroform is toxic and should be used in a ventilation hood. Store at 25°C.
Isopropanol: Store at 25°C.
Ethanol (75%): Prepare by mixing 37.5 mL ethanol and 12.5 mL dH2O (see Note 4). Store at 25°C.
RQ1 RNase-Free DNase (Promega, Madison, WI). Store at −20°C.
Phenol/Chloroform: Prepare phenol/chloroform/isoamyl alcohol at 25:24:1. Store at 4°C.
Ammonium acetate (NH4OAc): Prepare a 5 M stock in dH2O and store at 25°C.
2.9. cDNA Preparation
Reverse Transcription System (Promega, Madision, WI). Store at −20°C.
2.10. Quantitative Real Time PCR
iQ SYBR Green Supermix (BioRad, Hercules, CA).
iCycler Thermal Cycler (BioRad, Hercules, CA).
3. Methods
Performing RNA interference (RNAi) experiments in mammalian cells requires (1) a double-stranded siRNA that specifically targets the RNA transcript of interest, (2) an effective siRNA delivery system, (3) controls to ensure the specificity of the siRNA treatment, and (4) a biological assay to detect the effect of knocking down the target RNA. The amount of siRNA needed and the length of time required for destruction of the target RNA must be determined empirically and may vary with different cell lines and the confluency of the cells. A protein’s turnover rate will also affect the length of time required to decrease its expression. We have found that knocking down expression of estrogen receptor α (ERα) associated proteins in MCF-7 breast cancer cells, which endogenously express ERα and the coregulatory proteins needed for estrogen-responsive gene expression, generally requires ~24 h but sometimes can take as long as 96 h (11–16).
The effectiveness of target siRNA in knocking down a specific mRNA transcript can be determined using quantitative real-time PCR (qRT-PCR). This is a straight-forward method in which cellular RNA is isolated and reverse transcribed to produce complementary DNA (cDNA). Amplification of the cDNA with gene-specific primers is then used to detect the levels of cDNA present. In addition to monitoring target mRNA levels, the target protein levels should also be examined to ensure that the biological moiety responsible for mediating a cellular response has been knocked down. Western blot analysis is useful for this purpose. Protein expression should be monitored in cells that have been exposed to target siRNA and to control siRNA.
Once the conditions needed to knock down a protein’s expression have been determined, the biological consequence of reducing the target mRNA and protein can be defined. One common endpoint is monitoring gene expression, but protein activity can also be assessed. We typically monitor the expression of a number of estrogen-responsive genes in MCF-7 breast cancer cells that have been exposed to control or target siRNA in the absence or in the presence of 17β-estradiol (E2, refs. 11–16). In this manner, we can determine the effect of ERα-associated proteins on gene expression in a biologically-relevant cellular environment.
3.1. Preparation of Cultured Cells
MCF-7 breast cancer cells are maintained in MEM in 75 cm2 flasks and are passaged at confluency with 1× trypsin/EDTA.
Two days before transfection, the cells are washed twice with HBSS and media is replaced with PRF.
The day before transfection, cells are washed twice with HBSS, trypsinized, resuspended in PRF, and seeded into a 12-well plate at 5 × 105 cells per well. The number of wells seeded will depend on the conditions being tested.
3.2. siRNA Transfection
Prepare a 5 mL polysterene tube for the target siRNA and another for control siRNA. For each well to be transfected, mix 50 pmol siRNA (see Note 5) in 50 µL PRF-AS (see Note 1). Prepare enough reagent for all of the target siRNA wells in one tube and all of the control siRNA wells in another tube in order to minimize well-to-well variation. For example, to transfect 6 wells of a 12-well plate with target siRNA, add 300 pmol target siRNA to 300 µL PRF-AS. In another tube, add 300 pmol control siRNA to 300 µL PRF-AS to transfect the other half of the 12-well plate.
In another polysterene tube, combine 1.5 µL SiLentFect Lipid with 52.5 µL PRF-AS for each well to be transfected (see Note 1) and swirl gently. In order to limit well-to-well variation, prepare enough reagent for the target wells and the control wells in one tube. For example, to prepare enough reagent for one 12-well plate, combine 18 µL SiLentFect Lipid and 630 µL PRF-AS.
Add half of the diluted SiLentFect to the target siRNA tube and add the other half of the diluted SilentFect to the control siRNA tube. Mix by swirling gently. DO NOT VORTEX! Incubate the mixture for 20 min at 25°C. The solutions will become turbid during this time.
During the incubation, carefully aspirate the media from the wells, wash the cells once with PRF-A and add 500 µL fresh PRF-A to each well (see Note 1).
After the 20 min incubation, dispense 100 µL of the siRNA/ SiLentFect mixture dropwise into each well. The small amount of reagent remaining can be discarded.
Gently rock the plate back and forth to mix and place in a 37°C CO2 incubator for the length of time needed to knock down target mRNA and protein levels (see Note 6). For example, although nonmetastatic protein 23 homolog 1 (NM23-H1) protein levels decrease slightly 24 h after siRNA transfection, 96 h is required to achieve an appreciable decrease in NM-3-H1 expression (Fig. 1).
Fig. 1.
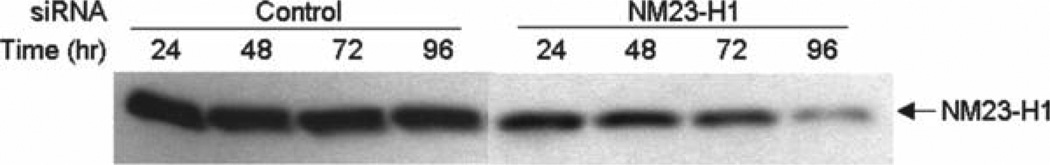
Decreasing endogenous NM23-H1 protein levels. MCF-7 breast cancer cells were transfected with control siRNA, which targets renilla luciferase mRNA (which is not expressed in these cells) or with NM23-H1 siRNA, that targets NM23-H1 mRNA for destruction. After 24–96 h, cells were lysed and subjected to Western blot analysis with an NM23-H1 specific antibody.
3.3. Preparation of Cell Lysate
Remove the media from each well. Add 500 µL TNE to each well. Pipet up and down 4–5 times to displace the cells. Transfer the cells to a 1.5 mL microcentrifuge tube.
Centrifuge at 900 × g for 5 min. Discard the supernatant and resuspend the cells in 30 µL Lysis Buffer.
Place the resuspended cells on dry ice for 5 min or until frozen. Then incubate the cells at 25°C for 5 min or until thawed. Repeat the freeze/thaw step and spin down the cell debris by centrifugation at 20,000 × g for 10 min.
Transfer the cell lysate to a new tube and discard the pellet.
3.4. Protein Assay
Set up 11 disposable test tubes and add 1 mL of 1× protein assay solution to each tube. One tube is reserved for a blank. 10 µL of the 5 diluted BSA standards (12.5, 10, 7.5, 5, or 2.5 µg) are dispensed in duplicate. Vortex.
Incubate at 25°C for 5 min.
Determine the absorbance of each protein standard at 595 nm using a spectrophotometer. Plot the standards on a curve with protein concentration on the x-axis and absorbance at 595 nm on the y-axis.
Bring 1–2 µL cell lysate to a total volume of 10 µL with dH2O and combine with 1 mL 1× protein assay solution. Prepare in duplicate and vortex.
Incubate at 25°C for 5 min.
Measure the absorbance of each sample at 595 nm and use the standard curve to determine the protein concentration of each cell lysate.
3.5. SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE)
These instructions assume the use of a Mini-PROTEAN 3 gel system, but can be adapted to other systems. Before use, wash the glass plates with detergent, rinse extensively with deionized water, and allow the plates to dry completely.
Slide the dry glass plates into the casting frame with the short plate in front. Ensure that both plates are flush at the bottom and lock into place.
Prepare a 1.0-mm thick, 10% acrylamide gel by mixing 5 mL Separating Gel Mix with 15 µL of 30% APS and 5 µL of TEMED. Immediately pour the separating gel, leaving space for the stacking gel, and carefully overlay with dH2O. Allow the gel to polymerize.
Once the gel has polymerized, pour off the dH2O.
Prepare the stacking gel by mixing 2 mL Stacking Gel Mix with 5 µL of 30% APS and 2 µL of TEMED. Immediately pour the Stacking Gel Mix on top of the polymerized separating gel and insert the comb. Allow the gel to polymerize.
Once the stacking gel has polymerized, remove the gel from the casting frame and pull out the comb. Gently rinse the wells with dH2O. Snap the gel into the electrode assembly with the short plate facing inward. Slide the gel and the electrode assembly into the clamping frame and lock into place.
Lower the assembly into the mini tank and fill the inner chamber with ~125 mL SDS Running Buffer. Add ~200 mL SDS Running Buffer to the lower buffer chamber of the mini tank. Using a syringe, rinse the wells with SDS Running Buffer.
Bring 20 µg cell lysate to 15 µL with 1× Lysis Buffer. Add 5 µL of 4× SDS Sample Buffer. Heat samples at 95°C for 3 min.
Load the prestained molecular weight markers into the first well and 20 µL of each sample into subsequent wells.
Place the lid on the mini tank and connect it to a power supply. Run the gel at 200 V for 45 min or until the dye front reaches the bottom of the gel.
3.6. Western Blot
Several hours before beginning the transfer process, fill the plastic cooling unit with dH2O and store at −20°C until frozen. The filled cooling unit can also be stored at −20°C between runs.
Prepare to transfer the proteins from the gel to a nitrocellulose membrane. These instructions assume the use of the Mini-Trans Blot cell system. However, they can easily be adapted to other systems.
Place 2 Scotchgard pieces and 4 pieces of Whatman paper (cut to the size of the cassette) in cold Transfer Buffer.
Turn off the electrophoresis power supply and disconnect the gel system. Pour off the buffer and disassemble the apparatus. Remove the gel from the plate and discard the stacking gel.
Cut a piece of nitrocellulose to the size of the gel and carefully slide the nitrocellulose into the cold Transfer Buffer starting with one edge. Be sure to use clean gloves when handling the nitrocellulose.
Assemble the transfer sandwich. Place the black side of the gel holder cassette down. Place one piece of Scotchgard on the black side of the gel holder cassette, then place two pieces of Whatman paper on the Scotchgard. Lay the gel on the Whatman paper and position the nitrocellulose on the gel. Place two pieces of Whatman paper on the nitrocellulose and complete the sandwich by positioning the last piece of Scotchgard on the Whatman paper. Remove any bubbles from the layers by rolling a Pasteur pipette back and forth over the sandwich. Close the cassette firmly and place the cassette in the module (see Note 7).
Remove the cooling unit from the freezer and place it in the tank. Position the module in the tank and add a small stir bar to the bottom. Fill the tank completely with cold Transfer Buffer.
Place the Mini-Trans Blot cell system on a magnetic stirrer. Set the magnetic stirrer on medium high to keep the ion distribution and temperature even in the tank. Place the lid on the tank and plug the cables into a power supply. Transfer for 1 h at 100 V at 4°C.
Disconnect the system from the power supply and disassemble the apparatus. Remove the nitrocellulose. The gel can be discarded or stained to verify that the proteins have transferred. Transfer of the prestained markers to the nitrocellulose membrane is also a good indicator that the proteins have been effectively transferred.
Place the blot in a 5% Milk Solution for ~30 min at 25°C with vigorous shaking. This process will block nonspecific binding of antibody to the membrane.
Prepare the primary antibody to detect the protein of interest by diluting the antibody in 5% Milk Solution (see Note 8). Incubate at 25°C for 1 h to overnight with vigorous shaking.
Discard the primary antibody and quickly rinse the blot with Wash Buffer (pour, swirl, and discard). Add 5 mL of Wash Buffer and place the blot on the shaker for 5 min at 25°C. Repeat the wash two times.
Dilute the secondary antibody in 5% Milk Solution (see Note 8). Incubate with the blot at 25°C for 1 h with vigorous shaking.
Discard the secondary antibody, rinse and wash as described in Subheading 3.6, step 12.
During the final wash, prepare the chemiluminescent substrate. We have found Supersignal West Femto Substrate (Pierce, Rockford, IL) to be extremely sensitive in detecting low abundance proteins (see Note 9).
Add the substrate to the blot and incubate at 25°C for 5 min.
Carefully remove the substrate mix with a pipette and wick away any remaining substrate with an absorbent tissue. Wrap the blot with one layer of plastic wrap. Tape the blot in the corner of an X-ray film cassette.
In a dark room, expose the blot to film for ~2–10 min depending on the intensity of the signal and develop the film. If possible, it is useful to demonstrate that the siRNA knocks down the target, but not a related protein. As seen in Fig. 2, NM23-H1 siRNA effectively reduces the level of NM23-H1 protein, but does not affect the expression of the related homolog NM23-H2.
Fig. 2.

Effect of knocking down NM23-H1 on protein levels. MCF-7 cells were transfected with control or NM23-H1 siRNA for 96 h. Cells were then treated with ethanol vehicle or 17β-estradiol (E2) for 24 h, lysed, and subjected to Western blot analysis with an antibody that recognizes NM23-H1 and NM23-H2, ERα, the progesterone receptors PR-A and PR-B, or GAPDH.
3.7. Stripping and Reprobing Blots
The blot can be stripped and reprobed to determine the effect of siRNA knockdown on the expression of other proteins. It is necessary to verify that the target siRNA does not affect the expression of other proteins and that equal amounts of cell lysate have been added (see Note 10). As seen in Fig. 2, neither the NM23-H1 siRNA nor the E2 alters expression of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Furthermore, the similarity in GAPDH levels in each of the lanes confirms that the lysates were evenly loaded.
Wash the blot with Wash Buffer for 10 min to fully rehydrate and remove any residual chemiluminescent substrate.
Transfer the blot to a small glass container with a tight seal. Add 5–10 mL of Stripping Buffer. Incubate with rotation at 37°C for 10 min.
Discard the Stripping Buffer and quickly rinse twice with Wash Buffer. Extensively wash the blot with Wash Buffer while vigorously shaking for 10 min at 25°C. Repeat twice.
Block the membrane in 5% Milk Solution for 30 min at 25°C. The membrane is then ready to be reprobed with another antibody as described in Subheading 3.6, steps 11–17.
Monitor the expression of proteins of interest when cells have been exposed to control and target siRNA. As shown in Fig. 2, the levels of ERα are decreased in the presence of E2, but are unaffected by the NM23-H1 siRNA. In contrast, expression of the estrogen-responsive progesterone receptors A and B (PR-A and PR-B) are increased when NM23-H1 is knocked down. The relative expression of a protein can be determined by using phosphorimager or infrared imaging analysis.
3.8. RNA Harvest and Isolation
Aspirate the media from the 12-well plate and replace with 500 µL Trizol per well. Incubate for 5 min and collect the cells by pipetting up and down 4–5 times. Transfer the cell lysate to a 1.5 mL microcentrifuge tube (see Note 11).
Add 100 µL chloroform and vortex at high speed for 30 s. Centrifuge at 20,000 × g for 15 min at 4°C.
Following centrifugation, transfer the colorless upper aqueous phase to a fresh tube being careful to leave the interface undisturbed.
Precipitate the RNA by adding an equal volume of isopropanol. Vortex at high speed for 30 s and incubate for 10 min at 25°C. Centrifuge at 20,000 × g for 10 min at 4°C. The RNA precipitate forms a gel-like pellet at the bottom of the tube.
Remove the supernatant and wash the pellet with 500 µL of 75% ethanol. Vortex and centrifuge at 7000 × g for 5 min at 4°C.
Remove the supernatant and air dry the pellet for ~10 min at 25°C. DO NOT VACUUM DRY! Drying the RNA pellet completely will decrease its solubility.
Add 56.5 µL dH2O to dissolve the RNA pellet (see Note 12). Place on ice.
Digest any residual DNA by adding 6.5 µL of 10× DNase buffer and 2 µL RQ RNase-free DNase I enzyme to each tube. Incubate at 37°C for 30 min.
Add 35 µL dH2O to bring to a final volume of 100 µL. Add an equal volume of phenol/chloroform. Vortex at high speed for 30 s. Centrifuge at 20,000 × g for 2 min at 4°C. Transfer the upper phase to a new 1.5 mL microcentrifuge tube and discard the lower phase in a chemical waste container.
Add 15 µL of 5 M NH4OAc and 287.5 µL ethanol and precipitate the RNA at −20°C overnight.
Centrifuge at 20,000 × g for 30 min at 4°C. Discard the supernatant.
Wash the pellet with 700 µL of 75% ethanol. Centrifuge at 20,000 × g for 10 min at 4°C. Air dry the pellet for 10–15 min at 25°C.
Resuspend the pellet in 24 µL dH2O (see Note 12). Store at −20°C.
3.9. cDNA Preparation
The isolated RNA is used for cDNA synthesis. Determine the absorbance of each RNA sample at 260 nm using a spectrophotometer (see Note 13).
To prepare the RNA samples for cDNA synthesis, aliquot 0.5 µg RNA from each sample into a 0.5 mL microcentrifuge tube, and add dH20 to a final volume of 10.5 µL. If hormone or another treatment is used, a total of 4 tubes will be needed for the control siRNA in the absence and in the presence of hormone and target siRNA in the absence and in the presence of hormone.
Prepare a cDNA sample, which will be used to derive a standard curve for each primer set during qRT-PCR. Aliquot 2.0 µg RNA from a control siRNA sample into another 0.5 mL microcentrifuge tube and add dH20 to a final volume of 10.5 µL.
Vortex, spin briefly in a microcentrifuge and then place all samples on ice.
To limit variability, make a master mix with enough reagent for all of the samples. Each sample will include 4 µL of 25 mM MgCl22 µL of 10× Reverse Transcription buffer, 1 µL of 10 mM dNTP, 0.5 µL of RNasin Ribonuclease Inhibitor, 15 U of AMV Reverse Transcriptase, 0.5 µL of Random Primers and dH2O to 9.5 µL. The reagents should be combined in the order given in a single 0.5 mL microcentrifuge tube.
Aliquot 9.5 µL of the master mix into each RNA-containing tube.
Incubate the reaction mixture at 42°C for 1 h and then at 95°C for 5 min (see Note 14). Chill on ice and then store at −20°C.
3.10. Quantitative Real Time PCR
These instructions assume the use of iQ SYBR Green Supermix and the iCycler Thermal Cycler, but can be adapted to other systems.
Design primers for the mRNA of interest (see Note 15) and dissolve in dH2O to a concentration of 10 µM. Primers should be designed for the target mRNA to confirm that the siRNA has knocked down the transcript. As seen in Fig. 3, NM23-H1 mRNA levels are significantly decreased in the presence of the target siRNA containing NM23-H1 mRNA sequence, but not in the presence of control siRNA containing renilla mRNA sequence.
Primers should also be designed to assess the mRNA levels of genes of interest (see Note 16). As seen in Fig. 3, the estrogen-responsive PR gene is significantly enhanced when NM-3-H1 expression is decreased, which is consistent with the Western analysis shown in Fig. 2. In contrast, neither pS2 nor ERα mRNA levels are significantly altered by the NM23-H1 siRNA.
Dilute the cDNA samples prepared in Subheading 11.3.9, step 2 (0.5 µg) 1:5 in dH2O (see Note 17).
Dilute the cDNA prepared in Subheading 3.9, step 3 (2.0 µg) 1:5 in dH2O. Next, make three 1:10 serial dilutions in dH2O (see Note 18).
Make a master mix with enough reagents for all of the qRT-PCR standards and samples, which are run in triplicate. Each reaction consists of 12.5 µL of 2× iQ SYBR Green Supermix, 9.5 µL dH2O, 1 µL of 10 µM Forward primer and 1 µL of 10 µM Reverse primer.
Prepare a 96-well plate for qRT-PCR. Aliquot 24 µL of the master mix per well and add 1 µL cDNA into each well. Make sure to keep track of the placement of each sample.
- Place the plate into the iCycler and start the program.
- Cycle 1, 3 min 95°C
- Cycle 2, 40 times
- 10°s 95°C Denaturation
- 30 s 56°C Annealing (see Note 19)
- 10 s 72°C Extension
- Cycle 3, 1 min 95°C
- Cycle 4, 1 min 50°C
- Cycle 5, 45 times Melt Curve 1°C/10 s 50°C
Analyze the data using the standard curve to ensure that the efficiency of the primers is sufficient (see Note 19). The melting curve of each amplicon should be examined to help ensure that a single product has been produced. Southern blot analysis can also be used to verify that the correct target sequence has been amplified.
The comparative Ct method (see Note 20) is useful in analyzing qRT-PCR data because it takes into consideration an internal housekeeping gene (see Note 16). Each of the samples shown in Fig. 3 has been normalized for 36B4 expression using the comparative Ct method.
Fig. 3.

Effect of decreased NM23-H1 on cellular mRNA levels. MCF-7 cells were transfected with control or NM23-H1 siRNA for 96 h, treated with ethanol vehicle (light gray bars) or E2 (dark gray bars) for 24 h, and lysed. RNA was isolated, cDNA was synthesized, and qRT-PCR was carried out with gene-specific primers. Relative fold change was determined using the comparative Ct method and the housekeeping gene, 36B4, as the internal control. Data from one experiment, which had been done in triplicate, is presented as the mean ± SD.
Acknowledgments
We are grateful to J. Bonéy Montoya, A. Rao, D. Thorngren, and Y. Ziegler for assistance in the preparation of this manuscript. This work was supported by NIH grant R01 DK 53884 (to AMN).
Footnotes
Antibiotic free media (PRF-A and PRF-AS) should be used just prior to the time of siRNA addition until the time of harvest.
The control siRNA should not be present in any endogenous cellular transcript and is used to demonstrate that neither the transfection reagent nor the procedure alters gene expression. A number of pretested control siRNAs such as renilla luciferase (Ambion, Austin, TX), which is used in Figs. 1–3, are available commercially. A scrambled siRNA sequence can also be used as a negative control, but care must be taken to ensure that the scrambled sequence will not recognize any endogenously-expressed mRNA sequence. BLAST searches (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) should be used to determine whether other mRNA or genomic sequences might be targeted. If overlap does exist, the sequence of the target siRNA and the mRNA transcript should have at least 2–3 mismatches.
Guidelines for designing target siRNA duplexes have been described in detail by the Tuschl laboratory (http://www.rockefeller.edu/labheads/tuschl/sirna.html) and on various company Websites. While it was previously necessary to design and order target siRNAs for each gene of interest, there are now several commercial suppliers that offer validated siRNAs, which have been tested for effectiveness in reducing target mRNA levels, and predesigned siRNAs, which have not been tested experimentally. All of the validated siRNAs we have used to date have been effective in knocking down mRNA and protein expression. If nonvalidated, predesigned siRNAs are used, two different target siRNAs should be tested.
Solutions used for RNA and cDNA procedures should be prepared with deionized (resistivity of 18.2 MΩ-cm and total organic content < 10 ppb) and autoclaved water, which is referred to as “dH2O” in this text. All solutions, reagents, and supplies used for RNA work should be RNase free.
Fifty picomole per well (100 nM) of a 21mer is generally a good starting point, but may need to be increased to effectively knock down the protein of interest or decreased so that it does not have unintended effects on the expression of other mRNA species.
The length of time needed to knock down the target mRNA and protein levels must be determined empirically and may range from 24 to 96 h. Although qRT-PCR can determine whether the target mRNA level has been reduced, it is important to ensure that the target protein is effectively knocked down as well. If used, hormone is added to the media after the protein has been effectively knocked down, and the plate is incubated for the length of time required for the biological endpoint being examined. Although siRNAs are effective in reducing target protein expression for ~3–5 days, it may be necessary to knock down expression of a protein for a week or more. In this case, short hairpin RNA (shRNA) expression vectors with antibiotic resistance can be utilized (17–19). Interpretation of these experiments may be difficult; however, since a change in the biological endpoint may not necessarily be attributed to the absence of the target protein, but may reflect a change in expression of another cellular protein whose presence or activity is affected by the target protein.
Make certain that the nitrocellulose membrane is placed between the gel and the anode. If the sandwich is not appropriately oriented or is inserted into the apparatus backward, the proteins will be eluted into the buffer rather than being transferred to the nitrocellulose membrane.
Primary and secondary antibody dilutions and incubation conditions must be determined individually. We typically start with primary antibody dilutions of 1:2,000–40,000 and secondary antibody dilutions of 1:5,000–100,000.
The Pierce chemiluminescent substrate is prepared at a 1:1 ratio of solutions 1 and 2. Approximately 25 µL of this chemiluminescent substrate mix is needed for each cm2 of nitrocellulose membrane. For example, a 5 × 7 cm blot = 35 cm2 × 25 µL/cm2 = 875 µL. Thus, use 437.5 µL each of Solution 1 and 2.
A protein (such as GAPDH or β-actin) that is unaffected by siRNA knockdown of the protein of interest should be used to verify that samples have been equally loaded. This loading control also helps to ensure that the target siRNA is specific.
Cell lysate may be stored in Trizol at −80°C for up to 1 month.
Pass the RNA pellet through a 200 µL pipette tip 8–10 times to dissolve. If the pellet is difficult to resuspend, incubate at 55–60°C for 10 min, and draw through a pipette 8–10 times.
- RNA concentration (µg/µL) = [(Abs260)(43 µg/mL) (dilution factor)]/1,000
A thermocycler can be programmed to carry out the reverse transcription reaction.
It is very important to use the mRNA or cDNA sequence when designing primers and not the DNA sequence which may contain an intronic sequence that is not transcribed. Amplicons of ~100 bp in length are efficiently produced. Longer length amplicons are acceptable, but generally should not exceed 250 bp.
Gene-specific primers are used to monitor endogenous gene expression after cells have been exposed to the target or control siRNA. It is also essential to monitor the expression of a gene that is unaffected by the siRNA treatment to ensure that the effects of the target siRNA are specific. When choosing a control gene, it is important to keep in mind that the gene should not be affected by any treatment used. Commonly used housekeeping genes might include GAPDH, β-actin or 36B4, which was used in the experiments shown in Fig. 3.
cDNA concentration = 0.5 µg/20 µL. After 1:5 dilution, cDNA concentration = 0.5 µg/100 µL or 5 ng/µL.
For the standard curve: cDNA concentration = 2.0 µg/20 µL. After 1:5 dilution, cDNA concentration = 2.0 µg/100 µL or 20 ng/µL. Three 1:10 serial dilutions will produce standards of 2, 0.2 and 0.02 ng/µL. By running standards in parallel, it is possible to determine the initial number of RNA transcripts present in the experimental wells.
The annealing temperature of individual primer sets will vary, but optimal annealing temperatures are generally provided by primer design software. The ideal primer set will have a standard curve r-value > 0.980 and an efficiency of 90–110%. An efficiency value significantly larger than 100% indicates the formation of primer dimers.
In qRT-PCR, the threshold cycle (Ct) refers to the point where the fluorescence generated within a reaction reaches threshold, indicating that the number of copies has accumulated significantly. Each well is assigned a Ct value and it can be used to compare copy number between samples. Thus, ΔΔCt = ΔCtt,sample − ΔCtt,referencewhere ΔCtt,sample is the Ct value for any sample normalized to the housekeeping gene and ΔCtt,reference is the Ct value for the control siRNA vehicle treated sample also normalized to the housekeeping gene. Relative fold change is 2−ΔΔCt.
References
- 1.Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci. 2001;114:4557–4565. doi: 10.1242/jcs.114.24.4557. [DOI] [PubMed] [Google Scholar]
- 2.Harborth J, Elbashir SM, Vandenburgh K, Manninga H, Scaringe SA, Weber K, Tuschl T. Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense Nucleic Acid Drug Dev. 2003;13:83–105. doi: 10.1089/108729003321629638. [DOI] [PubMed] [Google Scholar]
- 3.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in. Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 4.Montgomery MK, Fire A. Double- stranded RNA as a mediator in sequence-specific genetic silencing and co-suppression. Trends Genet. 1998;14:255–258. doi: 10.1016/s0168-9525(98)01510-8. [DOI] [PubMed] [Google Scholar]
- 5.Tuschl T, Zamore PD, Lehmann R, Bartel DP, Sharp PA. Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev. 1999;13:3191–3197. doi: 10.1101/gad.13.24.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 7.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 8.Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 10.Schwarz DS, Tomari Y, Zamore PD. The RNA-induced silencing complex is a Mg2 + -dependent endonuclease. Curr Biol. 2004;14:787–791. doi: 10.1016/j.cub.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Curtis CD, Likhite VS, McLeod IX, Yates JR, Nardulli AM. Interaction of nonmetastatic protein 23 homolog H1 and estrogen receptor alpha alters estrogen- responsive gene expression and DNA nicking. Cancer Res. 2007;67:10600–10607. doi: 10.1158/0008-5472.CAN-07-0055. [DOI] [PubMed] [Google Scholar]
- 12.Marzouk S, Schultz-Norton J, McLeod I, Yates J, Nardulli A. Rho GDP dissociation inhibitor alpha interacts with estrogen receptor alpha and influences estrogen responsiveness. J Mol Endocrinol. 2007;39:249–259. doi: 10.1677/JME-07-0055. [DOI] [PubMed] [Google Scholar]
- 13.Rao AK, Ziegler YS, McLeod IS, Yates JR, Nardulli AM. Effects of Cu/Zn Superoxide Dismutase (SOD1) on estrogen responsiveness and oxidative stress in human breast cancer cells. Mol Endocrinol. 2008;22:1113–1124. doi: 10.1210/me.2007-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schultz-Norton JR, McDonald WH, Yates JR, Nardulli AM. Protein disulfide isomerase serves as a molecular chaperone to maintain estrogen receptor α structure and function. Mol Endocrinol. 2006;20:1982–1995. doi: 10.1210/me.2006-0006. [DOI] [PubMed] [Google Scholar]
- 15.Schultz-Norton JR, Walt KA, Ziegler YS, McLeod IX, Yates JR, Raetzman LT, Nardulli AM. The DNA repair protein flap endonuclease-1 (FEN-1) modulates estrogen-responsive gene expression. Mol Endocrinol. 2007;21:1569–1580. doi: 10.1210/me.2006-0519. [DOI] [PubMed] [Google Scholar]
- 16.Schultz-Norton JR, Gabisi VA, Ziegler YS, McLeod IX, Yates JR, Nardulli AM. Estrogen receptor alpha interaction with the DNA repair protein proliferating cell nuclear antigen (PCNA) Nucleic Acids Res. 2007;35:5028–5038. doi: 10.1093/nar/gkm533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2:243–247. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- 18.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 19.Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]