

Abstract
IGF-1 and insulin promote β-cell expansion by inhibiting β-cell death and stimulating β-cell proliferation, and the phosphatidylinositol (PI) 3-kinase/Akt pathway mediates insulin and IGF-1 action. Impaired β-cell expansion is a risk factor for type 2 diabetes. Here, we identified SH2B1, which is highly expressed in β-cells, as a novel regulator of β-cell expansion. Silencing of SH2B1 in INS-1 832/13 β-cells attenuated insulin- and IGF-1–stimulated activation of the PI 3-kinase/Akt pathway and increased streptozotocin (STZ)-induced apoptosis; conversely, overexpression of SH2B1 had the opposite effects. Activation of the PI 3-kinase/Akt pathway in β-cells was impaired in pancreas-specific SH2B1 knockout (PKO) mice fed a high-fat diet (HFD). HFD-fed PKO mice also had increased β-cell apoptosis, decreased β-cell proliferation, decreased β-cell mass, decreased pancreatic insulin content, impaired insulin secretion, and exacerbated glucose intolerance. Furthermore, PKO mice were more susceptible to STZ-induced β-cell destruction, insulin deficiency, and hyperglycemia. These data indicate that SH2B1 in β-cells is an important prosurvival and proproliferative protein and promotes compensatory β-cell expansion in the insulin-resistant state and in response to β-cell stress.
Introduction
Insulin, which is secreted from pancreatic β-cells, decreases blood glucose by stimulating glucose uptake into skeletal muscle and adipose tissue as well as by suppressing hepatic glucose production. Plasma insulin levels are determined largely by β-cell mass and β-cell secretory function, and β-cell failure is a causal factor for both type 1 and type 2 diabetes (1,2).
Obesity is the primary risk factor for type 2 diabetes. In the prediabetes state, obesity-induced insulin resistance promotes adaptive β-cell expansion and hyperinsulinemia. Once compensatory β-cell expansion and hyperinsulinemia are insufficient to overcome insulin resistance, glucose intolerance and hyperglycemia ensue. Glucose, insulin, and IGF-1 are key factors that promote β-cell expansion by both decreasing death and increasing proliferation of β-cells (3–7). IGF-1 and insulin promote β-cell survival and growth at least in part by activating the phosphatidylinositol (PI) 3-kinase/Akt pathway (8–13).
SH2B1 is a PH and SH2 domain–containing adapter protein (14,15). It mediates/modulates insulin, IGF-1, leptin, platelet-derived growth factor, fibroblast growth factor, nerve growth factor, and growth hormone signaling in cultured cells (14,15). SH2B1 binds to both insulin and IGF-1 receptors (16,17), and it also binds to IRS1 and IRS2, two upstream activators of the PI 3-kinase pathway (18,19). We previously reported that disruption of the SH2B1 gene in mice results in severe obesity and type 2 diabetes (20–22). SH2B1 enhances leptin signaling by binding to and activating JAK2 (23). Neuronal SH2B1 protects against obesity in mice at least in part by enhancing leptin sensitivity (24). In agreement with our findings in mice, single nucleotide polymorphisms in SH2B1 are linked to obesity in European, American, and Asian populations (25–35). Chromosomal deletion of SH2B1 as well as SH2B1 missense mutations is associated with obesity and disproportional diabetes in humans (36–38).
SH2B1 is also expressed in peripheral tissues in addition to the brain (19,39). We previously reported that mice lacking SH2B1 in peripheral tissues are predisposed to high-fat diet (HFD)-induced diabetes (19); however, the peripheral targets of SH2B1 were unknown. In this study, we demonstrate that SH2B1 is expressed in β-cells at high levels. SH2B1 directly enhances insulin- and IGF-1–stimulated activation of the PI 3-kinase/Akt pathway in β-cells and promotes β-cell survival. We further demonstrate that pancreas-specific knockout of SH2B1 (PKO) impairs β-cell expansion in PKO mice fed an HFD, leading to impaired insulin secretion and glucose intolerance. Our data suggest that SH2B1 in β-cells is a previously unrecognized regulator of glucose homeostasis and promotes β-cell survival and islet expansion in the insulin-resistant state or under β-cell stress conditions.
Research Design and Methods
SH2B1 KO mice have previously been described (22). PKO mice were generated using the Cre/loxP system. Briefly, one loxP site was inserted into the intron between the second and third exons, and a second loxP site was inserted into the intron between the fifth and sixth exons in the SH2B1 gene. Exons 2–5 encode amino acids 1–436 of all four SH2B1 isoforms. A neo cassette flanked by unidirectional Flp-recombinase recognition sites was inserted 3′ of the first loxP, and a thymidine kinase expression cassette was included at the 3′ end of the targeting vector. A HindIII restriction site was introduced after the 3′ loxP site to facilitate detection of homologous recombination by Southern blot analysis. The linearized targeting construct was electroporated into R1 ES cells (129/Svx129/Sv-CP F1) at the University of Michigan Transgenic Animal Model Core, and homologous recombination was confirmed by PCR and Southern blot analysis. Two positive ES cell clones were expanded and microinjected into C57BL/6 blastocysts to generate chimeric SH2B1flox-neo/+ founder mice. The neo cassette was deleted from the germ line by crossing SH2B1flox-neo/+ mice to mice expressing Flp recombinase under the control of the human actin promoter (TgACTFLPe strain; The Jackson Laboratory). The Flp transgene was subsequently removed by backcrossing with wild-type (WT) C57BL/6 mice to generate SH2B1f/+ mice. PKO mice (genotype: SH2B1f/f;Cre+/−) were generated by crossing SH2B1f/f with Pdx1-Cre mice in which Cre recombinase is expressed under the control of the mouse Pdx1 promoter (40).
All mice were generated and maintained on a congenic C57BL/6 background. Mice were housed on 12-h light/12-h dark cycles in the Unit for Laboratory Animal Medicine at the University of Michigan. Mice were fed either a standard rodent chow diet (9% fat; Laboratory Diet, St. Louis, MO) or an HFD (45 or 60% fat; Research Diets, New Brunswick, NJ) ad libitum with free access to water. Animal experiments were conducted following animal protocols approved by the University Committee on Use and Care of Animals at the University of Michigan.
Streptozotocin-Induced Diabetes
Streptozotocin (STZ) was dissolved in 0.1 mol/L citrate buffer (pH 4.5) and injected (25 mg/kg body wt i.p.) daily for 5 days. Random-fed (9:00–10:00 a.m.) blood glucose levels were determined using glucometers (Bayer Corp., Pittsburgh, PA).
Glucose and Insulin Tolerance Tests
Glucose tolerance tests (GTT) and insulin tolerance tests (ITT) were conducted as previously described (41).
Plasma Insulin Levels, Insulin Secretion, and Total Pancreatic Insulin Content
Tail blood was collected at the indicated times, and plasma insulin levels were measured using a rat insulin ELISA kit (Crystal Chem Inc., Downers Grove, IL). For measurement of glucose-stimulated insulin secretion (GSIS), mice were fasted for 24 h before d-glucose (3 g/kg body wt i.p.) was injected. Plasma insulin levels were measured at the indicated time points by ELISA. Pancreata were isolated and homogenized in acid ethanol (1.5% HCl in 70% EtOH) to extract total pancreatic insulin. Pancreatic insulin content was measured using a rat insulin RIA kit (Linco Research, St. Charles, MO) and normalized to pancreas weight.
Immunoprecipitation and Immunoblotting
Mice were killed under anesthesia. Tissues were harvested and rapidly frozen in liquid nitrogen. Frozen tissue samples were homogenized in ice-cold lysis buffer (50 mmol/L Tris HCl, pH 7.5; 0.5% Nonidet P-40;150 mmol/L NaCl; 2 mmol/L EGTA; 1 mmol/L Na3VO4; 100 mmol/L NaF; 10 mmol/L Na4P2O7; 1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin; and 10 μg/mL leupeptin). Tissue extracts were immunoprecipitated and immunoblotted with the indicated antibodies. Antibody dilutions were as follows: pAkt (pSer473) (cat. no. 9271; Cell Signaling), 5,000; pAkt (pSer473) (4060; Cell Signaling), 10,000; pAkt (pThr308) (sc-16646-R; Santa Cruz), 3,000; Akt (sc-1618; Santa Cruz), 3,000; Akt (4691; Cell Signaling), 10,000; tubulin (sc-5286; Santa Cruz), 8,000; SH2B1 (laboratory generated), 10,000; phospho-p44/42 MAPK (Thr202/Tyr204) (9106; Cell Signaling), 10,000; and ERK1 (sc-94, Santa Cruz), 500.
Immunostaining
Pancreata were fixed in 4% paraformaldehyde for 3 h and then in 30% sucrose overnight. Frozen pancreatic sections (5–8 μm) were stained with the indicated antibodies and visualized using a BX51 microscope equipped with a DP70 Digital Camera (Olympus, Tokyo, Japan). For measurement of islet area, pancreatic sections were stained with hematoxylin-eosin (H-E). Islets were identified and total islet area was measured using ImageJ software. Islet area was normalized to total section area. Average islet area was determined for each mouse by taking the normalized islet area of four to five sections spaced >200 μm apart. Average fluorescence intensity was used as an index to quantify phosphorylation of Akt and FOXO-1. Antibody dilution ratios were as follows: pAkt (pThr308) (cat. no. sc-16646-R; Santa Cruz), 100; pFOXO1 (pSer256) (sc-101681; Santa Cruz), 50; insulin (A0564; Dako), 2,000; glucagon (G 2654; Sigma), 3,000; and Ki67 (VP-RM04; Vector Laboratories), 100.
SH2B1 Knockdown and Overexpression in INS-1 832/13 Cells
INS-1 832/13 cells were cultured at 37°C and 5% CO2 in RPMI-1640 medium supplemented with 10% FBS and 50 μmol/L β-mercaptoethanol (42). For knockdown of SH2B1, INS-1 832/13 cells were infected with a retroviral vector encoding SH2B1 short hairpin RNA (shRNA) (5′-CATCTGTGGTTCCAGTCCA-3′); a retroviral vector encoding a scrambled shRNA served as control. Stable lines were generated by puromycin selection. INS-1 832/13 cells were infected with lentiviral vectors, and stable lines were generated by puromycin selection.
Cell Viability Assays
Cell viability was measured by the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays. INS-1 832/13 cells were treated with STZ for 16 h in serum-free RPMI-1640 medium, and then MTT (75 μg/mL) was added for an additional 1 h. After extensive washes in PBS, cells were solubilized in DMSO. Absorbance (570 nm) of cell extracts was measured using a microplate reader and used as a viability index. The viability of untreated cells was defined as 100%.
TUNEL Assays
Frozen pancreatic sections (5–8 μm) or INS-1 832/13 cells were fixed with 4% paraformaldehyde and subjected to TUNEL assays using cell death detection kits (Roche Diagnostics) following the manufacturer’s recommended procedure. The samples were stained with DAPI to visualize total cells. Pancreatic sections were also immunostained with anti-insulin antibody to identify β-cells. TUNEL-positive cells were counted and normalized to total islet cells. In separate experiments, islets were isolated and incubated with interleukin (IL)-1β (1 ng/mL), tumor necrosis factor (TNF)-α (5 ng/mL), and interferon-γ (5 ng/mL) in serum-free RPMI-1640 for 16–17 h and then subjected to TUNEL assays.
Statistical Analysis
Data are presented as means ± SEM. Differences between groups were determined by two-tailed Student t tests. P < 0.05 was considered significant.
Results
SH2B1 Is Highly Expressed in Pancreatic β-Cells
We examined SH2B1 expression in the pancreas by immunostaining pancreatic sections with an anti-SH2B1 antibody. SH2B1 expression was detected in islets from WT but not in SH2B1 knockout (KO) mice (Fig. 1A). SH2B1 protein was predominantly colocalized with insulin in β-cells, but there was detectable SH2B1 in insulin-negative cells in the mantle of the islet of WT mice (Fig. 1A). Notably, there was minimal SH2B1 staining observed in acinar tissue throughout the pancreas (Fig. 1A). Mice express four isoforms of SH2B1 (α, β, γ, and δ) via alternative mRNA splicing (43). Multiple forms of SH2B1 were detected in protein extracts from whole pancreas and isolated islets from C57BL/6 mice (Fig. 1B). This is consistent with our previous report that multiple isoforms of SH2B1 can be detected in the mouse brain using our anti-SH2B1 antibodies (24). Consistent with the immunostaining results, SH2B1 protein levels were relatively higher in islet extracts than in total pancreatic extracts (Fig. 1B). SH2B1 protein was also detected in the rat INS-1 832/13 β-cell line (Fig. 2A).
Figure 1.

SH2B1 is highly expressed in pancreatic β-cells. A: Pancreas sections from WT and KO mice were immunostained with anti-SH2B1 (red) and anti-insulin (green) antibodies. Scale bar: 250 μm. B: Pancreas and islets extracts were immunoprecipitated (IP) and immunoblotted (IB) with anti-SH2B1 antibody. Extracts were also blotted with anti–β-actin antibody.
Figure 2.

SH2B1 cell-autonomously promotes β-cell survival. A–C: INS-1 832/13 cells were infected with SH2B1 shRNA or scramble retroviral vectors to generate stable lines. A: SH2B1 in cell extracts were immunoprecipitated (IP) and immunoblotted (IB) with anti-SH2B1 antibody. Cell extracts were also immunoblotted with anti–β-tubulin antibody. B: Cells were treated with STZ for 16 h, and cell viability was measured using MTT. Scramble: n = 4; shRNA: n = 4. C: Cells were treated with STZ (0.2 mmol/L) for 16 h, and apoptosis was measured using TUNEL assays. Scramble: n = 5; shRNA: n = 5. D–F: INS-1 832/13 cells were infected with SH2B1β or empty lentiviral vectors to generate stable cell lines. D: Cell extracts were immunoblotted with anti-SH2B1 and anti–β-tubulin antibodies. E: Cells were treated with STZ for 16 h, and cell viability was measured using MTT. Control (Con): n = 4; SH2B1: n = 4. a.u., arbitrary units. F: Cells were treated with STZ (0.5 mmol/L) for 16 h, and apoptosis was measured by TUNEL assays. Control: n = 5; SH2B1: n = 6. Data are means ± SE. *P < 0.05. a.u., arbitrary units.
SH2B1 Protects INS-1 832/13 β-Cells Against Injury and Death
To gain insight into the potential function of SH2B1 in β-cells, we knocked down SH2B1 in INS-1 832/13 β-cells using shRNA-mediated gene silencing. INS-1 832/13 cells were infected with SH2B1 shRNA or scramble retroviral vectors to generate stable cell lines. SH2B1 protein levels were ∼60% less in INS-1 832/13 cells infected with SH2B1 shRNA retroviral vectors (Fig. 2A). For determination of whether SH2B1 promotes β-cell survival, INS-1 832/13 cells with SH2B1 silencing were treated with STZ, a β-cell toxin, and cell viability was measured using MTT assays. Silencing of SH2B1 significantly decreased the viability of STZ-treated INS-1 832/13 cells (Fig. 2B). We also measured apoptosis using TUNEL assays. Silencing of SH2B1 significantly increased STZ-induced apoptosis (Fig. 2C). To determine whether overexpression of SH2B1 has the opposite effects, we stably introduced rat SH2B1β into INS-1 832/13 β-cells (Fig. 2D). Overexpression of SH2B1 significantly increased the viability of STZ-treated INS-1 832/13 cells (Fig. 2E). Overexpression of SH2B1 also protected INS-1 832/13 cells from STZ-induced apoptosis (Fig. 2F). These data indicate that SH2B1 is a prosurvival protein in β-cells.
SH2B1 Promotes Activation of the PI 3-Kinase/Akt Pathway in INS-1 832/13 β-Cells
In β-cells, the PI 3-kinase pathway is the major prosurvival pathway and is activated by nutrients, hormones, and growth factors. For determination of whether SH2B1 is required for the activation of the PI 3-kinase pathway in β-cells, SH2B1 was silenced in INS-1 832/13 cells by shRNA retroviral vectors as described above. The cells were treated with IGF-1 or insulin, and phosphorylation of Akt was measured by immunoblotting with anti–phospho-Akt (pThr308 or pSer473) antibodies. IGF-1 rapidly stimulated Akt phosphorylation in control cells transduced with scramble shRNA, but silencing of SH2B1 significantly decreased Akt phosphorylation on both Thr308 (by 17%) and Ser473 (by 43%) (Fig. 3A). Silencing of SH2B1 also inhibited IGF-1–stimulated phosphorylation of ERK1 and ERK2 (Fig. 3B). Insulin also stimulated Akt phosphorylation in INS-1 832/13 cells, and silencing of SH2B1 significantly decreased insulin-stimulated phosphorylation of Akt (Fig. 3C). For determination of whether SH2B1 directly augments IGF-1 and insulin signaling in β-cells, SH2B1β was stably overexpressed in INS-1 832/13 cells as described above. Stable cell lines were treated with IGF-1 or insulin, and Akt and ERK1/2 phosphorylation was measured by immunoblotting. Overexpression of SH2B1 not only increased IGF-1–stimulated Akt phosphorylation on both Thr308 (by 87%) and Ser473 (by 37%) (Fig. 3D) but also increased IGF-1–stimulated phosphorylation of ERK1/2 (Fig. 3E). SH2B1 overexpression also significantly increased insulin-stimulated phosphorylation of Akt (Fig. 3F). These data demonstrate that in β-cells, SH2B1 is an endogenous enhancer of the PI 3-kinase/Akt pathway and integrates multiple prosurvival signals, including those from IGF-1 and insulin.
Figure 3.

SH2B1 promotes the PI 3-kinase/Akt pathway in β-cells. A–D: INS-1 832/13 cells were stably infected with scramble or shRNA vectors as described for Fig. 2A. A–C: Cells were deprived of serum overnight and stimulated with 50 nmol/L IGF-1 (A and B) or 100 nmol/L insulin (C) for 15 min. Cell extracts were immunoblotted with the indicated antibodies. Akt phosphorylation was quantified and normalized to total Akt levels, whereas phosphorylation of ERK1/2 was normalized to total ERK1/2 levels. Basal: n = 4–6; IGF-1: n = 4–6; insulin: n = 4. D–F: SH2B1β was stably overexpressed in INS-1 832/13 cells as described for Fig. 2D. Cells were stimulated with 50 nmol/L IGF-1 (D and E) or 100 nmol/L insulin (F) for 15 min, and Akt and ERK1/2 phosphorylation was measured as described above. Basal: n = 4; IGF-1: n = 4; insulin: n = 4; 2.8 mmol/L: n = 4–8. Data are means ± SE. *P < 0.05. a.u., arbitrary units; Con, control.
Generation of PKO Mice
To determine whether SH2B1 in β-cells has similar prosurvival functions in vivo, we generated PKO mice using the Cre/loxP system. Two loxP sites flanking coding exons 1–4 were engineered into the SH2B1 gene in ES cells (Fig. 4A). Targeted ES cells were used to generate floxed SH2B1 mice (designated as SH2B1f/f), which were subsequently backcrossed (>6 generations) on the C57BL/6 congenetic background. SH2B1f/f mice were crossed with Pdx1-Cre transgenic mice to generate PKO mice (genotype: SH2B1f/f;Cre+/−). Pdx1-Cre mice have been widely used to generate PKO mice (40). As predicted, SH2B1 protein was detected in the pancreata of SH2B1f/f and Pdx1-Cre, but not PKO, mice (Fig. 4B). SH2B1 protein levels in the brain and liver were similar between SH2B1f/f and PKO mice (Fig. 4C), indicating that the SH2B1 gene is disrupted specifically in the pancreata of PKO mice. General inspection revealed that PKO mice were healthy with normal body weight (Fig. 4D). Body weight gain was also comparable between SH2B1f/f, Pdx1-Cre, and PKO mice when fed an HFD (Fig. 4E).
Figure 4.
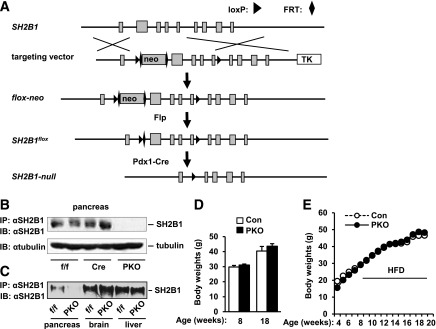
Generation of PKO mice. A: A schematic representation of gene targeting strategies to generate conditional SH2B1 KO mice. Squares represent individual exons, and the first is a noncoding exon. B: Pancreatic extracts were prepared from SH2B1f/f, Pdx1-Cre, and PKO male mice (12 weeks) and immunoprecipitated (IP) and immunoblotted (IB) with anti-SH2B1 antibody. Pancreatic extracts were also immunoblotted with anti–β-tubulin antibody. C: Tissue extracts were immunoprecipitated and immunoblotted with anti-SH2B1 antibody. D: Body weights of chow-fed male mice at 8 (control: n = 7; PKO: n = 8) and 18 (control: n = 4; PKO: n = 5) weeks of age. E. Body weights of male mice fed an HFD (45% fat) at 8 weeks of age. Control group includes both SH2B1f/f (n = 17–29) and Pdx1-Cre (n = 4–11) mice; PKO: n = 10–21. Con, control; FRT, Flp-recombinase recognition; TK, thymidine kinase.
Pancreas-Specific Deletion of SH2B1 Impairs Insulin Secretion and Glucose Tolerance
To determine whether islet SH2B1 is involved in the regulation of glucose metabolism, we measured blood glucose and examined glucose and insulin tolerance in PKO mice fed a normal chow diet. A combination of SH2B1f/f and Pdx1-Cre mice was used as a control group. PKO mice had relatively normal blood glucose levels (Fig. 5A). Their insulin sensitivity and glucose tolerance were also normal, as revealed by ITTs (Fig. 5B) and GTTs (Fig. 5C), respectively.
Figure 5.
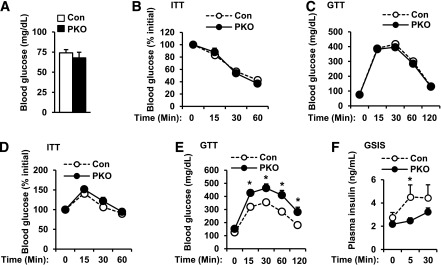
Pancreas-specific deletion of SH2B1 impairs glucose tolerance. A–C: PKO (n = 16–19) and control male mice (n = 29) were fed a normal chow diet. Overnight fasting blood glucose (A) was measured and ITTs (insulin: 1 unit/kg body wt) (B) and GTTs (d-glucose: 2 g/kg body wt) (C) were conducted at 7–8 weeks of age. D–F: Male mice (7–8 weeks) were fed an HFD (60% fat) for 10 weeks. ITTs (insulin: 1 unit/kg body wt) (D) and GTTs (d-glucose: 1 g/kg body wt) (E) were performed. Control: n = 10; PKO: n = 9. F: Mice were fasted for 24 h and injected with d-glucose (3 g/kg). Plasma insulin was measured 0, 5, and 30 min after injection. Control: n = 6; PKO: n = 8. Data are means ± SE. *P < 0.05. Con, control.
In prediabetes and insulin-resistant states, compensatory β-cell expansion and hyperinsulinemia are able to restrain blood glucose within a relatively normal range. For determination of whether SH2B1 in β-cells is involved in the regulation of compensatory β-cell expansion, PKO mice were fed an HFD to induce obesity and insulin resistance. As noted previously, body weight gain was similar between HFD-fed PKO and control mice (Fig. 4E). Obese PKO and control mice developed insulin resistance to a degree similar to that revealed by ITT (Fig. 5D). However, PKO mice exhibited greater glucose intolerance than control mice (Fig. 5E). For determination of whether pancreas-specific deletion of SH2B1 impairs insulin secretion, HFD-fed PKO and control mice were injected with glucose, and plasma insulin levels were measured after glucose injection. GSIS was impaired in obese PKO mice (Fig. 5F). Collectively, these data suggest that SH2B1 deficiency in the pancreas impairs GSIS in HFD-fed PKO mice, contributing to increased glucose intolerance.
Pancreas-Specific Deletion of SH2B1 Impairs β-Cell Survival, Proliferation, and Islet Expansion
For determination of whether impaired GSIS in HFD-fed PKO mice is caused by reductions in β-cell number and islet mass, pancreatic sections from HFD-fed PKO and control mice were stained with H-E or with antibodies against insulin and glucagon. Compared with the control groups, both the size of individual islets (Fig. 6A and B) and total islet area (Fig. 6C) were significantly reduced in PKO mice. Relative β-cell mass, determined by insulin immunoreactivity, was lower in PKO mice (Fig. 6B). Accordingly, pancreatic insulin content was 39% lower in PKO than in control mice (Fig. 6D). To determine whether cell death contributes to reduced β-cell mass in PKO mice, we measured islet apoptosis by TUNEL staining. Pancreatic sections were coimmunostained with anti-insulin antibody to visualize β-cells. The number of TUNEL-positive β-cells was more in PKO than in control mice (Figs. 6E and F). To determine whether deletion of SH2B1 in β-cells directly increases β-cell susceptibility to cytokine-induced death, we isolated islets and treated them with IFN-γ, TNF-α, and IL-1β in vitro. SH2B1-deficient islets were much more sensitive to cytokine-induced cell death (Fig. 6G). We also examined β-cell proliferation by immunostaining pancreatic sections with anti-Ki67 antibody (Fig. 6H). Islets from HFD-fed PKO mice had significantly fewer Ki67-positive β-cells (Figs. 6H and I). Together, these data suggest that increased apoptosis and decreased proliferation of β-cells may restrict compensatory β-cell expansion in HFD-fed PKO mice, contributing to reduced islet mass and impaired GSIS.
Figure 6.

Pancreas-specific deletion of SH2B1 decreases islet mass, pancreatic insulin content, and islet function. Male mice fed an HFD (60% fat) for 10–12 weeks. A: H-E staining of pancreas sections. B: Pancreatic sections were immunostained with anti-insulin or anti-glucagon antibodies. C: Islet area was quantified and normalized to pancreatic section area. Control: n = 10; PKO: n = 11. D: Pancreatic insulin content in fasted (16 h) male mice was measured and normalized to pancreas weights. Control: n = 8; PKO: n = 6. E and F: Islet cell apoptosis measured by TUNEL assays. Control: n = 6; PKO: n = 5. G: Islets were isolated from SH2B1f/f (control, n = 3) and PKO (n = 4) male mice (14–16 weeks) treated with cytokines (IL-1β: 1 ng/mL; TNF-α: 5 ng/mL; and INF-γ: 5 ng/mL) for 16–17 h. Apoptosis was analyzed using TUNEL assays. TUNEL-positive cells were normalized to total islet cells. H and I: Pancreatic sections were costained with anti-Ki67 and anti-insulin antibodies. Control: n = 8; PKO: n = 11. J–L: Males (9–10 weeks) were fed an HFD (45% fat) for 1 week and then injected with STZ (25 mg/kg) daily for 5 days. J: Blood glucose was monitored weekly. Control: n = 29; PKO: n = 16. K: Plasma insulin levels 5 weeks after STZ treatment. Control: n = 28; PKO: n = 16. L: Pancreatic insulin content 5 weeks after STZ treatment. Control: n = 25; PKO: n = 11. Data are means ± SE. *P < 0.05. Con, control.
In INS-1 832/13 cells, silencing of SH2B1 increased STZ-induced apoptosis (Fig. 2C). Similarly, islets isolated from PKO mice were more sensitive to cytokine-induced apoptosis (Fig. 6G). To determine whether SH2B1-deficient β-cells in mice are also predisposed to injury, we injected PKO and control mice with multiple low-dose STZ administrations (25 mg/kg body wt for 5 consecutive days). STZ treatments induced progressive hyperglycemia in both PKO and control mice; however, blood glucose levels were significantly higher in PKO mice (Fig. 6J). Plasma insulin levels (Fig. 6K) and pancreatic insulin content (Fig. 6L) were 38 and 47% lower, respectively, in STZ-treated PKO than in control mice. These data further support the conclusion that SH2B1 in β-cells protects against β-cell injury and death and is required for compensatory β activity in obesity and in response to β-cell stress.
Pancreas-Specific Deletion of SH2B1 Impairs the PI 3-Kinase/Akt Pathway in β-Cells
In INS-1 832/13 β-cells, SH2B1 directly enhanced IGF-1– and insulin-stimulated activation of the PI 3-kinase/Akt pathway (Fig. 3). To determine whether SH2B1 similarly enhances the activation of the PI 3-kinase pathway in β-cells in mice, we measured phosphorylation of Akt and its physiological substrate, FOXO1, in the β-cells of PKO mice that were fed an HFD for 10–12 weeks. Frozen pancreatic sections were immunostained with anti–phospho-Akt (pThr308) or anti–phospho-FOXO1 (pSer256). β-Cells were identified by immunostaining with anti-insulin antibodies. Akt phosphorylation was 31% lower in the islets of PKO mice (Fig. 7A). FOXO1 phosphorylation in β-cells was also significantly lower in PKO mice (Fig. 7B). For verification of reduction in Akt phosphorylation in the β-cells of PKO mice, male mice were fed an HFD for 5 weeks. Islets were isolated and stimulated with or without insulin, and islet extracts were immunoblotted with anti–phospho-Akt (pSer473) antibody. Akt phosphorylation was lower in the islets of PKO mice under both basal and insulin-stimulated conditions (Fig. 7C). These results indicate that SH2B1 directly enhances activation of the PI 3-kinase/Akt pathway not only in cultured INS-1 832/13 cells but also in β-cells in vivo.
Figure 7.

SH2B1 promotes the Akt/FOXO1 pathway in islets. A: Pancreas sections were prepared from PKO and control mice fed an HFD (60% fat) for 10–12 weeks and immunostained with anti–phospho-Akt (pThr308) and anti-insulin antibodies. Akt phosphorylation was quantified using ImageJ software. a.u., arbitrary units. Control: n = 12; PKO: n = 10. B: Pancreatic sections were prepared from PKO (n = 11) and control (n = 11) mice fed an HFD for 10–12 weeks and coimmunostained with anti–phospho-FOXO1 (pSer256) and anti-insulin antibodies. C: Male mice (7–10 weeks) were fed an HFD (60% fat) for 5 weeks and fasted for 18–22 h. Islets were isolated and treated with insulin (100 nmol/L) for 15 min, and islet extracts were immunoblotted with anti–phospho-Akt (pSer473) or Akt, respectively. Phosphorylation of Akt was quantified and normalized to total Akt levels. SH2B1f/f (control): n = 8; PKO: n = 8. Data are means ± SE. *P < 0.05. Con, control.
Discussion
β-Cell dysfunction is a hallmark of both type 1 and type 2 diabetes, and numerous genetic and environmental factors promote diabetes by impairing β-cell survival, growth, and activity. β-Cell growth, survival, and death are tightly regulated by various nutritional, hormonal, and neuronal signals, and β-cell sensitivity to these extracellular signals is likely to be closely regulated by β-cell intrinsic factors. In this study, we have identified SH2B1 as a β-cell intrinsic enhancer of the PI 3-kinase pathway. SH2B1 cell-autonomously protects against β-cell injury and death and promotes β-cell proliferation by integrating multiple external signals, including those from insulin and IGF-1 and other factors.
We have provided multiple lines of evidence demonstrating that SH2B1 in β-cells is a novel prosurvival and proproliferative signaling molecule. First, we showed that SH2B1 was expressed in β-cells at the high levels. Second, silencing of SH2B1 in INS1 832/13 β-cells increased STZ-induced apoptosis, whereas overexpression of SH2B1 was protective. Third, pancreas-specific deletion of SH2B1 increased β-cell apoptosis in HFD-fed PKO mice and exacerbated STZ-induced β-cell injury and hyperglycemia in PKO mice. Fourth, β-cell proliferation rates were lower in HFD-fed PKO mice than in HFD-fed control mice. Fifth, SH2B1 in β-cells directly promoted activation of the PI 3-kinase/Akt pathway in vitro and in vivo. The PI 3-kinase pathway is the major survival pathway in β-cells, and activation of this pathway also stimulates β-cell proliferation (44). In rat INS1 832/13 β-cells, silencing of SH2B1 attenuated the activation of the PI 3-kinase pathway by multiple survival factors (e.g., insulin and IGF-1); conversely, overexpression of SH2B1 enhanced insulin- and IGF-1–stimulated activation of the PI 3-kinase pathway. Furthermore, pancreas-specific deletion of SH2B1 attenuated phosphorylation of both Akt and FOXO1 in the β-cells of PKO mice.
SH2B1 has been reported to directly bind to both IGF-1 and insulin receptors in multiple cell types (16,19,45). SH2B1-insulin receptor interactions markedly enhance insulin receptor catalytic activity (19,46). SH2B1 also binds to both IRS1 and IRS2 to facilitate the formation of insulin receptor/SH2B1/IRS signaling complexes (18). Furthermore, SH2B1-IRS interactions protect IRS proteins from tyrosine dephosphorylation, thus sustaining the ability of IRS proteins to activate the PI 3-kinase pathway (19). Accordingly, SH2B1 markedly enhances insulin-stimulated tyrosine phosphorylation of IRS1 and IRS2 (19,22,47). We propose that in β-cells, SH2B1 directly enhances the activation of the PI 3-kinase/Akt pathway by enhancing insulin receptor and IGF-1 receptor activation, recruiting IRS proteins into insulin receptor and IGF-1 receptor signaling complexes, or protecting IRS proteins from tyrosine dephosphorylation. Additionally, SH2B1 in β-cells may also enhance activation of the PI 3-kinase pathway indirectly through stimulating insulin secretion. In agreement with this idea, deletion of SH2B1 markedly reduced basal Akt phosphorylation in the islets of PKO mice in the absence of exogenous insulin stimulation. Although the PI 3-kinase pathway is likely to mediate the prosurvival and proproliferative effects of SH2B1 in β-cells, our data do not exclude the possibility that SH2B1 may promote β-cell expansion by additional mechanisms.
β-Cell expansion is an important adaptation to obesity-associated insulin resistance, and impaired β-cell expansion is a risk factor for type 2 diabetes. Here, we report for the first time that pancreatic SH2B1 is required for β-cell expansion in obese mice. Lean PKO mice are able to maintain normal blood glucose, insulin sensitivity, and glucose tolerance, indicating that SH2B1 in β-cells is dispensable for the maintenance of glucose homeostasis in mice fed a normal chow diet. By contrast, PKO mice with HFD-induced obesity had reduced β-cell mass and pancreatic insulin content and impaired GSIS, indicating that compensatory β-cell expansion and function are impaired in these mice. PKO mice displayed more severe glucose intolerance after HFD feeding and developed more severe hyperglycemia after STZ treatment. As discussed above, SH2B1 directly promotes proliferation and inhibits apoptosis of β-cells; therefore, SH2B1 in β-cells promotes β-cell expansion in the setting of obesity or β-cell stress at least in part by increasing proliferation-to-apoptosis ratios. However, our data do not exclude the possibility that SH2B1 in β-cells may regulate systemic glucose homeostasis by other mechanisms in addition to promoting β-cell expansion, and this possibility should be further investigated in the future.
In summary, we observed that SH2B1 directly promotes insulin- and IGF-1–stimulated activation of the PI 3-kinase/Akt pathway in INS-1 832/13 β-cells and in islets from mice. SH2B1 also cell-autonomously promotes β-cell survival and proliferation, as well as GSIS, in mice fed an HFD. Moreover, disruption of pancreatic SH2B1 worsens STZ-induced diabetes in mice. Collectively, these findings suggest that SH2B1 in β-cells regulates glucose homeostasis by promoting compensatory β growth and function in obesity and in response to β stress. Therefore, β-cell SH2B1 may serve as a new therapeutic target for the treatment of diabetes.
Article Information
Acknowledgments. The authors thank Drs. Decheng Ren, Shin-ichi Oka, and Kae Won Cho and Ms. Suqing Wang (University of Michigan) for their helpful discussions. The authors thank Dr. Richard M. Mortensen (University of Michigan) for providing Pdx1-Cre mice and Dr. Christopher Newgard (Duke University) for providing INS-1 832/13 cells.
Funding. This study was supported by the National Institutes of Health (NIH) grants RO1 DK-065122, RO1 DK-091591, RO1 DK-094014 (to L.R.) and F31 NS-056575 (to D.L.M.). This work used cores supported by the Michigan Diabetes Research and Training Center (funded by NIH 5P60 DK-20572), the University of Michigan's Cancer Center (funded by NIH 5P30 CA-46592), the University of Michigan Nathan Shock Center (funded by NIH P30 AG-013283), and the University of Michigan Gut Peptide Research Center (funded by NIH DK-34933).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. Z.C. and D.L.M. researched data and wrote the manuscript. L.J. researched data. Y.L. and L.R. wrote the manuscript. L.R. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in oral form at the 73rd Scientific Sessions of the American Diabetes Association, Chicago, IL, 21–25 June 2013.
References
- 1.Marchetti P, Lupi R, Del Guerra S, Bugliani M, Marselli L, Boggi U. The beta-cell in human type 2 diabetes. Adv Exp Med Biol 2010;654:501–514 [DOI] [PubMed] [Google Scholar]
- 2.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest 2006;116:1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arumugam R, Horowitz E, Lu D, et al. The interplay of prolactin and the glucocorticoids in the regulation of beta-cell gene expression, fatty acid oxidation, and glucose-stimulated insulin secretion: implications for carbohydrate metabolism in pregnancy. Endocrinology 2008;149:5401–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bai L, Meredith G, Tuch BE. Glucagon-like peptide-1 enhances production of insulin in insulin-producing cells derived from mouse embryonic stem cells. J Endocrinol 2005;186:343–352 [DOI] [PubMed] [Google Scholar]
- 5.Kulkarni RN, Holzenberger M, Shih DQ, et al. beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat Genet 2002;31:111–115 [DOI] [PubMed] [Google Scholar]
- 6.Xu GG, Rothenberg PL. Insulin receptor signaling in the beta-cell influences insulin gene expression and insulin content: evidence for autocrine beta-cell regulation. Diabetes 1998;47:1243–1252 [DOI] [PubMed] [Google Scholar]
- 7.Xuan S, Kitamura T, Nakae J, et al. Defective insulin secretion in pancreatic beta cells lacking type 1 IGF receptor. J Clin Invest 2002;110:1011–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitamura T, Nakae J, Kitamura Y, et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest 2002;110:1839–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kushner JA, Ye J, Schubert M, et al. Pdx1 restores beta cell function in Irs2 knockout mice. J Clin Invest 2002;109:1193–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakae J, Biggs WH, 3rd, Kitamura T, et al. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 2002;32:245–253 [DOI] [PubMed] [Google Scholar]
- 11.Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat Genet 1999;23:32–40 [DOI] [PubMed] [Google Scholar]
- 12.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev 2008;29:351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin X, Taguchi A, Park S, et al. Dysregulation of insulin receptor substrate 2 in beta cells and brain causes obesity and diabetes. J Clin Invest 2004;114:908–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris DL, Rui L. Recent advances in understanding leptin signaling and leptin resistance. Am J Physiol Endocrinol Metab 2009;297:E1247–E1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maures TJ, Kurzer JH, Carter-Su C. SH2B1 (SH2-B) and JAK2: a multifunctional adaptor protein and kinase made for each other. Trends Endocrinol Metab 2007;18:38–45 [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Riedel H. Insulin-like growth factor-I receptor and insulin receptor association with a Src homology-2 domain-containing putative adapter. J Biol Chem 1998;273:3136–3139 [DOI] [PubMed] [Google Scholar]
- 17.Nelms K, O’Neill TJ, Li S, Hubbard SR, Gustafson TA, Paul WE. Alternative splicing, gene localization, and binding of SH2-B to the insulin receptor kinase domain. Mamm Genome 1999;10:1160–1167 [DOI] [PubMed] [Google Scholar]
- 18.Duan C, Li M, Rui L. SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. J Biol Chem 2004;279:43684–43691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris DL, Cho KW, Zhou Y, Rui L. SH2B1 enhances insulin sensitivity by both stimulating the insulin receptor and inhibiting tyrosine dephosphorylation of insulin receptor substrate proteins. Diabetes 2009;58:2039–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M, Ren D, Iseki M, Takaki S, Rui L. Differential role of SH2-B and APS in regulating energy and glucose homeostasis. Endocrinology 2006;147:2163–2170 [DOI] [PubMed] [Google Scholar]
- 21.Ren D, Li M, Duan C, Rui L. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab 2005;2:95–104 [DOI] [PubMed] [Google Scholar]
- 22.Duan C, Yang H, White MF, Rui L. Disruption of the SH2-B gene causes age-dependent insulin resistance and glucose intolerance. Mol Cell Biol 2004;24:7435–7443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Zhou Y, Carter-Su C, Myers MG, Jr, Rui L. SH2B1 enhances leptin signaling by both Janus kinase 2 Tyr813 phosphorylation-dependent and -independent mechanisms. Mol Endocrinol 2007;21:2270–2281 [DOI] [PubMed] [Google Scholar]
- 24.Ren D, Zhou Y, Morris D, Li M, Li Z, Rui L. Neuronal SH2B1 is essential for controlling energy and glucose homeostasis. J Clin Invest 2007;117:397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bauer F, Elbers CC, Adan RA, et al. Obesity genes identified in genome-wide association studies are associated with adiposity measures and potentially with nutrient-specific food preference. Am J Clin Nutr 2009;90:951–959 [DOI] [PubMed] [Google Scholar]
- 26.Hester JM, Wing MR, Li J, et al. Implication of European-derived adiposity loci in African Americans. Int J Obes (Lond) 2012;36:465–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hotta K, Kitamoto T, Kitamoto A, et al. Computed tomography analysis of the association between the SH2B1 rs7498665 single-nucleotide polymorphism and visceral fat area. J Hum Genet 2011;56:716–719 [DOI] [PubMed] [Google Scholar]
- 28.Ng MC, Tam CH, So WY, et al. Implication of genetic variants near NEGR1, SEC16B, TMEM18, ETV5/DGKG, GNPDA2, LIN7C/BDNF, MTCH2, BCDIN3D/FAIM2, SH2B1, FTO, MC4R, and KCTD15 with obesity and type 2 diabetes in 7705 Chinese. J Clin Endocrinol Metab 2010;95:2418–2425 [DOI] [PubMed] [Google Scholar]
- 29.Prudente S, Morini E, Larmon J, et al. The SH2B1 obesity locus is associated with myocardial infarction in diabetic patients and with NO synthase activity in endothelial cells. Atherosclerosis 2011;219:667–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Renström F, Payne F, Nordström A, et al. GIANT Consortium Replication and extension of genome-wide association study results for obesity in 4923 adults from northern Sweden. Hum Mol Genet 2009;18:1489–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandholt CH, Vestmar MA, Bille DS, et al. Studies of metabolic phenotypic correlates of 15 obesity associated gene variants. PLoS ONE 2011;6:e23531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi J, Long J, Gao YT, et al. Evaluation of genetic susceptibility loci for obesity in Chinese women. Am J Epidemiol 2010;172:244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Speliotes EK, Willer CJ, Berndt SI, et al. MAGIC. Procardis Consortium Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010;42:937–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeuchi F, Yamamoto K, Katsuya T, et al. Association of genetic variants for susceptibility to obesity with type 2 diabetes in Japanese individuals. Diabetologia 2011;54:1350–1359 [DOI] [PubMed] [Google Scholar]
- 35.Willer CJ, Speliotes EK, Loos RJ, et al. Wellcome Trust Case Control Consortium. Genetic Investigation of ANthropometric Traits Consortium Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 2009;41:25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bochukova EG, Huang N, Keogh J, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature 2010;463:666–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doche ME, Bochukova EG, Su HW, et al. Human SH2B1 mutations are associated with maladaptive behaviors and obesity. J Clin Invest 2012;122:4732–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volckmar AL, Bolze F, Jarick I, et al. Mutation screen in the GWAS derived obesity gene SH2B1 including functional analyses of detected variants. BMC Med Genomics 2012;5:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rui L, Mathews LS, Hotta K, Gustafson TA, Carter-Su C. Identification of SH2-Bbeta as a substrate of the tyrosine kinase JAK2 involved in growth hormone signaling. Mol Cell Biol 1997;17:6633–6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002;129:2447–2457 [DOI] [PubMed] [Google Scholar]
- 41.Chen Z, Sheng L, Shen H, et al. Hepatic TRAF2 regulates glucose metabolism through enhancing glucagon responses. Diabetes 2012;61:566–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 2000;49:424–430 [DOI] [PubMed] [Google Scholar]
- 43.Yousaf N, Deng Y, Kang Y, Riedel H. Four PSM/SH2-B alternative splice variants and their differential roles in mitogenesis. J Biol Chem 2001;276:40940–40948 [DOI] [PubMed] [Google Scholar]
- 44.Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest 2001;108:1631–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kotani K, Wilden P, Pillay TS. SH2-Balpha is an insulin-receptor adapter protein and substrate that interacts with the activation loop of the insulin-receptor kinase. Biochem J 1998;335:103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang M, Deng Y, Tandon R, Bai C, Riedel H. Essential role of PSM/SH2-B variants in insulin receptor catalytic activation and the resulting cellular responses. J Cell Biochem 2008;103:162–181 [DOI] [PubMed] [Google Scholar]
- 47.Li M, Li Z, Morris DL, Rui L. Identification of SH2B2beta as an inhibitor for SH2B1- and SH2B2alpha-promoted Janus kinase-2 activation and insulin signaling. Endocrinology 2007;148:1615–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]