

Abstract
Lymphotoxin (LT) is a member of the tumour necrosis factor (TNF) superfamily that was originally thought to be functionally redundant to TNF, but these proteins were later found to have independent roles in driving lymphoid organogenesis. More recently, LT mediated signalling has been shown to actively contribute to effector immune responses. LT regulates dendritic cell and CD4+ T cell homeostasis in the steady state and determines the functions of these cells during pathogenic challenges. The LT receptor pathway is essential for controlling pathogens and even contributes to the regulation of the intestinal microbiota, with recent data suggesting that LT induced changes in the microbiota promote metabolic disease. In this Review, we discuss these newly defined roles for LT, with a particular focus on how the LT receptor pathway regulates innate and adaptive immune responses to microorganisms.
Lymphotoxin-α (LTα; also known as TNFSF1) is a member of the tumour necrosis factor (TNF) super-family and was originally identified as a product of lymphocytes that was capable of exerting cytotoxic effects on tumour cells in vitro (hence the name lymphotoxin)1,2. LTα forms a homotrimer that binds to herpes virus entry mediator (HVEM; also known as TNFRSF14) with low affinity, and this has unknown biological effects. Homotrimers of LTα also bind to TNF receptor 1 (TNFR1; also known as TNFRSF1A) and TNFR2 (also known as TNFRSF1B) with high affinity, but they have a much less prominent role in driving TNFR signalling than does TNF itself 2–4. It was later shown that LTα-mediated signalling is essential for the development of secondary lymphoid tissues, whereas TNF-mediated signalling alone has only a minor role in this process5–7. Indeed, without LTα, animals lack lymph nodes and Peyer’s patches. LTα is also essential for the organization of lymphoid organs, such as the spleen and thymus4,8–10.
Notably, LTα-mediated induction of TNFR signalling is not the major pathway for promoting lymphoid tissue development; this process is instead dependent on lymphotoxin-β receptor (LTβR; also known as TNFRSF3)-mediated signalling. LTα forms a heterotrimer with LTβ (a product of the nearby LTB gene (also known as TNFSF3)), and this heterotrimer binds to LTβR4,6,11,12. LTα1β2 is produced by cells of the lymphocytic lineage, including B cells, T cells and innate lymphoid cells (ILCs) that express the transcription factor retinoic acid receptor-related orphan receptor-γt (RORγt)2,13,14. LTα-, LTβ- and LTβR-deficient mice have been shown to have similarly defective lymphoid organogenesis, which indicates that LTβR signalling in response to LTα1β2 binding is crucial for this process. LTβR is expressed by fetal stromal cells that are found in the lymph node anlagen during development, as well as by cells of the myeloid lineage, hepatocytes, intestinal epithelial cells and endothelial cells2,14,15. Therefore, this cytokine–receptor pair serves as a bridge between lymphoid cells and non-lymphoid cells, including parenchymal and stromal cells.
Although the role of LT was once thought to be restricted to promoting lymphoid tissue development7, it is now known that LT also maintains the structure of the postnatal spleen16. LT regulates endothelial cell expression of mucosal vascular addressin cell adhesion molecule 1 (MADCAM1), an adhesion molecule that is recognized by lymphocytes that express L-selectin. MADCAM1 is detectable 3–5 days after birth in the spleens of Lta−/− and Ltbr−/− mice, but its expression is lost at later time points16. Thus, in addition to promoting lymphoid tissue development, LT is also essential for maintaining these structures postnatally.
As LT has such a crucial role in lymphoid tissue organogenesis, it has been more difficult to define roles for LT in effector immune responses that are independent of tissue organogenesis. However, recent studies have shown that LT contributes to the effector responses of both the innate and the adaptive immune systems (discussed below). Several reviews have covered the functions of LT in prenatal lymphoid tissue development2,14,17; therefore, in this Review, we focus on the immune functions of the LT pathway in adult mice. In particular, we emphasize the new roles that have been identified for LT in protecting the host against pathogenic insults and in regulating the composition of the host microbiota.
DC regulation depends on the LT receptor
DC derived LT modulates the cellularity of secondary lymphoid tissues
Although endothelial cells are known to be the targets of LT in secondary lymphoid tissues, the cellular source of the LT is less clear. Conventionally, lymphocytes have been considered to be the only producers of LTα1β2, but this has been challenged by recent data that indicate that dendritic cells (DCs) also express LT and can control secondary lymphoid tissue cellularity18 (FIG. 1). Furthermore, another LTβR ligand, LIGHT (also known as TNFSF14), is expressed by human DCs19,20.
Figure 1. The LT pathway regulates DC fates.
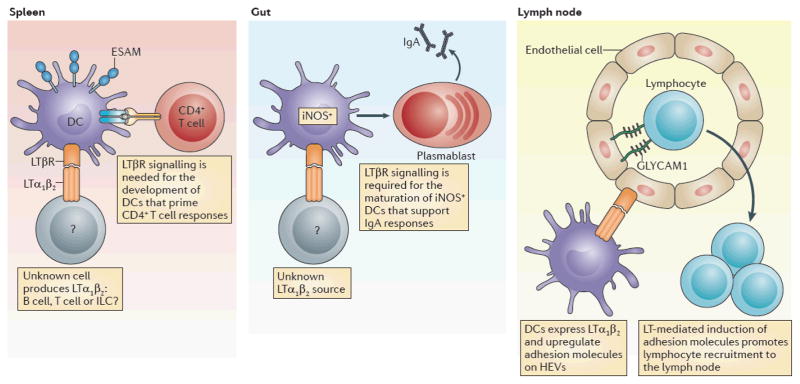
Dendritic cells (DCs) require lymphotoxin-α1β2 (LTα1β2) that is provided by a currently undefined cell type. Endothelial cell selective adhesion molecule (ESAM)hi splenic DCs, which are essential for priming CD4+Tcells, require T signaling for their development29,30. In the gut, LTβR induces the maturation of inducible nitric oxide synthase (iNOS)+ DCs, which then induce the development of IgA producing plasmablasts36. In both of these cases, further studies are required to define the cellular source of the LT. Additionally, in the lymph nodes, DCs have recently been shown to express LTα1β2 (REF. 18), which is recognized by high endothelial venules (HEVs) and leads to their upregulation of adhesion molecules, such as glycosylation dependent cell adhesion molecule 1 (GLYCAM1), and is important for maintaining lymph node cellularity in the steady state. ILC, innate lymphoid cell.
The classic model for DC function is that DCs patrol tissues in search of invasive pathogens, that they mature in response to ‘danger’ or ‘damage’ signals that they receive in situ and that they subsequently initiate adaptive immune responses by migrating to secondary lymphoid organs where they function as antigen-presenting cells (APCs)21; however, the role of DCs in uninfected hosts is less clear22. Using the Cd11c–DTR system, one group observed a decrease in overall lymph node cellularity after DC ablation18. This finding extended previous work that showed that antigen-pulsed DCs increase lymph node cellularity, but only if the DCs express CC-chemokine receptor 7 (CCR7) and are therefore able to home to the lymph nodes23. DC depletion correlated with the reversion of high endothelial venules (HEVs), which regulate lymphocyte entry into lymphoid tissues, from a mature to an immature phenotype18, in which they lacked expression of high-affinity cell-surface receptors that facilitate lymphocyte recruitment24–26 (FIG. 1). The phenotype of HEVs that are found in DC-depleted mice is reminiscent of the phenotypes observed in animals that lack the LTβR18,24,27. Further analysis showed that classical DCs (CD11chiMHC class IIint) that reside in the lymph node can express LT, although migratory DCs (CD11cintMHC class IIhi) do not express this protein18. The same study showed that the elimination of DCs capable of LTα expression also resulted in decreased lymph node cellularity18.
One potential caveat to the experiments addressing DC expression of LT is that the membrane-bound version of LT was never stained to demonstrate that it was actually expressed by DCs18,28. Therefore, more work is needed to confirm the expression of LT by DCs. Classical DCs are a rapidly turning over population of cells21, and so additional work is needed to determine whether it is DC development or DC function that is directly dependent on LT.
LT is required for DC homeostasis
The relationship between DCs and LT is actually quite complex. Ltbr−/− animals lack specific subsets of splenic DCs29,30, and mixed chimaeras show that this defect cannot be rescued by the presence of wild-type stromal cells. This indicates that LTβR signalling is a cell intrinsic requirement for DC homeostasis29.
It was recently shown that Notch-dependent endothelial cell-selective adhesion molecule (ESAM)hi DCs require LTβR for their development, whereas the development of ESAMlow DCs is unperturbed in the absence of LTβR30 (FIG. 1). The newly characterized population of ESAMhi DCs proliferates at a greater rate than their ESAMlow counterparts, and ESAMhi DCs are essential for CD4+ T cell priming (FIG. 1). OT-II T cells — CD4+ T cells with a transgenic T cell receptor (TCR) that is specific for ovalbumin — divide at a much lower rate in animals lacking the LTβR-dependent ESAMhi DC population than in wild-type animals, which indicates that LTβR signalling might promote CD4+ T cell priming by regulating the maturation status of APCs30. Although DC subsets are reduced in Ltbr−/− mice, the mechanisms behind these selective DC deficiencies are still unclear. Early studies suggested that chemokine production is diminished in the absence of LT signalling, preventing the entry of DCs into tissues31. However, mixed bone marrow chimaeras indicate that this explanation is unlikely to be correct29. Instead, it seems that the LT pathway is essential for promoting the maturation of DC subsets. In this scenario, one would envision the agonism of LTβR signalling on DCs to alter their cell fate through transcription factors downstream of the LT pathway (BOX 1). This might be a mechanism that enables the maturation of DCs once they have migrated into certain host tissues, such as lymphoid organs or intestinal tissue, where a large proportion of LTα1β2-expressing lymphocyte populations reside22,32–34.
Box 1. LTβR signalling that enables effector functions.
Known ligands for the lymphotoxin-β receptor (LTβR) include LTα1β2 and LIGHT19. Agonism of LTβR by either of these ligands initiates both canonical and non canonical nuclear factor κB (NF-κB) signalling105. Tumour necrosis factor receptor 1 (TNFR1) signalling preferentially, but not exclusively, results in the translocation of p50–RELA (also known as p50–p65) complexes into the nucleus, whereas LTβR signalling preferentially results in the translocation of p52–RELB complexes into the nucleus, in addition to the aforementioned canonical NF-κB complex19,105. The direct cytotoxic effects of LTβR signalling can be initiated by both LTα1β2 and LIGHT in conjunction with interferon γ (IFNγ)106,107. Other effector consequences that are initiated by the LT pathway (such as the expression of certain cytokines) are influenced by either canonical or non canonical NF-κB activity. LTα3 is a secreted homotrimer that can initiate cytotoxic functions through TNFR1 (REF. 2).
DC derived LT regulates IgA production
The production of IgA is essential for maintaining symbiotic relationships between the host and its microbiota1,35. Recent work has indicated that the effects of LT on DCs might explain why the LT pathway is necessary for IgA production36. Animals lacking LTβR or LTα are devoid of both serum IgA and intestinal IgA37; remarkably, the defect in IgA production seen in LT-deficient animals is more severe than that observed in μMT−/− mice38. The inability of Ltbr−/− or Lta−/− mice to produce IgA was originally attributed to defects in stromal cell populations in the small intestine that might have provided signals for B cells to class switch to IgA37. Support for this model came from a putative role for RORγt+ ILCs in providing membrane-bound LTα1β2 to stromal cells and in inducing their production of factors that regulate IgA39,40. However, it was recently shown that inducible nitric oxide synthase (iNOS)-producing DC subsets, which are absent in the gastrointestinal tract of Ltb−/− and Ltbr−/− mice, contribute to intestinal IgA production and carry out this function independently of LTβR-expressing stromal cells36 (FIG. 1).
DCs residing in mucosa-associated lymphoid tissue (MALT) express iNOS, and mice lacking iNOS expression have defects in IgA production41–43. As mentioned above, it was previously thought that the defect in IgA production seen in Ltbr−/− animals was due to the absence of LTβR on stromal cells in the intestinal lamina propria; this original finding was supported by data which demonstrated that intestinal tissue from recombination-activating gene 1 (RAG1)-deficient mice could rescue IgA production in Ltbr−/− hosts37. However, it is possible that iNOS+ DCs in the graft could also have been responsible for rescuing IgA production in these earlier experiments. In light of new findings, the main role of ILCs in mediating IgA production might be to promote the organization of structures in which iNOS+ DCs can interact with B cells. Thus, the role of LT in IgA production might be multifaceted: LT might recruit B cells and DCs by inducing chemokine expression, it might initiate lymphoid structure organization and it might even function as a signal to induce the development of iNOS+ DC subsets. Perturbing iNOS-dependent IgA production, or eliminating class-switch recombination and somatic hypermutation, in mice alters the composition of the host microbiota36,44. Clearly, future work that aims to address how LT contributes to DC homeostasis in the intestine and in lymphoid tissues will be crucial for appreciating host–microorganism symbiosis.
Roles for LT in adaptive immunity
LT supports CD4+ T cell differentiation
In addition to showing deficiencies in IgA production, mice deficient in LT signalling fail to produce IgE45. This inability to produce IgE might reflect a broader imbalance in CD4+ T helper (TH) cell subsets that occurs in Lta−/− and Ltbr−/− mice. Defective CD4+ T cell responses have been observed in LT-deficient mice subjected to various immune challenges28,46–48. Mice lacking LT express higher levels of TH1-type cytokines in their spleen and lungs45. Although these data indicate that LT might be essential for TH2 cell development, LT is traditionally thought of as a hallmark cytokine in the TH1 cell response49,50. The generation of TH2 cells is not completely understood51,52, and the contribution of LT to TH1- or TH2-type immunity reveals how the host creates unique CD4+ T cell effector programmes.
Citrobacter rodentium and Leishmania major are pathogens that require TH1-type immunity to be cleared from the body46,47. Blocking LT signalling in wild-type mice results in an increased susceptibility to C. rodentium. Intriguingly, although wild-type splenocytes produce interferon-γ (IFNγ) in response to C. rodentium challenge, splenocytes from Ltbr−/− animals produce interleukin-4 (IL-4) and IL-10 (REF. 46). In addition, Ltbr−/− mice show defective humoral responses during C. rodentium infection and generate higher levels of pathogen-specific IgG1 and lower levels of pathogen-specific IgG2a compared with wild-type mice. However, Ltbr−/− mice are particularly susceptible to C. rodentium during the first 10 days of infection, which indicates that LT might be important for innate responses; and this might complicate the interpretation of these studies.
Experiments using L. major have also indicated that LT might contribute to TH cell differentiation47. Animals deficient in LT signalling fail to control L. major infection, and, similar to the defect seen during C. rodentium infection, Ltbr−/− mice favour the development of IL-4-producing CD4+ T cells, rather than IFNγ producing T cells, in the spleen. In the case of infection with L. major, blocking peripheral lymph node development by treating animals in utero with soluble LTR-Ig and TNFR-Ig recapitulated the TH2-type skewing of the immune response observed in the Ltbr−/− and Lta−/− mice, even though animals generated using this technique could still signal through LTβR.
Whether lymph nodes contribute to the immune defence against C. rodentium infection by preventing TH2-type immune skewing or by another mechanism remains unclear. This is difficult to determine as the LT pathway has dual roles during C. rodentium challenge: hosts require LT for both TH1-type cytokine expression and innate IL-22 production in response to this pathogen76,77.
Why does LT deficiency affect T cell polarization?
One might conclude from the data discussed above that LT promotes TH1-type immune responses by supporting lymphoid tissue development, and that the absence of lymph nodes causes pathogens to evoke an inappropriate TH2 response. However, other data have demonstrated that Lta−/− mice generate increased levels of TH1-type cytokines in response to schistosome egg challenge experiments, which normally elicit a TH2-type immune bias45. This is a surprising result considering the data discussed above that indicate that LT promotes TH1-type immunity against various pathogens. The idea that LT influences TH cell education through lymphoid organogenesis does not explain why one would develop opposing T helper cell biases for both TH1- and TH2-type immunity.
It is probably the case that active LT signalling supports both lymph node development and the complex cellular interactions that occur in a fully developed lymph node (BOX 2; FIG. 2). This idea is supported by a recent study that explored LT deficiency in the setting of Heligmosomoides polygyrus infection53, a pathogen that is known to induce IL-4 and IgE production28. The authors found that LT signalling is essential for the generation of IL-4-producing CD4+ T cells in response to this infection. H. polygyrus infection results in considerable DC migration to the perifollicular and interfollicular regions of the lymph node28. These DCs express CXC-chemokine receptor 5 (CXCR5), which enables them to migrate in response to CXC-chemokine ligand 13 (CXCL13) that is produced by follicular DCs (FDCs)28.
Box 2. Lymphoid organogenesis directed by the LT pathway.
During embryonic development, retinoic acid is produced by neurons to recruit retinoic acid receptor related orphan receptor-γt (RORγt)+ fetal lymphoid tissue inducer (LTi) cells to the location of future lymph nodes14. Once LTi cells have arrived, they express membrane bound lymphotoxin-α1β2 (LTα1β2), which binds to the LT-β receptor (LTβR) on fetal stromal cells; stromal cells undergo transcriptional changes in response to LT signalling. Agonism of the LT pathway results in the local production of vascular cell adhesion molecule 1 (VCAM1), intercellular adhesion molecule 1 (ICAM1), mucosal vascular addressin cell adhesion molecule 1 (MADCAM1), CC chemokine ligand 19 (CCL19), CCL21 and CXC chemokine ligand 13 (CXCL13) to recruit a vast range of cells that ultimately form and populate the lymph node14. After birth, the LT pathway has an essential role in the maintenance of lymphoid tissues. For example, active LT signalling, mostly from B cells, is required to maintain splenic architecture in adult mice108. In the colon, B cells must express LTα to induce isolated lymphoid follicle expansion109, and the lack of LTα on B cells results in atrophic follicles109. Animals that lack LTi cells as a result of the absence of the transcription factor RORγt lack secondary lymphoid tissues, which includes isolated lymphoid follicles, but can develop tertiary lymphoid follicles from chronic inflammatory stimuli in similar locations110.
Figure 2. LT coordinates adaptive immunity.

The lymphotoxin (LT) pathway organizes multicellular interactions in the lymph node. B cell-derived LTα1β2 is recognized by the LTβ receptor (LTβR), which is expressed on follicular dendritic cells (FDCs), and this interaction promotes the secretion of chemokines that recruit antigen presenting dendritic cells (DCs) and CD4+ T cells to facilitate the development of T helper cell responses28,47. In the germinal centre, miR-155 expression in B cells maintains LT expression. Ligation of inducible T cell co-stimulator (ICOS) on T cells to ICOS ligand (ICOSL) on B cells and the CD40–CD40 ligand (CD40L) interaction are also essential for the upregulation of LT on B cells that enables a germinal centre response 55,56. CXCL13, CXC chemokine ligand 13; CXCR5, CXC chemokine receptor 5; TCR, T cell receptor.
In addition, follicular helper T (TFH) cells are also recruited to the perifollicular regions during helminth challenge28. The authors speculated that LT might contribute to this multicellular organization process in the lymph node, as it does in the spleen16,31. Blockade of LT signalling results in diminished CXCL13 production, and it was also shown that B cells need to express LT to coordinate interactions between T cells, DCs and themselves in the lymph node in order to promote the development of a TH2-type immune response following helminth infection28 (FIG. 2). The role of B cells in this coordination may be antigen nonspecific as both pathogen-specific and pathogen-nonspecific B cells upregulate LT in response to helminth infection28. Understanding the exact role of LT during helminth infection might be essential for appreciating the general mechanisms that are involved in the generation of TH2 cells. Furthermore, it is unclear why LT has seemingly opposing effects in different infections.
Accounting for B cell defects in LT deficient mice
Reconsidering the humoral defects in LT-deficient animals, it is possible that LT might contribute to B cell class-switching responses through its effects on lymph node architecture. One explanation for why animals that lack the LT signalling pathway have defective humoral immunity is that LT is essential for the formation of FDCs and germinal centres54. The microRNA miR-155 directs the expression of TNF and LTα in mature B cells, and deficiencies in miR-155 or LTα result in the abrogation of the germinal centre response55 (FIG. 2). Inducible T cell co-stimulator (ICOS)-deficient animals also have abnormal germinal centres and experience defects in humoral immunity similar to those seen in LT-deficient animals56. Interestingly, ICOS crosslinking results in T cell expression of LTα1β2, which is essential for the chemokine expression that is necessary to organize the germinal centre56. Induction of LT expression via the ICOS pathway is also dependent on CD40, but the overall contribution of CD40 to this pathway is unclear. However, it is clear that both B cells and T cells have unique mechanisms to induce LT expression and that this is essential for normal germinal centre responses55,56.
Roles for LT in immunity to viruses
LT supports SCS macrophages
The ability of B cells to produce LT is essential to control viral infections. During vesicular stomatitis virus (VSV) infection, virions are normally captured by subcapsular sinus (SCS) macrophages, which produce type I IFNs and which prevent the systemic spread of the pathogen57 (FIG. 3). The lymph nodes of B cell-deficient mice have significantly lower numbers of SCS macrophages, and these mice are more susceptible to infection with VSV. B cell-derived LT has now been linked to SCS macrophage development and the generation of B cell-specific immunity58,59. Mice lacking conventional B cells fail to survive following VSV challenge; however, DHLMP2A animals, in which B cells contribute to normal lymphoid tissue development but do not generate antigen-specific B cell responses, are resistant to VSV infection60. The role of B cells in VSV immunity was therefore attributed to their ability to express LT. Mice with a B cell-restricted deficiency in LT expression are susceptible to VSV infection owing to defects in SCS macrophage development60. Therefore, LT produced by B cells affects SCS macrophages and contributes to their ability to prevent the systemic spread of pathogens60 (FIG. 3).
Figure 3. Regulation of viral infections with LT.
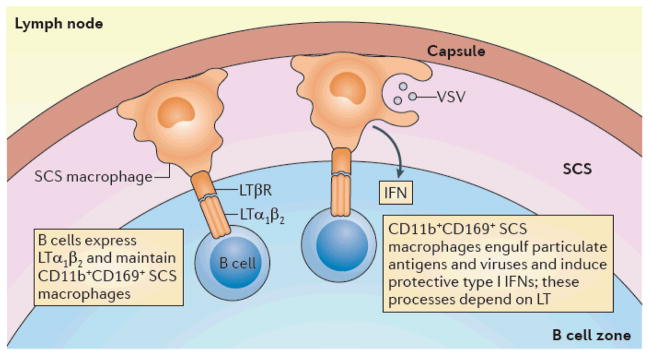
In the fully developed host, B cells provide lymphotoxin (LT), which is recognized by the macrophages that populate the subcapsular sinus (SCS) of the lymph node and causes them to adopt an SCS macrophage phenotype (CD11b+CD169+)60. These macrophages engulf particulate antigens, including vesicular stomatitis virus (VSV), and prevent their spread to the periphery57,70. The macrophages also produce protective type I interferons (IFNs). During lymphocytic choriomeningitis (LCMV) infection, active LT signalling is required to organize B cell follicles; the cell type sensing this LT signalling is currently uncharacterized, but might be the high endothelial venule (HEV)64 (not shown). The LT pathway is also essential for the production of type I IFNs from radio-resistant cells in the spleen that protect the host during viral infection63,65 (not shown). In addition, dendritic cells (DCs) that produce type I IFN in response to LTβ receptor (LTβR) agonism and Toll like receptor (TLR) signalling promote CD8+ T cell homeostasis in the spleen63,68 (not shown).
LT supports type I IFN responses
The LT signalling pathway has different protective functions in other viral infection models. The importance of type I IFNs in viral immunity has been known for some time61, and a recent study has demonstrated that LT is part of a dual mechanism which results in IFNβ production in response to murine cytomegalovirus (MCMV)62. Lta−/−, Ltb−/− and Ltbr−/− mice fail to control MCMV infection and, compared with wild-type mice, their viral loads increase by nearly 100-fold63. It is thought that plasmacytoid DCs (pDCs), which are activated by virally derived Toll-like receptor 9 (TLR9) ligands, are the main source of IFNβ during viral infection. During MCMV infection, serum levels of IFNβ exhibit a biphasic expression pattern, with peak levels occurring at 8 hours and 36–72 hours after MCMV infection. Mice lacking LTβ and LIGHT do not produce IFNβ 8 hours after MCMV infection, but animals lacking TLR9 signalling are still able to produce IFNβ at this early time point. B cells must express LTα1β2 for the early IFNβ peak in response to MCMV infection62, and radio-resistant cells in the spleen are the source of the type I IFN peak at 36–72 hours during infection with MCMV62 (FIG. 3).
Considering a different viral infection model, it was shown that, in mice infected with lymphocytic choriomeningitis (LCMV), B cell-derived LT is important not only for type I IFN production, but also for the reorganization of the lymphoid architecture64,65. In the lymph node, LTβR is required for LCMV-induced B cell follicle formation, and the absence of B cell-derived LT results in unorganized follicles64. In response to LCMV challenge, Lta−/− mice produce only 3% of wild-type levels of type I IFN65. It is thought that the effects of LT on splenic macrophages is necessary for the production of type I IFNs, in a similar manner to the way in which LT induces type I IFN production by SCS macrophages in the lymph node65 (FIG. 3).
LT promotes antiviral T cell responses
The induction of IFNβ is crucial for protective antiviral CD8+ T cell responses66. LT-mediated regulation of DCs might provide an important source of type I IFN for CD8+ T cell viability during viral infection. Both LTα and LTβR are required to prevent CD8+ T cell apoptosis in response to MCMV challenge67, and it was recently demonstrated that LT might contribute to CD8+ T cell homeostasis by inducing the expression of type I IFNs68. In this study, blockade of LT signalling with LTβR-Ig resulted in a decreased capacity of the OVA-specific CD8+ T (OT-I) cell population to expand in response to cognate antigen delivery68. Using the CD11c–DTR system, investigators demonstrated that DCs were required for CD8+ T cell proliferation68. They speculated that DCs might produce type I IFNs in response to LT signalling to maintain adequate CD8+ T cell homeostasis. In vitro experiments showed that, in conjunction with lipopolysaccharide (LPS; a TLR4 ligand), agonist antibodies against LTβR induce IFNβ production from bone marrow-derived DCs68. Ltbr−/− DCs are less efficient at inducing the expansion of the CD8+ T cell population in vivo, and this defect can be rescued by the exogenous delivery of type I IFNs68.
Why the LT pathway regulates type I IFNs during viral infection is not immediately obvious, but it might be linked to the role of LT in host defence mechanisms against viruses. It is important to note that the LT pathway has a non-redundant role in promoting protective immunity in the fully developed lymph node, where LT prevents the spread of viruses into the blood57,69,70. The ability of the host to induce type I IFN production by this pathway is of dual importance in that it helps to initiate adaptive immune responses, and it can also prevent the sudden death of T cells in response to viral challenge67.
The role of LT in immunity is specific
One might erroneously conclude from the work on the role of LT in lymphoid tissue development (BOX 2) and the extensive work detailing the multicellular interactions coordinated by the LT pathway (FIGS 2,3) that LT only contributes to the adaptive immune response by supporting lymphoid tissue development and organization. In fact, the importance of the studies highlighted above is that they demonstrate the unique requirement of LT in active immune processes, and show the distinct and specific roles for the LT receptor. LT is not required for the development of all lymphoid tissues; for example, bronchus-associated lymphoid tissue (BALT) develops in the absence of LT and is able to support immunity to influenza71. However, even in the lung, the importance of LT is still apparent, though poorly understood, because host survival relies on the LT receptor pathway during challenge with other airborne pathogens, such as Mycobacterium tuberculosis72. The above studies emphasize the important and unique roles of the LT receptor pathway in modulating adaptive immunity.
LT regulates mucosal immune responses
Regulation of immunity to mucosal pathogens
The LT pathway is essential for the control of enteric pathogens73–78. Investigators studying MALT found that Ltbr−/− mice have a greater systemic spread of Salmonella typhimurium73,74, but the mechanisms responsible for this have not been well defined. It was found that B cell localization in response to S. typhimurium is dependent on the LT pathway73, but how B cell localization might contribute to host defence is unclear because wild-type and Ltbr−/− animals seem to be equally susceptible to initial infection with S. typhimurium. A major caveat to these studies on S. typhimurium is that they rely on the pretreatment of the mice with streptomycin, which may alter the composition of the commensal microbiota and change undetermined variables in both wild-type and Ltbr−/− mice.
Toxoplasma gondii, a neuroinvasive pathogen that infects the host via the terminal ileum, induces fatal infection in hosts lacking the LT pathway78. However, as in the case of S. typhimurium infection, it is not clear how the LT receptor pathway protects the host from T. gondii. By contrast, we have a better understanding of the role of the LT pathway in response to C. rodentium infection, a mucosal pathogen that is controlled by ILCs and elements of the adaptive immune response (discussed below).
The role of LT in immunity to C. rodentium
In the past few years, there has been much interest in understanding the development and the function of ILCs that express the transcription factor RORγt79–83 and that contribute to tissue immunity in response to IL-23 (REF. 84). Although there has been great debate about the origin of these cells, a consensus is beginning to be reached, that they might be an adult version of lymphoid tissue-inducer cells; however, whether these cells differentiate from their fetal counterparts is still a contentious topic85–87. Although LT does not contribute to the development of ILCs88, RORγt+ ILCs have crucial effector functions, including IL-22 production, via the LT pathway76,77,89.
During C. rodentium infection, IL-22 from RORγt+ ILCs is required for the clearance of the bacteria. The role of the LT pathway in the response to C. rodentium challenge was originally attributed to the capacity of LT to regulate the TH1 and TH2 cell balance. However, Ltbr−/− mice are ill as early as 7 days following initial challenge with C. rodentium and some succumb to C. rodentium infection 10 days after challenge75. This early onset of disease indicated a possible innate role for LT in this infection model. Indeed, it was found that LTβR expression was required by both myeloid cells and intestinal epithelial cells for mice to control C. rodentium75. Blockade of LT renders Rag1−/− hosts more susceptible to C. rodentium infection, and RORγt+ ILCs are the innate source of the LTα1β2 that promotes host resistance to C. rodentium75.
It was initially reported that Ltbr−/− mice have normal levels of IL-22 but fail to clear C. rodentium88, which caused confusion as to what role LT has in promoting innate immunity to this pathogen. As indicated above, numbers of RORγt+ ILCs are not decreased in Ltbr−/− mice. It was later suggested that the susceptibility of Ltbr−/− mice to C. rodentium might, after all, occur because these animals fail to express IL-22 (REFS 76,77). Treatment of animals with an agonist antibody against LTβR can rescue IL-22 production in Ltb−/− mice infected with C. rodentium77. Furthermore, addition of exogenous IL-23 induces IL-22 production in mice infected with C. rodentium when LT signalling is blocked and rescues these animals from death76. The ability of IL-23 to rescue this defect indicates that agonism of the LT signalling pathway might control IL-22 by regulating the production of IL-23.
Microscopy revealed that providing exogenous IL-22 to mice after blockade of LTβR signalling restored normal lymphoid structures in these animals during C. rodentium infection76. Furthermore, isolated lymphoid follicles (ILFs) were identified as the structures containing protective ILCs, and RORγt+IL-22+ ILCs were found in immediate proximity to DCs in ILFs77 (FIG. 4). Mice in which LTβ deficiency was restricted to ILCs had lower expression of IL-22, but treatment with exogenous IL-22 rescued the ability of these mice to resist C. rodentium. This was an interesting result because RORγt+ ILCs were required to express membrane-bound LT to promote IL-22 production, but these were the same cells that produced the IL-22 that was necessary to control C. rodentium infection77. Furthermore, LTβR is not expressed by ILCs. This confusion was resolved when it was discovered that, in the absence of LTβR, DCs could not express wild-type levels of IL-23 in response to C. rodentium77. Conditional deficiency of LTβR on DCs renders animals more susceptible to C. rodentium and reduces IL-22 production by ILCs77.
Figure 4. LT regulates responses to microorganisms at mucosal surfaces.

a|During Citrobacter rodentium infection, the lymphotoxin (LT) pathway is part of an innate feedback loop in which retinoic acid receptor related orphan receptor γt(RORγt) innate lymphoid cells (ILCs)induce interleukin 12(IL 12) and IL 23production by local dendritic cells (DCs) via the LT pathway76,77. In response to IL 23, these ILCs produce IL 22, which promotes gut epithelial cells to produce antimicrobial peptides, such as RegIIIβ and RegIIIγ77. These antimicrobial peptides have direct killing functions on C. rodentium and members of the commensal microbiota. LT dependent interactions between ILCs and stromal cells can also support the formation of isolated lymphoid follicles (ILFs) during infections. b | In response to a high fat diet, LT signalling is increased within the colon (V.U. and Y. X.F., unpublished observations) and this increases IL 23 and IL 22 production96. This then induces the production of antibacterial proteins, which are essential for the clearance of some microorganisms (such as segmented filamentous bacteria (SFB)) in response to high fat diet administration and which contribute to the alteration of the microbiota, which in turn confers greater weight gain96. IL 22R, interleukin 22 receptor; LTβR, LTβ receptor.
These studies have resulted in the development of a model to describe the roles of the LT pathway in mucosal defences. It seems that there is an ILC–DC feedback loop in ILFs, in which LT is produced by ILCs and induces IL-23 production by local DCs, which in turn induces IL-22 production by the ILCs (FIG. 4). As the LT pathway also regulates TH cells, it is important to determine whether ILCs regulate TH cells that contribute to similar infection models.
Regulation of the microbiota by LT
There is growing evidence that in addition to promoting immunity to intestinal pathogens, such as C. rodentium, LT signalling also contributes to the regulation of the intestinal microbiota. The role of the microbiota in human disease is complex and multifold. This is perhaps best demonstrated by the landmark discovery of an ‘obesity-associated microbiome’ in 2006, which demonstrated that specific changes in the host microbiota could contribute to the development of obesity111.
Obesity is also significantly linked to natural polymorphisms that occur in LTA itself90. The idea that LT contributes to metabolic disease is not without precedent91–93. LTβR has previously been reported to regulate hepatic lipid production and atherosclerosis91,92; this occurs in the setting of the pathological overexpression of LIGHT by T cells91 or in mice that have abnormalities in lipid homeostasis through other genetic mechanisms92. However, despite epidemiological studies that indciate that there might be a link between obesity and LT signalling, the exact mechanisms by which LT contributes to weight gain are unclear.
A link between LT and metabolic disease
One group previously identified single nucleotide polymorphisms in the TNF–LTA gene locus in the Pima Indians94. Their study reports the significant association between a single nucleotide polymorphism in the dinucletotide repeat marker TNFir24, which is located 10 kilobases away from the TNF gene, and changes in body fat percentage and body mass index (BMI)94. However, sequencing of the TNF gene in obese and lean subjects of Pima Indian origin did not reveal any differences in TNF alleles between the obese and lean subjects. At the time, it was suggested that this polymorphism could indicate a link between the nearby LTA gene and obesity.
Nearly a decade later, these findings were revisited by another group, who hypothesized that the LT signalling pathway might contribute to obesity95. It was discovered that the Thr60Asn mutation in LTα significantly correlated with an increased incidence of type 2 diabetes in a Danish cohort of patients. The incidence of type 2 diabetes in Thr60Asn/Thr60Asn homozygous individuals was 1.24-fold higher than in control subjects who had a Thr60Thr/Thr60Thr genotype. Closer examination revealed that this allele did not influence BMI, but other indicators of increased adiposity, including waist-to-hip ratio, were significantly increased in individuals who were homozygous for the mutation compared with those who were either heterozygous for the mutation or had two normal alleles95.
Three important considerations caused investigators to later consider the possibility that LT might regulate changes to the microbiota that are causative of obesity: the link between the LT pathway and obesity, the role of the microbiota in metabolic disease, and the regulation of gut immunity by LT96. Indeed, two groups independently demonstrated that LT-deficient animals are resistant to diet-induced obesity96,97. One group found that Lta−/− animals resisted excess weight gain that is induced by a high-fat diet, and that deficiency in the LT pathway resulted in increased lymphocyte infiltration into the perigonadal fat pads of animals, regardless of the diet they received97. In this study, the role of LTα in weight gain was considered to be redundant to that of TNF97. The second study showed that animals deficient in LTα, LTβ or LTβR resisted high-fat diet-induced weight gain96. However, which cells usually express LT in this setting remain to be determined. It is also possible that more than one type of cell is involved.
LT regulates the commensal microbiota
Although animals lacking elements of the LT pathway resist excessive weight gain induced by a high-fat diet, they still consume similar amounts of food96,97. This is a unique finding because excess weight gain is driven by a positive energy imbalance induced by a high-fat diet98. Given the role of the LT pathway in regulating C. rodentium and other mucosal pathogens73–78, it was speculated that the LT pathway might influence host weight gain by regulating the commensal microbiota of the host.
16S rRNA sequencing revealed that the microbial communities of Ltbr−/− mice differ from those of their Ltbr+/− siblings, both on a normal chow diet or on a high-fat diet96. Notable differences include the sustained overgrowth of segmented filamentous bacteria (SFB) in Ltbr−/− mice on a high-fat diet, whereas SFB are cleared by Ltbr+/− mice fed on the same diet. Furthermore, members of the Erysipelotrichi class were found to be enriched in Ltbr+/− animals fed a high-fat diet, but are not enriched in Ltbr−/− mice on the same diet96. Importantly, outgrowth of Erysipelotrichi class members was previously reported to occur in response to a high-fat diet, and this bacterial class might contribute to obesity99. Which species have essential roles for LT-controlled diet-induced obesity remain to be determined. Co-housing Ltbr−/− mice with their obesity-prone littermates actually rescued their capacity to gain weight and restored control of the SFB population96. This confirms that the LT pathway influences weight gain by regulating the composition of the microbiota. LT is essential for the production of IL-23 and IL-22 in response to a high-fat diet, and restoring IL-22 levels in Ltbr−/− mice rescued the defective SFB homeostasis and the metabolic abnormalities, including perigonadal fat depot expansion, that occur in Ltbr−/− mice96.
Although the role of the LT pathway in promoting protection against invasive pathogens such as C. rodentium, S. typhimurium and T. gondii might originally have been thought to be its primary immune function at mucosal sites, these recent data show that the LT pathway is also involved in regulating the symbiotic relationship between the host and their commensal microbiota. Notably, some of the mechanisms by which the LT pathway contributes to protective immune defences could also contribute to weight gain through regulation of the microbiota (FIG. 4).
Breaking (mucosal) barriers with the LT receptor
The LT pathway has an important role at mucosal surfaces, where it facilitates the formation of unique, dynamic lymphoid structures, such as ILFs, in the gut76. ILFs help to coordinate the symbiotic relationship between the host and their commensal microbiota1,100. Although the role of the LT receptor pathway in ILF formation is well understood in healthy host physiology, this process might also highlight the ability of the LT pathway to coordinate the dynamic lymphoid tissue neogenesis that can occur at all epithelial surfaces. This would be an unappreciated property of epithelial surfaces, but would explain why many autoimmune disorders are characterized by lymphoid aggregation near epithelia, and would also highlight modulation of the LT receptor pathway as a new avenue of therapeutics for people with autoimmune diseases. For example, in a mouse model of Sjögren’s syndrome, lymphoid tissue aggregates form near the ductal epithelium as a result of CXCL13 overexpression, and the use of LTβR-Ig decreases lymphocyte recruitment to these sites101.
Furthermore, recent work in mouse models has shown that autoimmunity might be partly driven by alterations in the commensal bacteria present at mucosal surfaces, and therefore breaks in immune tolerance could be induced by previously unappreciated factors, such as milk fat-containing diets102–104. Given the unique position of the LT receptor pathway in regulating both the lymphoid follicle formation and the commensal microbiota, it is intriguing to consider possibilities such as a combination of biological and probiotic therapies. For example, beneficial probiotic bacteria could be reseeded into hosts when disease is driven by ectopic lymphoid follicles and dysbiosis. The combination of LTβR-Ig in conjunction with this probiotic agent might be more effective than either agent in isolation at tackling complex human diseases. The ability to block ectopic lymphoid follicle formation and to control microbial communities by modulating the LT receptor pathway might, therefore, be a future avenue to explore to treat autoimmune diseases, or other inflammatory host states, such as obesity.
Concluding remarks
The history of our understanding of the role of LT has many chapters. It was initially considered a redundant cytokine, and was later thought to be the focus of lymphoid organogenesis. In recent years, it has become clear that LT is an important component of effector immune responses. LT is essential for host defences against specific pathogens and even contributes to the regulation of the gut microbiota. It is now widely appreciated that the LT receptor pathway is essential for protective innate and adaptive immune responses, but LT signalling may also contribute to the development of metabolic disease through the regulation of commensal microbiota. It will be interesting to explore whether manipulation of the LT receptor pathway could be useful in the clinic.
Acknowledgments
The authors would like to thank A. Tumanov and M. Zhu for their contribution to the lymphotoxin project. This work is partially supported by DK080736 and CA141975 to Y. X.F.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Eberl G. A new vision of immunity: homeostasis of the superorganism. Mucosal Immunol. 2010;3:450–460. doi: 10.1038/mi.2010.20. [DOI] [PubMed] [Google Scholar]
- 2.Ware CF, VanArsdale TL, Crowe PD, Browning JL. The ligands and receptors of the lymphotoxin system. Curr Top Microbiol Immunol. 1995;198:175–218. doi: 10.1007/978-3-642-79414-8_11. [DOI] [PubMed] [Google Scholar]
- 3.Josefowicz SZ, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. 2012;482:395–399. doi: 10.1038/nature10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lathrop SK, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geuking MB, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806. doi: 10.1016/j.immuni.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 6.De Togni P, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 7.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- 8.Zhu M, Brown NK, Fu YX. Direct and indirect roles of the LTβR pathway in central tolerance induction. Trends Immunol. 2010;31:325–331. doi: 10.1016/j.it.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chin RK, et al. Lymphotoxin pathway directs thymic Aire expression. Nature Immunol. 2003;4:1121–1127. doi: 10.1038/ni982. [DOI] [PubMed] [Google Scholar]
- 10.Iizuka K, et al. Requirement for membrane lymphotoxin in natural killer cell development. Proc Natl Acad Sci USA. 1999;96:6336–6340. doi: 10.1073/pnas.96.11.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fütterer A, Mink K, Luz A, Kosco Vilbois MH, Pfeffer K. The lymphotoxin β receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- 12.Crowe PD, et al. A lymphotoxin β specific receptor. Science. 1994;264:707–710. [PubMed] [Google Scholar]
- 13.Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal fetal conflict. Cell. 2012;150:29–38. doi: 10.1016/j.cell.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nature Rev Immunol. 2010;10:664–674. doi: 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- 15.Sokol H, et al. Faecalibacterium prausnitzii is an anti inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zindl CL, et al. The lymphotoxin LT1β2 controls postnatal and adult spleen marginal sinus vascular structure and function. Immunity. 2009;30:408–420. doi: 10.1016/j.immuni.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mebius RE. Organogenesis of lymphoid tissues. Nature Rev Immunol. 2003;3:292–303. doi: 10.1038/nri1054. [DOI] [PubMed] [Google Scholar]
- 18.Moussion C, Girard JP. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature. 2011;479:542–546. doi: 10.1038/nature10540. [DOI] [PubMed] [Google Scholar]
- 19.Schneider K, Potter KG, Ware CF. Lymphotoxin and LIGHT signaling pathways and target genes. Immunol Rev. 2004;202:49–66. doi: 10.1111/j.0105-2896.2004.00206.x. [DOI] [PubMed] [Google Scholar]
- 20.Tamada K, et al. LIGHT, a TNF like molecule, costimulates T cell proliferation and is required for dendritic cell mediated allogeneic T cell response. J Immunol. 2000;164:4105–4110. doi: 10.4049/jimmunol.164.8.4105. [DOI] [PubMed] [Google Scholar]
- 21.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 22.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 23.MartIn Fontecha A, et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198:615–621. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Girard JP, Moussion C, Förster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nature Rev Immunol. 2012;12:762–773. doi: 10.1038/nri3298. [DOI] [PubMed] [Google Scholar]
- 25.Uchimura K, et al. A major class of L selectin ligands is eliminated in mice deficient in two sulfotransferases expressed in high endothelial venules. Nature Immunol. 2005;6:1105–1113. doi: 10.1038/ni1258. [DOI] [PubMed] [Google Scholar]
- 26.Kawashima H, et al. N acetylglucosamine 6 O sulfotransferases 1 and 2 cooperatively control lymphocyte homing through L selectin ligand biosynthesis in high endothelial venules. Nature Immunol. 2005;6:1096–1104. doi: 10.1038/ni1259. [DOI] [PubMed] [Google Scholar]
- 27.Browning JL, et al. Lymphotoxin β receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity. 2005;23:539–550. doi: 10.1016/j.immuni.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 28.León B, et al. Regulation of TH2 development by CXCR5+ dendritic cells and lymphotoxin expressing B cells. Nature Immunol. 2012;13:681–690. doi: 10.1038/ni.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabashima K, et al. Intrinsic lymphotoxin β receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity. 2005;22:439–450. doi: 10.1016/j.immuni.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 30.Lewis KL, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity. 2011;35:780–791. doi: 10.1016/j.immuni.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ngo VN, et al. Lymphotoxin α/β and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med. 1999;189:403–412. doi: 10.1084/jem.189.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chow A, Toomre D, Garrett W, Mellman I. Dendritic cell maturation triggers retrograde MHC class II transport from lysosomes to the plasma membrane. Nature. 2002;418:988–994. doi: 10.1038/nature01006. [DOI] [PubMed] [Google Scholar]
- 33.Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 34.Kaisho T, Akira S. Dendritic cell function in Toll like receptor and MyD88 knockout mice. Trends Immunol. 2001;22:78–83. doi: 10.1016/s1471-4906(00)01811-1. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki K, Fagarasan S. How host bacterial interactions lead to IgA synthesis in the gut. Trends Immunol. 2008;29:523–531. doi: 10.1016/j.it.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Fritz JH, et al. Acquisition of a multifunctional IgA+ plasma cell phenotype in the gut. Nature. 2011;481:199–203. doi: 10.1038/nature10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang HS, et al. Signaling via LTβR on the lamina propria stromal cells of the gut is required for IgA production. Nature Immunol. 2002;3:576–582. doi: 10.1038/ni795. [DOI] [PubMed] [Google Scholar]
- 38.Macpherson AJS, et al. IgA production without μ or δ chain expression in developing B cells. Nature Immunol. 2001;2:625–631. doi: 10.1038/89775. [DOI] [PubMed] [Google Scholar]
- 39.Tsuji M, et al. Requirement for lymphoid tissue inducer cells in isolated follicle formation and T cell independent immunoglobulin A generation in the gut. Immunity. 2008;29:261–271. doi: 10.1016/j.immuni.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 40.Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell dependent and T cell independent IgA synthesis. Annu Rev Immunol. 2010;28:243–273. doi: 10.1146/annurev-immunol-030409-101314. [DOI] [PubMed] [Google Scholar]
- 41.Lee MR, Seo GY, Kim YM, Kim P. H iNOS potentiates mouse Ig isotype switching through AID expression. Biochem Biophys Res Commun. 2011;410:602–607. doi: 10.1016/j.bbrc.2011.06.035. [DOI] [PubMed] [Google Scholar]
- 42.Tezuka H, et al. Regulation of IgA production by naturally occurring TNF/iNOS producing dendritic cells. Nature. 2007;448:929–933. doi: 10.1038/nature06033. [DOI] [PubMed] [Google Scholar]
- 43.Cerutti A. The regulation of IgA class switching. Nature Rev Immunol. 2008;8:421–434. doi: 10.1038/nri2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fagarasan S, et al. Critical roles of activation induced cytidine deaminase in the homeostasis of gut flora. Science. 2002;298:1424–1427. doi: 10.1126/science.1077336. [DOI] [PubMed] [Google Scholar]
- 45.Kang HS, et al. Lymphotoxin is required for maintaining physiological levels of serum IgE that minimizes Th1 mediated airway inflammation. J Exp Med. 2003;198:1643–1652. doi: 10.1084/jem.20021784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spahn TW, et al. The lymphotoxin β receptor is critical for control of murine Citrobacter rodentium induced colitis. Gastroenterology. 2004;127:1463–1473. doi: 10.1053/j.gastro.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 47.Ehrchen JM, et al. The absence of cutaneous lymph nodes results in a Th2 response and increased susceptibility to Leishmania major infection in mice. Infect Immun. 2008;76:4241–4250. doi: 10.1128/IAI.01714-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dohi T, et al. Elimination of colonic patches with lymphotoxin β receptor Ig prevents Th2 cell type colitis. J Immunol. 2001;167:2781–2790. doi: 10.4049/jimmunol.167.5.2781. [DOI] [PubMed] [Google Scholar]
- 49.Gramaglia I, Mauri DN, Miner KT, Ware CF, Croft M. Lymphotoxin αβ is expressed on recently activated naive and Th1 like CD4 cells but is down regulated by IL 4 during Th2 differentiation. J Immunol. 1999;162:1333–1338. [PubMed] [Google Scholar]
- 50.Morel PA, Oriss TB. Crossregulation between Th1 and Th2 cells. Crit Rev Immunol. 1998;18:275–303. doi: 10.1615/critrevimmunol.v18.i4.10. [DOI] [PubMed] [Google Scholar]
- 51.Trinchieri G. Interleukin 12 and the regulation of innate resistance and adaptive immunity. Nature Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 52.Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Res. 2009;20:4–12. doi: 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Else KJ, Finkelman FD. Intestinal nematode parasites, cytokines and effector mechanisms. Int J Parasitol. 1998;28:1145–1158. doi: 10.1016/s0020-7519(98)00087-3. [DOI] [PubMed] [Google Scholar]
- 54.Matsumoto M, et al. Affinity maturation without germinal centres in lymphotoxin α deficient mice. Nature. 1996;382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- 55.Thai TH, et al. Regulation of the germinal center response by microRNA 155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. A demonstration that a miRNA-dependent programme regulates LT expression to contribute to germinal centre formation. [DOI] [PubMed] [Google Scholar]
- 56.Vu F, Dianzani U, Ware CF, Mak T, Gommerman JL. ICOS, CD40, and lymphotoxin β receptors signal sequentially and interdependently to initiate a germinal center reaction. J Immunol. 2008;180:2284–2293. doi: 10.4049/jimmunol.180.4.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Junt T, et al. Subcapsular sinus macrophages in lymph nodes clear lymph borne viruses and present them to antiviral B cells. Nature. 2007;450:110–114. doi: 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 58.Phan TG, Green JA, Gray EE, Xu Y, Cyster JG. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nature Immunol. 2009;10:786–793. doi: 10.1038/ni.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phan TG, Grigorova I, Okada T, Cyster JG. Subcapsular encounter and complement dependent transport of immune complexes by lymph node B cells. Nature Immunol. 2007;8:992–1000. doi: 10.1038/ni1494. [DOI] [PubMed] [Google Scholar]
- 60.Moseman EA, et al. B cell maintenance of subcapsular sinus macrophages protects against a fatal viral infection independent of adaptive immunity. Immunity. 2012;36:415–426. doi: 10.1016/j.immuni.2012.01.013. A mechanistic demonstration that LT from B cells maintains the sentinel function of SCS macrophages and prevents the dissemination of lethal pathogens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Platanias LC. Mechanisms of type I and type II interferon mediated signalling. Nature Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 62.Schneider K, et al. Lymphotoxin mediated crosstalk between B cells and splenic stroma promotes the initial type I interferon response to cytomegalovirus. Cell Host Microbe. 2008;3:67–76. doi: 10.1016/j.chom.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Banks TA, et al. A lymphotoxin IFN β axis essential for lymphocyte survival revealed during cytomegalovirus infection. J Immunol. 2005;174:7217–7225. doi: 10.4049/jimmunol.174.11.7217. [DOI] [PubMed] [Google Scholar]
- 64.Kumar V, et al. Global lymphoid tissue remodeling during a viral infection is orchestrated by a B cell lymphotoxin dependent pathway. Blood. 2010;115:4725–4733. doi: 10.1182/blood-2009-10-250118. [DOI] [PubMed] [Google Scholar]
- 65.Louten J, van Rooijen N, Biron CA. Type 1 IFN deficiency in the absence of normal splenic architecture during lymphocytic choriomeningitis virus infection. J Immunol. 2006;177:3266–3272. doi: 10.4049/jimmunol.177.5.3266. [DOI] [PubMed] [Google Scholar]
- 66.Heath WR, Carbone FR. Cross presentation in viral immunity and self tolerance. Nature Rev Immunol. 2001;1:126–135. doi: 10.1038/35100512. [DOI] [PubMed] [Google Scholar]
- 67.Banks TA, Rickert S, Ware CF. Restoring immune defenses via lymphotoxin signaling: lessons from cytomegalovirus. Immunol Res. 2006;34:243–254. doi: 10.1385/IR:34:3:243. [DOI] [PubMed] [Google Scholar]
- 68.Summers deLuca L, et al. LTβR signaling in dendritic cells induces a type I IFN response that is required for optimal clonal expansion of CD8+ T cells. Proc Natl Acad Sci USA. 2011;108:2046–2051. doi: 10.1073/pnas.1014188108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roozendaal R, et al. Conduits mediate transport of low molecular weight antigen to lymph node follicles. Immunity. 2009;30:264–276. doi: 10.1016/j.immuni.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iannacone M, et al. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature. 2010;465:1079–1083. doi: 10.1038/nature09118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moyron Quiroz JE, et al. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nature Med. 2004;10:927–934. doi: 10.1038/nm1091. [DOI] [PubMed] [Google Scholar]
- 72.Ehlers S, et al. The lymphotoxin β receptor is critically involved in controlling infections with the intracellular pathogens Mycobacterium tuberculosis and Listeria monocytogenes. J Immunol. 2003;170:5210–5218. doi: 10.4049/jimmunol.170.10.5210. [DOI] [PubMed] [Google Scholar]
- 73.Barthel M, et al. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun. 2003;71:2839–2858. doi: 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hapfelmeier S, et al. The Salmonella pathogenicity island (SPI) 2 and SPI 1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88 dependent and MyD88 independent mechanisms. J Immunol. 2005;174:1675–1685. doi: 10.4049/jimmunol.174.3.1675. [DOI] [PubMed] [Google Scholar]
- 75.Wang Y, et al. Lymphotoxin β receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity. 2010;32:403–413. doi: 10.1016/j.immuni.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ota N, et al. IL 22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nature Immunol. 2011;12:941–948. doi: 10.1038/ni.2089. [DOI] [PubMed] [Google Scholar]
- 77.Tumanov AV, et al. Lymphotoxin controls the IL 22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe. 2011;10:44–53. doi: 10.1016/j.chom.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schlüter D, et al. Both lymphotoxin α and TNF are crucial for control of Toxoplasma gondii in the central nervous system. J Immunol. 2003;170:6172–6182. doi: 10.4049/jimmunol.170.12.6172. [DOI] [PubMed] [Google Scholar]
- 79.Eberl G, et al. An essential function for the nuclear receptor RORγt in the generation of fetal lymphoid tissue inducer cells. Nature Immunol. 2003;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 80.Eberl G, Lochner M. The development of intestinal lymphoid tissues at the interface of self and microbiota. Mucosal Immunol. 2009;2:478–485. doi: 10.1038/mi.2009.114. [DOI] [PubMed] [Google Scholar]
- 81.Eberl G, Littman DR. Thymic origin of intestinal αβT cells revealed by fate mapping of RORγt+ cells. Sci Signal. 2004;305:248–251. doi: 10.1126/science.1096472. [DOI] [PubMed] [Google Scholar]
- 82.Langrish CL, et al. IL 23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL 17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 84.Takatori H, et al. Lymphoid tissue inducer like cells are an innate source of IL 17 and IL 22. J Exp Med. 2009;206:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sawa S, et al. Lineage relationship analysis of RORγt+ innate lymphoid cells. Science. 2010;330:665–669. doi: 10.1126/science.1194597. [DOI] [PubMed] [Google Scholar]
- 86.Cherrier M, Sawa S, Eberl G. Notch, Id2, and RORγt sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J Exp Med. 2012;209:729–740. doi: 10.1084/jem.20111594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vonarbourg C, et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor expressing RORγt+ innate lymphocytes. Immunity. 2010;33:736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Satoh Takayama N, et al. Lymphotoxin β receptor independent development of intestinal IL 22 producing NKp46+ innate lymphoid cells. Eur J Immunol. 2011;41:780–786. doi: 10.1002/eji.201040851. [DOI] [PubMed] [Google Scholar]
- 89.Tumanov AV, et al. Dissecting the role of lymphotoxin in lymphoid organs by conditional targeting. Immunol Rev. 2003;195:106–116. doi: 10.1034/j.1600-065x.2003.00071.x. [DOI] [PubMed] [Google Scholar]
- 90.Mahajan A, et al. Obesity dependent association of TNF LTA locus with type 2 diabetes in North Indians. J Mol Med. 2010;88:515–522. doi: 10.1007/s00109-010-0594-5. [DOI] [PubMed] [Google Scholar]
- 91.Lo JC, et al. Lymphotoxin β receptor dependent control of lipid homeostasis. Science. 2007;316:285–288. doi: 10.1126/science.1137221. [DOI] [PubMed] [Google Scholar]
- 92.Lotzer K, et al. Mouse aorta smooth muscle cells differentiate into lymphoid tissue organizer like cells on combined tumor necrosis factor receptor 1/Lymphotoxin β receptor NF κB signaling. Arterioscler Thromb Vasc Biol. 2010;30:395–402. doi: 10.1161/ATVBAHA.109.191395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tumanov AV, Christiansen PA, Fu YX. The role of lymphotoxin receptor signaling in diseases. Curr Mol Med. 2007;7:567–578. doi: 10.2174/156652407781695701. [DOI] [PubMed] [Google Scholar]
- 94.Norman R, Bogardus C, Ravussin E. Linkage between obesity and a marker near the tumor necrosis factor α locus in Pima Indians. J Clin Invest. 1995;96:158–162. doi: 10.1172/JCI118016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hamid YH, et al. The common T60N polymorphism of the lymphotoxin-α gene is associated with type 2 diabetes and other phenotypes of the metabolic syndrome. Diabetologia. 2005;48:445–451. doi: 10.1007/s00125-004-1659-1. [DOI] [PubMed] [Google Scholar]
- 96.Upadhyay V, et al. Lymphotoxin regulates commensal responses to enable diet induced obesity. Nature Immunol. 2012;13:947–953. doi: 10.1038/ni.2403. This paper demonstrates the ability of the LT pathway to regulate the commensal microbiota. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pamir N, McMillen TS, Edgel KA, Kim F, LeBoeuf RC. Deficiency of Lymphotoxin α does not exacerbate high fat diet induced obesity but does enhance inflammation in mice. Am J Physiol Endocrinol Metab. 2012;302:e961–e971. doi: 10.1152/ajpendo.00447.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–543. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 99.Turnbaugh PJ, et al. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bouskra D, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–510. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 101.Fava RA, Browning JL, Gatumu M, Skarstein K, Bolstad AI. LTBR pathway in Sjogren’s syndrome: CXCL13 levels and B cell enriched ectopic lymphoid aggregates in NOD mouse lacrimal glands are dependent on LTBR. Adv Exp Med Biol. 2011;691:383–390. doi: 10.1007/978-1-4419-6612-4_39. [DOI] [PubMed] [Google Scholar]
- 102.Wen L, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Berer K, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:1–5. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- 104.Devkota S, et al. Dietary fat induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dejardin E, et al. The lymphotoxin β receptor induces different patterns of gene expression via two NF κB pathways. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 106.Chang YH, Chao Y, Hsieh SL, Lin WW. Mechanism of LIGHT/interferon γ induced cell death in HT 29 cells. J Cell Biochem. 2004;93:1188–1202. doi: 10.1002/jcb.20282. [DOI] [PubMed] [Google Scholar]
- 107.Browning JL, et al. Signaling through the lymphotoxin β receptor induces the death of some adenocarcinoma tumor lines. J Exp Med. 1996;183:867–878. doi: 10.1084/jem.183.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mackay F, Majeau GR, Lawton P, Hochman PS, Browning JL. Lymphotoxin but not tumor necrosis factor functions to maintain splenic architecture and humoral responsiveness in adult mice. Eur J Immunol. 1997;27:2033–2042. doi: 10.1002/eji.1830270830. [DOI] [PubMed] [Google Scholar]
- 109.Lorenz RG, Chaplin DD, McDonald KG, McDonough JS, Newberry RD. Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin sufficient B lymphocytes, lymphotoxin β receptor, and TNF receptor I function. J Immunol. 2003;170:5475–5482. doi: 10.4049/jimmunol.170.11.5475. [DOI] [PubMed] [Google Scholar]
- 110.Lochner M, et al. Microbiota induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORγt and LTi cells. J Exp Med. 2011;208:125–134. doi: 10.1084/jem.20100052. This article demonstrates the LT-dependent formation of aberrant lymphoid structures near an epithelial surface. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Turnbaugh PJ, et al. An obesity associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1131. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]