

Abstract
The De Sanctis-Cacchione Syndrome is the rarest and most severe kind of xeroderma pigmentosum, characterized by microcephaly, hypogonadism, neurological disorders, mental and growth retardation, with very few cases published. The clinical findings compatible with De Sanctis-Cacchione Syndrome and the therapeutic approach used to treat a one year and nine months old child, with previous diagnosis of xeroderma pigmentosum, are reported.
Keywords: DNA repair, DNA Repair-Deficiency Disorders, Xeroderma pigmentosum, Xeroderma pigmentosum group A protein
Abstract
A síndrome de de Sanctis-Cacchione é a forma mais rara e grave do xeroderma pigmentoso e é caracterizada por microcefalia, hipogonadismo, alterações neurológicas e retardo mental e de crescimento, com poucos casos publicados. Relatam-se os achados clínicos compatíveis com essa síndrome e a terapêutica instituída em uma lactente de um ano e nove meses, com diagnóstico prévio de xeroderma pigmentoso.
INTRODUCTION
The De Sanctis-Cacchione Syndrome is the most rare and severe kind of xeroderma pigmentosum (XP). It occurs when XP type A, develops with microcephaly, hypogonadism, multiple neurological alterations and mental and growth retardation. On the other hand, xeroderma pigmentosum is an autosomal recessive disorder associated with failure of the DNA damage excision and repair mechanism, leading to cell hypersensitivity to ultraviolet radiation (UVR).1-4
As a result of sunlight exposure, hyperkeratotic lesions, ephelides, telangiectasias and solar lentigines appear on the skin. Benign tumors, pre-neoplastic and neoplastic lesions are frequent at an early age. The most frequently found malignant histological types are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC).1,3,4
Neurological alterations derive from fibroblast deterioration caused by UVR. Ataxia, hyporeflexia or areflexia may be observed, as well as alterations in motor coordination, microcephaly, mental retardation and sensorineural deafness. Various ophthalmologic alterations are also described.2,4
The treatment is primarily based on the education of the patients and their families regarding sun exposure , in addition to wearing photoprotective clothing, glasses and lotions. For the treatment of already established lesions, chemotherapeutic agents (5-fluorouracil), immunomodulators (imiquimod) and retinoids (acitretin) may be employed, depending on the location, severity and extension of the lesion. More interventionist possibilities are radiotherapy, dermabrasion, cryotherapy and surgical excision. The prognosis of the disease is poor.5,6,7
CASE REPORT
A child with a year and nine months, female, born in and from Água Branca - Alagoas, of mixed race, was admitted to the pediatric emergency of a tertiary hospital with complaint of skin tumors that appeared when she was six months of age.
The physical examination revealed important weight and height impairment, microcephaly, assessed with charts proposed by the World Health Organization (WHO) and the Brazilian Pediatric Society (SBP), with values below the 3rd percentile in every parameter. At the neurological examination, the absence of several neuropsychomotor developmental markers expected at her age were noted: she could not find the right words to use, presented difficulty to walk alone, interacted little with recreational objects and did not cooperate to get dressed.
Erythema and purulent secretion were observed in the conjunctivas and skin, which presented hypochromic and achromic spots interspersed with hyperchromic spots, mainly on the face and limbs, in addition to hair rarefaction (Figure 1).
FIGURE 1.
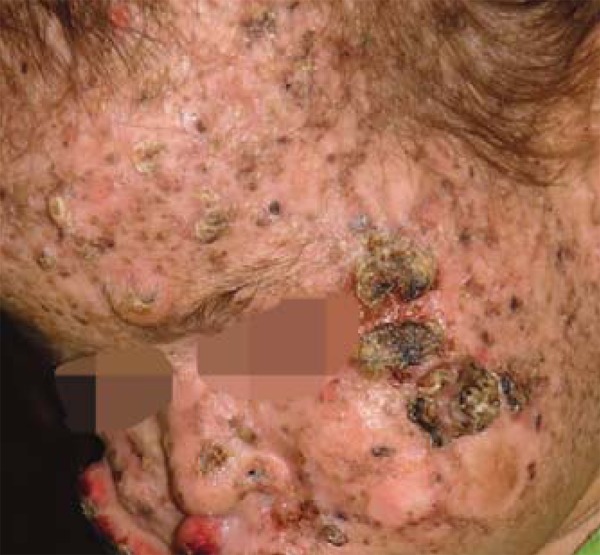
General aspect of the infant's face. Tumoral syndrome on left malar region and hair rarefaction
A map of skin lesions was drawn. The outstanding locations, by suspected malignancy aspect, were:
left malar region: three keratotic lesions, of tumoral aspect and well-defined limits, crusty surface and total area measuring 4.8 x 1.8 x 0.7 cm (Figure 1).
right malar region: tumor lesion with elevated borders and central ulceration, measuring 3.3 x 2.8 x 0.7cm (Figure 2).
FIGURE 2:
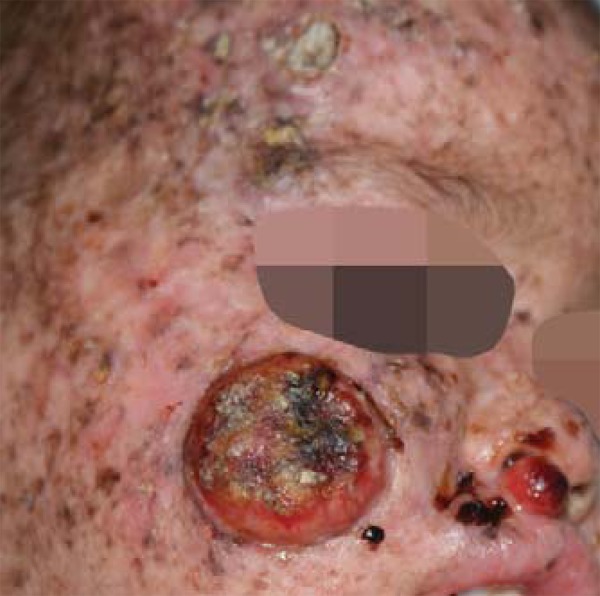
Lesions in right malar region and supralabial region
supralabial region: nodular-tumoral smooth surfaced lesion measuring 1.0 x 0.9 x 0.6 cm (Figure 2).
cervical and thoracic dorsal region: lesions disposed as elevated, keratotic lesions, three of them larger and two presenting central ulcerations (Figure 3).
FIGURE 3.
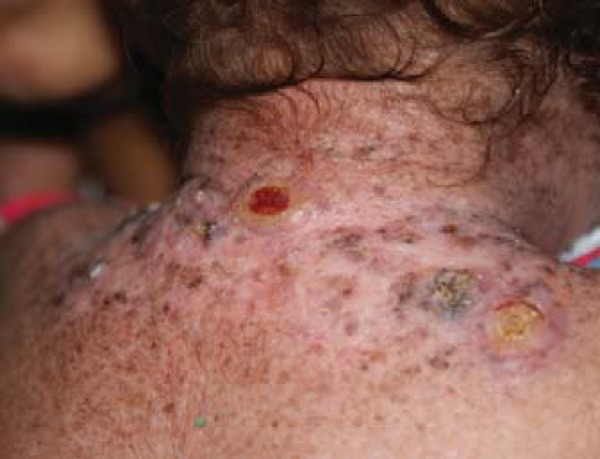
Lesions in cervical and posterior thoracic regions, characterized by elevated keratotic plaques and central ulceration
right forearm: keratotic tumoral lesion with a crusty surface and well-defined limits (Figure 4).
FIGURE 4.
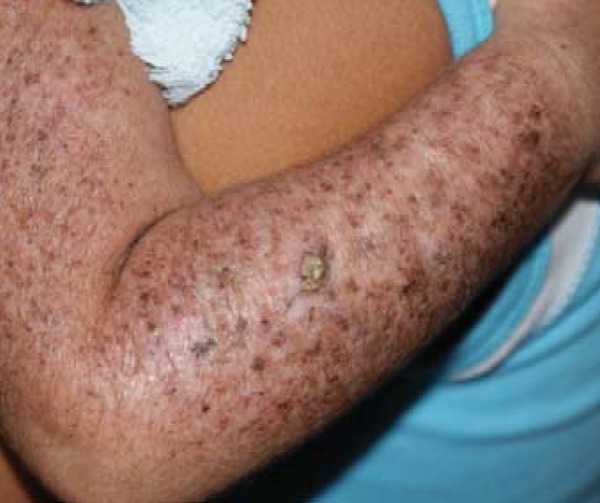
Right upper limb demonstrating the general aspect of the skin of the patient, with evidence hypochromic/ achromic, hyperchromic and lentiginous lesions, as well as tumoral lesion of keratotic surface
She underwent excision and histopathological examination of the tumoral lesions and a skin graft was performed on the two larger facial lesions. Daily local dressings with antibiotics (neomycin) were applied. There was graft loss during the postoperative period. After 15 days new grafting was performed on the raw areas, evolving with partial loss of both grafts.
The revaluation in the second month of the postoperative period revealed new facial tumor growth on the scar tissue of the left malar region and increased number of cervical lesions. As there was scarcity of donor areas for skin grafts, the removal of all lesions was not achieved, the poor prognosis and the unsatisfactory therapeutic result, the child was referred to the radiotherapy service for treatment of the remaining skin lesions.
The histopathological results for the excised lesions were invasive SCC and invasive SCC with sarcomatoid area for the right and left malar regions, respectively. For the upper lip tumor the result was atypical fibroxanthoma (AF).
Despite the symptoms, a head MRI scan did not reveal alterations.
The images were acquired and published with the authorization of the child's mother, by means of an informed consent form.
DISCUSSION
Xeroderma Pigmentosum carriers present a great spectrum of clinical manifestations, which vary within the eight existing XP groups (XPA-XPG and a type known as variant - XPV). In 1932, De Sanctis and Cacchione reported a syndrome with classical XP alterations type A, in addition to neurological and somatic findings.
On the De Sanctis-Cacchione Syndrome the peculiarity is the early onset and severity of lesions, which begin around the age of six months, in contrast with other XP forms, where age at onset averages 2 years. This is justified by alteration in DNA repair capacity after sun exposure, which depending on XP type is reduced by up to 7.5%, compared to 100% in the control group. When she was admitted, the patient already presented well marked tumor lesions.1,2,4,6,8
Hypogonadism investigation was compromised by hormonal and physiological immaturity typical of the age. As in other reports, there was severe weight and height impairment and microcephaly. No mention of these alterations was made in the referral letter and data recorded in the child's pediatric follow-up card. Such evaluation might have allowed an even earlier diagnosis.1,3,4,9
A more accurate neurological examination was hindered by lack of cooperation from the child. Although the first MRI was unaltered, other studies should be carried out in the future, since damage is progressive.2,4,8
Basal cell carcinoma (BCC) is the most frequent tumor. A SCC finding discloses a more aggressive clinical picture. AF, which is rare in healthy children, is reported in XP patients.1,3,4,10
Genetic counseling is essential for the limitations to treatment and prognosis. Meetings were held with family members for clarification about the disease and treatment, in accessible, colloquial language, taking into consideration that the mother of the patient was illiterate. A summary of the clinical history and treatments to which the child was submitted was given to the child's mother.1
Footnotes
Study carried out at the Institute of Integral Medicine (Instituto de Medicina Integral Professor Fernando Figueira - IMIP) – Recife (PE), Brazil.
Financial Support: none
Conflict of Interests: none
REFERENCES
- 1.Liy MDC, Durán-McKinster C, Orozco L, Sáez MDM, Carrasco D, Ruiz-Maldonado R. Xeroderma pigmentoso con retraso psicomotor: síndrome de De Sanctis Cacchione. Reporte de 2 casos de origen mexicano. Dermatol Pediatr Lat. 2004;2:50–53. [Google Scholar]
- 2.Oliveira CRD, Elias L, Barros ACM, Conceição DB. Anestesia em paciente com Xeroderma Pigmentoso: relato de caso. Rev Bras Anestesiol. 2000;53:46–51. [PubMed] [Google Scholar]
- 3.Falcón Lincheta L, Dorticós Balea A, Daniel Simón R, Garbayo Otaño E. Xeroderma pigmentoso. Síndrome de Sanctis Cacchione: Presentación de 1 caso. Rev Cubana Pediatr. 1998;70:113–116. [Google Scholar]
- 4.Kraemer KH, Lee MM, Scotto J. Xeroderma Pigmentosum: cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–250. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 5.Nelson BR, Fader DJ, Gillard M, Baker SR, Johnson TM. The role of dermabrasion and chemical peels in the treatment of patients with xeroderma pigmentosum. J Am Acad Dermatol. 1995;32:623–626. doi: 10.1016/0190-9622(95)90348-8. [DOI] [PubMed] [Google Scholar]
- 6.Rubió Casadevall J, Graña-Suárez B, Hernandez-Yagüe X, Vayreda Ribera J, Huc Grasa O, Brunet Vidal J. Xeroderma pigmentosum: neck lymph node metastasis of a squamous cell carcinoma of the skin treated with cetuximab. Eur J Dermatol. 2009;19:163–165. doi: 10.1684/ejd.2008.0574. [DOI] [PubMed] [Google Scholar]
- 7.Prieto GEA, Borroto LJM, Valdés GF, Pomares IY. Propuesta de manejo terapéutico en un caso de Xeroderma pigmentosum. Rev Cubana Invest Bioméd. 1999;18:40–42. [Google Scholar]
- 8.Cleaver JE, Feeney L, Tang JY, Tuttle P. Xeroderma Pigmentosum Group C in an Isolated Region of Guatemala. J Invest Dermatol. 2007;127:493–496. doi: 10.1038/sj.jid.5700555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.World Health Organization . WHO Multicentre Growth Reference Study Group.WHO Child Growth Standards: Length/height-for-age, weight-for-age, weight-for-length, weight-for-height and body mass index-for-age: Methods and development. Geneva: World Health Organization; 2006. 312 [Google Scholar]
- 10.Fleury LFF, Jr, Sanches JA., Jr Primary cutaneous sarcomas. An Bras Dermatol. 2006;81:207–221. [Google Scholar]