

Abstract
Study Objectives:
Obstructive sleep apnea (OSA) is strongly associated with cardiovascular disease, including stroke and acute coronary syndromes. Plasminogen activator inhibitor-1 (PAI-1), the principal inhibitor of tissue-type plasminogen activator (t-PA), has a pronounced circadian rhythm and is elevated in both OSA and cardiovascular disease and may be an important link between the two conditions. Endothelial dysfunction is one of the underlying pathophysiological mechanisms of cardiovascular disease, and may be altered in OSA. Our primary aim was to compare circadian variability of PAI-1 and t-PA in patients with OSA and normal controls by determining the amplitude (peak level) and mesor (rhythm adjusted mean) of PAI-1 and t-PA in serial blood samples over a 24-h period. The secondary aim was to measure markers of endothelial function (brachial and radial artery flow) in patients with OSA compared with normal controls.
Setting:
Cross-sectional cohort study.
Patients or Participants:
Subjects age 18 y or older, with a body mass index of 25-45 kg/m2, with or without evidence of untreated OSA.
Interventions:
Plasma samples were collected every 2 h, in OSA patients and matched controls, over a 24-h period. PAI-1 and t-PA antigen and activity were measured. The presence or absence of OSA (apnea-hypopnea index of 5 or greater) was confirmed by overnight polysomnography. Endothelial function was measured via brachial artery flow mediated vasodilatation and computerized arterial pulse waveform analysis.
Measurements and Results:
The rhythm-adjusted mean levels of PAI-1 antigen levels in the OSA group (21.8 ng/mL, 95% confidence level [CI], 18 to 25.7) were significantly higher as compared to the non-OSA group (16 ng/mL, 95% CI, 12.2 to 19.8; P = 0.03). The rhythm-adjusted mean levels of PAI-1 activity levels in the OSA group (23.9 IU/mL, 95% CI, 21.4 to 26.5) were also significantly higher than in the non-OSA group (17.2 IU/ mL, 95% CI, 14.6 to 19.9; P < 0.001).There were strong correlations between amplitude of PAI-1 activity and severity of OSA as measured by AHI (P = 0.02), and minimum oxygen levels during sleep (P = 0.04). Endothelial function parameters did not differ significantly between the two groups.
Conclusion:
The presence of obstructive sleep apnea adversely affects circadian fibrinolytic balance with higher mean plasminogen activator inhibitor-1 activity and antigen, and significantly lower mean tissue-type plasminogen activator activity compared with controls. This perturbation may be an important mechanism for increased cardiovascular events in patients with obstructive sleep apnea. Intermittent hypoxia and changes in circadian clock gene activity in obstructive sleep apnea may be responsible for these findings and warrant further study. Favorable changes in fibrinolytic balance may underlie the reduction in cardiovascular events observed with the treatment of obstructive sleep apnea.
Citation:
Bagai K; Muldowney JAS; Song Y; Wang L; Bagai J; Artibee KJ; Vaughan DE; Malow BA. Circadian variability of fibrinolytic markers and endothelial function in patients with obstructive sleep apnea. SLEEP 2014;37(2):359-367.
Keywords: OSA, fibrinolytic activity, endothelial function
INTRODUCTION
Obstructive sleep apnea (OSA) is a common disorder that results in frequent nocturnal arousals and hypoxemia due to recurrent episodes of upper airway collapse. The severity of OSA is indicated by the apnea-hypopnea index (AHI), defined as the average number of apneas and hypopneas per hour. Cardiovascular disease (CVD) is the leading cause of mortality in the United States. A recent American Heart Association Statistics Committee report1,2 demonstrated that nearly 2,300 Americans die of CVD each day at an average of one death every 38 sec. Coronary heart disease caused approximately one of every six deaths in the United States in 2006. Each year, approximately 795,000 people experience a new or recurrent stroke. Multiple studies have demonstrated an association between the presence of OSA, stroke,3–9 and fatal and nonfatal myocardial infarctions (MIs).10,11
The majority of strokes and MIs are due to atherothrombotic events, with impaired fibrinolytic activity increasing the propensity for these events.12,13 Plasminogen activator inhibitor-1 (PAI-1) is the principal physiologic inhibitor of the body's fibrinolytic system, acting as a suicide substrate for tissue-type plasminogen activator (t-PA). Plasma PAI-1 is derived from several sources, including the vascular endothelium, adipose tissue, and liver.14,15 Platelets store large quantities of PAI-1 that are secreted following platelet aggregation. In healthy individuals, PAI-1 circulates briefly in plasma in an active form (activity); only a fraction of the secreted active PAI-1 has the opportunity to react with plasma t-PA to form inert covalent complexes. Plasma PAI-1 antigen shows a marked circadian variation in the plasma of normal individuals. PAI-1 activity is directly related whereas t-PA activity is inversely related with PAI-1 antigen levels throughout the 24-h cycle.16–19 PAI-1 antigen levels are elevated in patients with MI and stroke20,21 and elevated levels are associated with an increased risk of death from MI.22 PAI-1 levels peak in the early morning hours, which may account for the early morning peak in the incidence of stroke and MI.21,23 Recent studies have shown that PAI-1 levels are elevated in patients with OSA and PAI-1 levels also correlate with severity of OSA,24–26 suggesting that PAI-1 potentially mediates the effects of OSA on thrombosis. In addition, increasing severity of obstructive sleep apnea is independently associated with higher PAI-1 concentration.24 Further, severity of OSA, as determined by the AHI, is a significant predictor of PAI-1 levels, independent of mean night-time oxygen desaturation, body mass index (BMI), and age.
Endothelial dysfunction plays an important role in the pathogenesis of CVDs27,28 and may underlie the association between OSA and CVD. Perturbation of endothelium-dependent vasomotor function is associated with OSA.29–31 Obesity, which is a common finding in OSA, is a known cause for elevation of PAI-1 and endothelial dysfunction.32,33
Our primary aim was to compare circadian variability of PAI-1 and t-PA in patients with OSA compared to normal controls by assessing amplitude (peak) and mesor (rhythm adjusted mean) levels. Specifically, we hypothesized that in patients with OSA, the mean levels of PAI-1 activity assessed by serial collection of blood samples over a 24-h period would be higher compared with that of normal controls. Our secondary aim was to measure changes in markers of endothelial function, including flow-mediated vasodilatation and radial artery tonometry, in patients with OSA compared to normal controls. In this observational study, we describe the circadian rhythm of both PAI-1 and t-PA antigen and activity, and measurements of endothelial function, in patients with OSA and controls. Our goal was to better understand the mechanisms underlying OSA and CVDs such as myocardial infarction and stroke.
METHODS
Subjects
Subjects age 18 y or older, with a BMI of 25-45 kg/m2, with or without evidence of untreated OSA (AHI ≥ 5 events/h), and capable of providing informed consent were included in this study. Subjects were excluded if they had a prior history of CVDs such as myocardial infarction, stroke, transient ischemic attack, or peripheral vascular disease; history of certain chronic diseases such as diabetes mellitus; uncontrolled hypertension; renal failure on dialysis; cancer; autoimmune or liver disease; significant history of medical or psychiatric disease that may impair participation in the trial; evidence of medical instability (cardiac arrhythmias, congestive heart failure, pulmonary disease) that required an expedited evaluation and treatment of OSA; history of alcohol or drug abuse during the 1-y period prior to trial participation; current use of tobacco products; current treatment with angiotensin converting enzyme-inhibitors and/or chronic use of nonsteroidal anti-inflammatory drugs; another primary sleep disorder that required intervention with medications or caused disrupted sleep; unusual sleep or wake habits, including shift work; transmeridian travel in the previous 3 mo; OSA previously treated with continuous positive airway pressure (CPAP), surgery, or an oral appliance; and pregnancy.
Study Protocol
The Vanderbilt University Institutional Review Board approved the study protocol, and all participants provided written informed consent. The study consisted of a screening visit and a study visit.
For the screening visit, potential participants who appeared to meet inclusion/exclusion criteria met the study team at the Vanderbilt Clinical Research Center (CRC), where informed consent was obtained. Patients were recruited from the community by advertisement and also by posting the study on the National Institutes of Health website, www.clinicaltrials.gov (NCT 00936481), to avoid the potential for referral bias. Participants had a comprehensive history (including sleep related complaints) and physical examination by the study physician. Blood pressure, pulse, weight, height, and BMI were recorded. Women of childbearing potential had a urine pregnancy test. Treatment of OSA was not delayed due to participation in the research protocol.
For the study visit, each subject was admitted to the CRC in the afternoon and had an intravenous saline lock placed to allow for serial blood collection. Sleep stages and breathing events were monitored with overnight polysomnography (PSG), performed in the Vanderbilt Sleep Research Core located in the CRC. The following morning, fasting lipid panel and glucose levels were obtained. Sequential blood samples for PAI-1 (antigen and activity) and t-PA (antigen and activity) were drawn at 2-h intervals over a 24-h period beginning in the evening of admission. Brachial artery flow mediated vasodilatation and Computerized Arterial Pulse Waveform Analysis (CAPWA) were performed on the morning after the PSG.
Study Procedures
Overnight Polysomnogram
A trained sleep technologist performed the overnight PSG, using standard protocols. The PSG was performed in digital video format consisting of six EEG channels (F3,F4,C3,C4,01,02 by the International 10-20 system), three chin electromyogram (EMG) leads, two electrooculogram (EOG) leads, two electrocardiogram (EKG) leads, snoring sound, respiratory effort over the chest and abdomen, airflow at the nose and mouth using thermocouples and nasal pressure cannulas, and bilateral surface EMG recording from the legs (with electrodes placed over the anterior tibialis muscles).Oxyhemoglobin saturation (SaO2) was monitored by pulse oximetry. All studied were staged according to standard guidelines. Apneas were scored if there was a > 90% decrement in the thermistor for at least 10 sec and hypopneas were scored if there was a 50-90% decrement in the nasal pressure transducer, for at least 10 sec, with concurrent oxyhemoglobin desaturation of 3% or greater or an EEG arousal.34
Serial Blood Samples
Plasma was obtained at 2-h intervals during the subjects' admission to the CRC for the measurement of PAI-1 and t-PA. One indwelling saline lock was placed into an arm vein of the subject, under local anesthesia (0.2 mL of 1% lidocaine subdermally) for drawing blood samples during the study. After each blood draw, the intravenous port was flushed with normal saline. Biopool stabilite highly acidic sodium citrate blood collection tubes were used for sample collection. Plasma was separated within 2 h, by centrifugation for 20 min at 3000 g. Plasma was stored at -70 C for not more than 5 mo prior to assay. Enzyme-linked immunosorbent assay technique was used for the quantitative determination of PAI-1 and t-PA antigen and activity. At least 1 mm of plasma was withdrawn and evenly distributed into four microcentrifuge tubes. To minimize awakening patients from sleep, the CRC nurses used a small flashlight when drawing blood samples during the night, rather than turning on overhead lights in the patient's room.
Endothelial Function Testing
Flow-Mediated Vasodilatation: The brachial artery was imaged using high-resolution ultrasound equipped with a 7.5 MHz linear-array transducer. The artery was imaged longitudinally, to allow visualization of the posterior wall intima-lumen interface and the anterior wall media-adventitial interface. Endothelial-dependent vasodilatation was assessed by measuring the change in caliber of the brachial artery during reactive hyperemia. The brachial artery was imaged continuously for 2 min following cuff deflation, as the artery diameter returned to baseline. Measurements were obtained at 30, 60, 90, and 120 sec. Baseline and hyperemic velocities were also recorded.35
Computerized Arterial Pulse Waveform Analysis: CAPWA was used to measure vascular compliance based on 30-sec recordings of signal-averaged arterial pulse waves, obtained by applanation plethysmography, using a surface-residing transducer over the radial pulse of supine subjects. Blood pressure was measured oscillometrically in the opposite arm (Hypertension Diagnostics, Inc., Eagan, MN; Nelicor N-500 arterial tonometer, Nellcor Inc, Colin, San Antonio, TX). The diastolic decay of the waveform was mathematically analyzed, and two components of arterial compliance were calculated based on a modified Windkessel model of the circulation: capacitive compliance (C1) was obtained for large conduit arteries, and oscillatory (or reflective) compliance (C2) was obtained for smaller, more peripherally located arteries and arterioles.36
Data Analysis
Baseline demographic and clinical variables, including risk factors obtained from the history and physical examination, were summarized for participants in each group of the study. Descriptive summaries of continuous baseline variables were presented as mean, standard deviation, and percentiles, whereas discrete variables were presented as frequencies and percentages. PAI-1 and t-PA circadian variations (oscillations) were evaluated by fitting the nonlinear mixed-effect model y = (M + μ1) + (A + μ2) sin(2π ÷ 24hr × time + B + μ3), where M was the rhythm-adjusted mean, A was the amplitude, and B was the acrophase, μ1 was the subject-specific rhythm-adjusted mean effect, μ2 was the subject-specific amplitude effect, μ3 was the subject-specific acrophase. Based on this model, we calculated the peak PAI-1 or t-PA oscillation time (time of the day at which the maximum level of the variable was reached) by setting sin [(2*π / 24hr) * time + B] = sin [0.5 π] = 1. To test for circadian variation, the null hypothesis (amplitude equals zero) was evaluated using a two-tailed t-test.
The primary study objective was to compare peak and mean levels of PAI-1 and t-PA in patients with OSA and normal controls. The Wilcoxon rank-sum test was used to assess the difference between two study arms, testing the null hypothesis that there was no difference in the levels of PAI-1 and t-PA antigen and activity, and endothelial function in patients with OSA, as compared with normal controls. Parameters of endothelial function included small and large artery capacitance and change in velocity and diameter. Prespecified potential confounders including age, sex, BMI, and blood pressure were adjusted using a general linear model.
The second objective of this study was to assess the association between PAI-1 and t-PA levels and severity of OSA and other polysomnographic parameters. The Spearman correlation coefficient between the PAI-1 and t-PA levels and AHI, minimum oxygen, mean oxygen levels, time with oxygen saturations below 90%, arousal index was calculated after adjusting for age.
We calculated the sample size based on a previous study,24 anticipating the average peak level of PAI-1 in the normal group to be 10 ng/mL, and that the average peak level of PAI-1of the OSA group would increase by 25 to 35 ng/mL. A sample size of 17 in each group provided 80% power using a two-group t-test with a 0.05 two-sided significance level.
Statistical analysis was performed using both SPSS for Windows (version 16, manufacturer IBM) and the statistical software R (www.r-project.org). CIs were calculated and P values less than 0.05 were considered significant.
RESULTS
Study Population
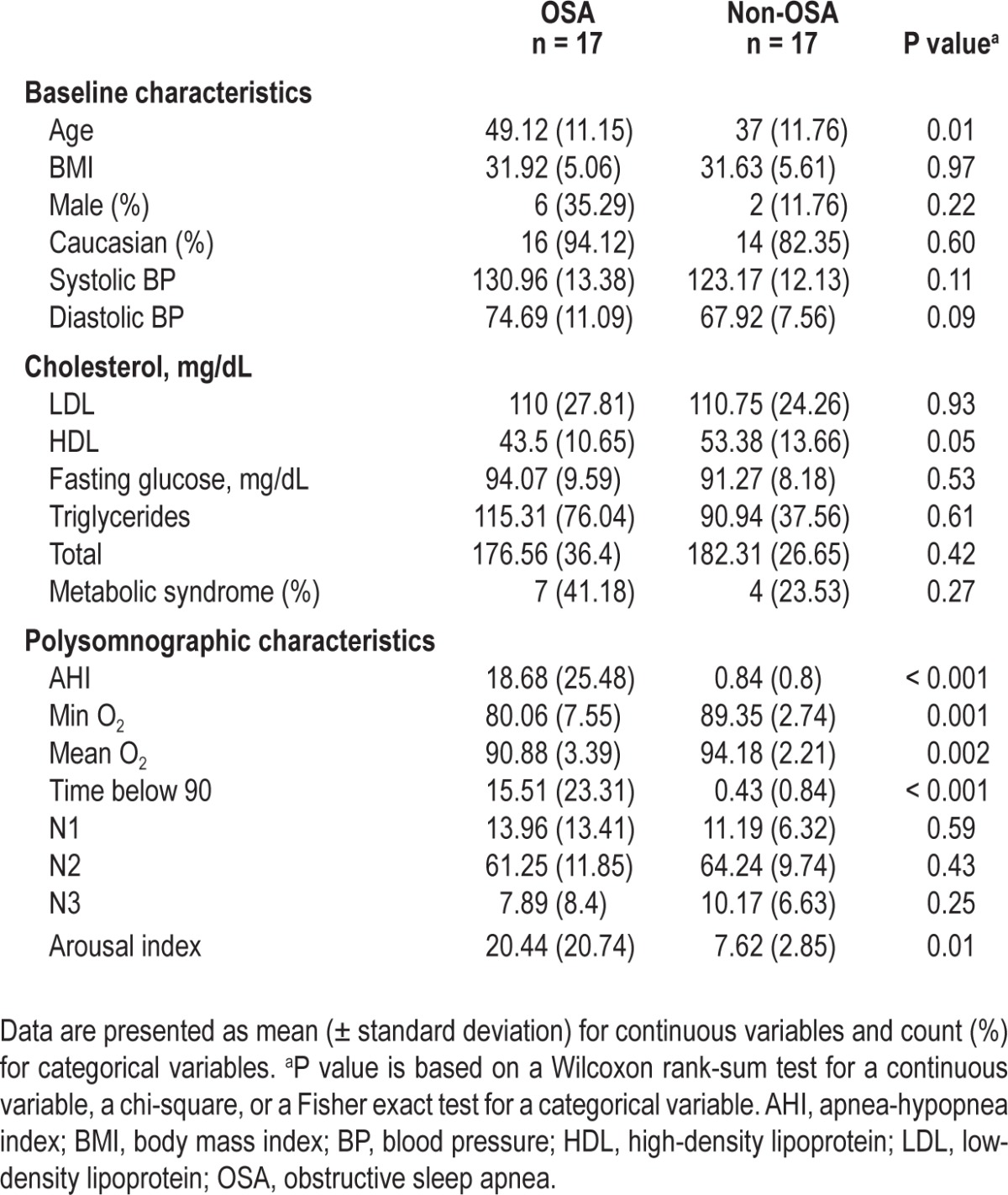
OSA was diagnosed in 17 patients, and 17 participants met inclusion criteria for the control group (Table 1). The mean AHI in the OSA group was 16.8 events/h and the mean AHI in the non-OSA group was 0.84 events/h (P < 0.001). The mean and minimum oxygen levels in the OSA group (90.8% and 80% respectively) were significantly lower than in the non-OSA group (94% and 89% respectively). As expected, the arousal index was higher in the OSA group (20.4/h) versus the non-OSA group (7.6/h; P = 0.01) (Table 1).
Table 1.
Clinical and polysomnographic characteristics of patients with OSA and control subjects using Wilcoxon rank-sum test for continuous variables
Effects of OSA on Markers of Fibrinolysis
The rhythm-adjusted mean levels of PAI-1 antigen levels in the OSA group (21.8 ng/mL, 95% CI, 18-25.7) were higher, as compared with the non-OSA group (16 ng/mL, 95% CI, 12.2-19.8; P = 0.03).The rhythm adjusted mean PAI-1 activity levels in the OSA group (23.9 IU/mL, 95% CI, 21.4-26.5) were also higher than in the non-OSA group (17.2 IU/mL, 95% CI, 14.6-19.9; P < 0.001). There was no difference in the rhythm adjusted mean t-PA antigen levels (Table 2). As expected, rhythm adjusted mean t-PA activity levels in the OSA group (0.4, 95% CI 0.3, 0.4) were lower than the non-OSA group (0.5, 95% CI 0.5, 0.6; P < 0.001).
Table 2.
Levels of fibrinolytic markers in patients with OSA and control subjects
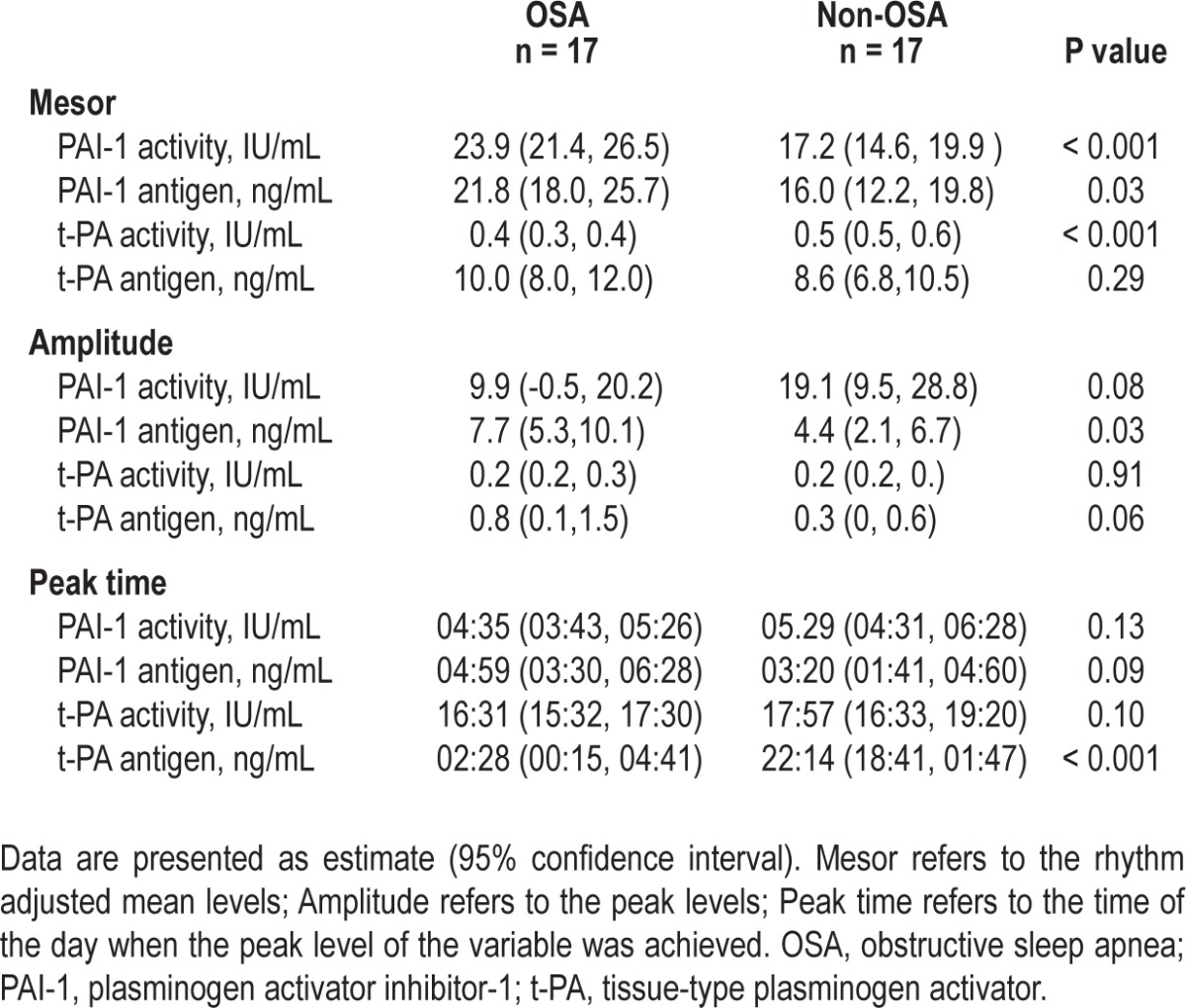
The amplitude of the PAI-1 antigen was higher in the OSA group (difference of 3.3 units; P = 0.02). However the amplitude of the PAI-1 activity was higher in the non-OSA group (difference of 9.3 units; P = 0.08), but did not reach significance. There was no difference in the peak timing of PAI-1 antigen and activity, and t-PA activity levels. There was a difference in timing of the t-PA antigen detected in OSA versus the non-OSA group (Table 2).
A circadian rhythm was detectable for PAI-1 antigen and activity, and t-PA antigen and activity. However, the amplitude for t-PA antigen was small (Table 3).
Table 3.
P-values of testing if amplitude is zero

Effects of OSA on Endothelial Function
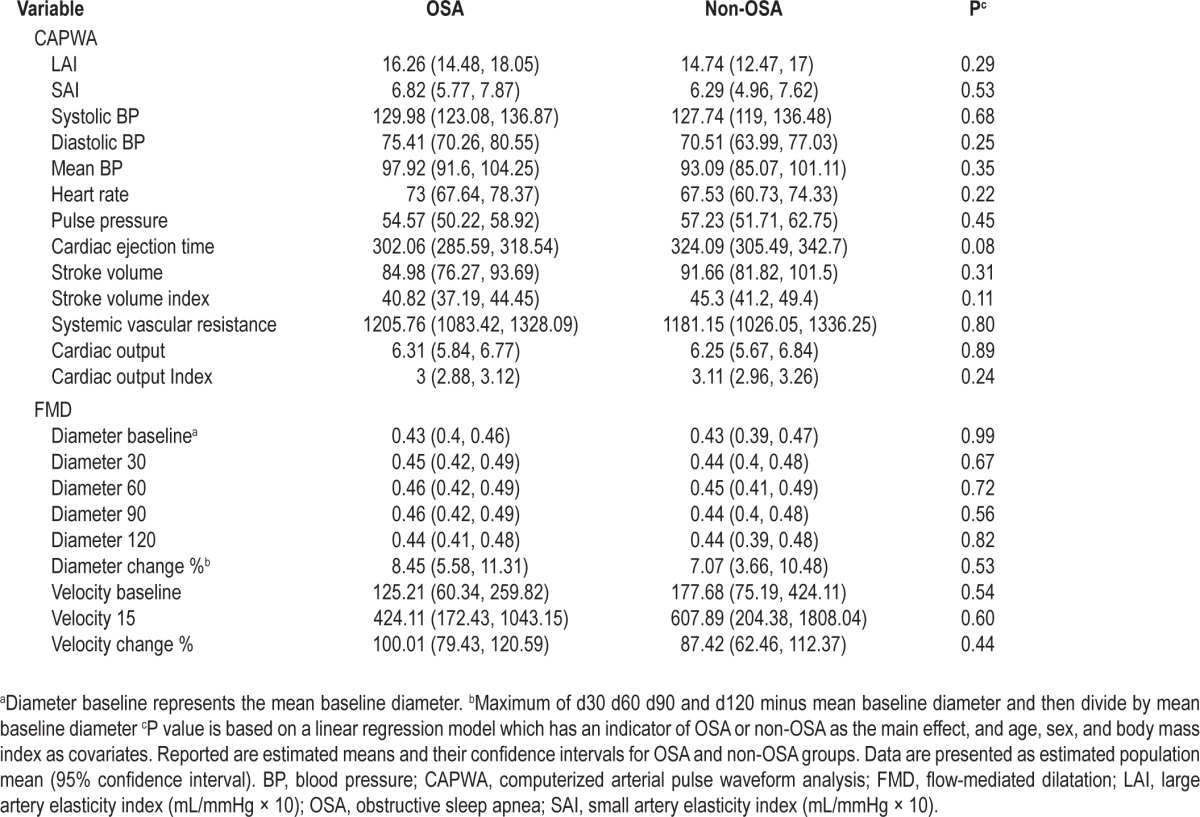
The brachial artery diameter and velocity at baseline and after reactive hyperemia was calculated in the patients with OSA and control subjects after adjusting for age and sex (Table 4). The mean brachial artery diameter at baseline did not differ in the OSA group (0.4 cm, 95% CI, 0.4-0.5) compared with the non-OSA group (0.4 cm, 95% CI, 0.4-0.5; P = 0.99). The mean % change in brachial artery diameter with reactive hyperemia in the OSA group (8.5, 95% CI, 5.6-11.3) was not different as compared with the non-OSA group (7.1, 95% CI, 3.7-10.5; P = 0.5). There were no differences in the large artery elasticity index in OSA (16.3, 95% CI, 14.5-18.1) versus non-OSA (14.8, 95% CI, 12.5-17; P = 0.3) and small artery elasticity index in the OSA (6.8, 95% CI, 5.8-7.9) and non-OSA groups (6.2, 95% CI, 5.0-7.6; P = 0.5), as measured by CAPWA (Table 4).
Table 4.
Results of endothelium-dependent flow-mediated dilation and computerized arterial pulse waveform analysis testing in patients with OSA and control subjects
Correlations between OSA and Markers of Fibrinolysis
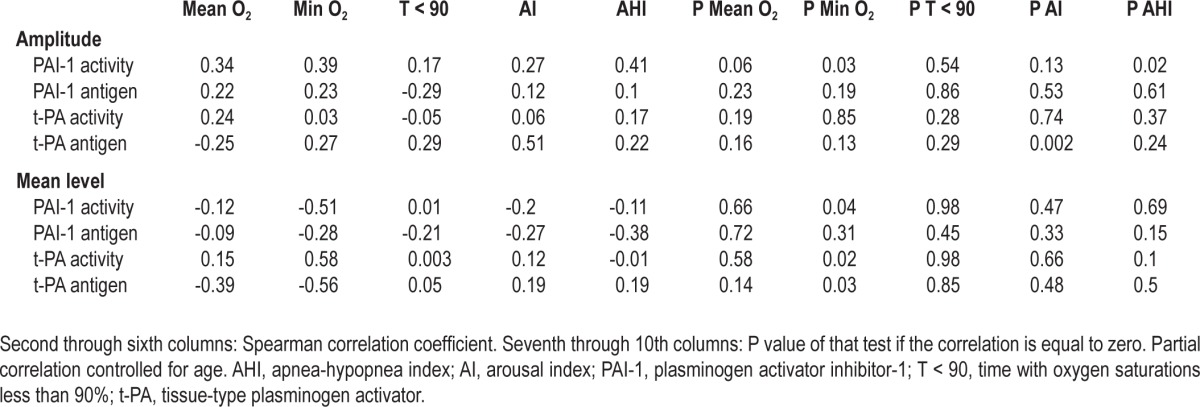
Spearman correlations were calculated between amplitude and mean levels of PAI-1 antigen and activity, t-PA antigen and activity, and sleep related variables including mean, minimum oxygen levels, time with oxygen saturations below 90%, arousal index, and severity of OSA. There were strong correlations between amplitude of PAI-1 activity and severity of OSA as measured by AHI (P = 0.02), and minimum oxygen levels during sleep (P = 0.03). There were strong correlations between mean PAI-1 activity levels and minimum oxygen levels (P = 0.04). There was also a strong correlation between mean t-PA antigen levels and arousal index (Table 5). There were no significant correlations between fibrinolytic markers and time with oxygen saturations below 90%. Rhythm-adjusted mean levels of PAI-1 antigen or activity or t-PA antigen or activity were not correlated with age of patients with OSA. Age was correlated with amplitude of PAI-1 activity but not the amplitude of PAI-1 antigen, or t-PA antigen or activity (supplemental Tables S1 and S2).
Table 5.
Spearman correlations between amplitude and mean levels of fibrinolytic markers in patients with obstructive sleep apnea and mean O2 levels, minimum O2 levels, time with oxygen saturations below 90%, arousal index and obstructive sleep apnea severity as measured by apnea-hypopnea index
DISCUSSION
Our results showed that the rhythm-adjusted mean levels of PAI-1 activity and antigen were higher in patients with OSA during the entire circadian cycle (Figures 1 and 2). To our knowledge, our study is the first to describe the complete circadian rhythm of these biomarkers in the OSA population. Another strength of our study is the stringent inclusion/exclusion criteria. The study is limited by small sample size, although we included the number of patients based on statistical power calculations. In an effort to increase recruitment and study efficiency, this study was not conducted in age/sex matched fashion. Because most of our subjects recruited in both groups were female; these data may not be generalizable to male patients with OSA. We did not measure plasma catecholamines and markers of oxidative stress and vascular inflammation, which would have contributed further to our understanding of putative mechanisms of cardiovascular events in OSA.
Figure 1.

Twenty-four-hour variability of PAI-1 and t-PA activity in patients with OSA and normal controls. PAI-1 and t-PA oscillations were evaluated by fitting nonlinear mixed-effect models y = (M + μ1) + (A + μ2) sin(2π ÷ 24hr × time + B + μ3), where M was the rhythm-adjusted mean, A was the amplitude, and B was the acrophase. The solid line is based on the model and it is an estimator of population mean PAI-1 or t-PA activity along the time. OSA, obstructive sleep apnea; PAI-1, plasminogen activator inhibitor-1; t-PA, tissue-type plasminogen activator.
Figure 2.

Twenty-four-hour variability of PAI-1 and t-PA antigen in patients with OSA and normal controls. PAI-1 and t-PA circadian variations (oscillations) were evaluated by fitting non- linear mixed-effect models y = (M + u1) + (A + u2) sin(2π ÷ 24hr × t + B + u3), where M was the rhythm-adjusted mean, A was the amplitude, and B was the acrophase. In the above formula, the peak time (time of the day at which the maximum level of the variable was reached) was calculated by setting sin [(2*π / 24hr) * time + B] = sin [0.5 π] = 1. The solid line is based on the model and it is an estimator of population mean PAI-1 or t-PA antigen along the time. The mean amplitude of PAI-1 antigen in the OSA group was 3.3 units higher than that of the non-OSA group (P = 0.03). Shaded portion shows circadian night. OSA, obstructive sleep apnea; PAI-1, plasminogen activator inhibitor-1; t-PA, tissue-type plasminogen activator.
Fibrinolytic Markers in OSA
PAI-1 antigen and activity have a well-established diurnal variation in normal controls with a nadir occurring during 17:00-23:00 and 33% variability between peak and trough plasma PAI-1 levels.19 This diurnal pattern of variation in PAI-1 antigen and activity is believed to be of clinical significance, as it mirrors the diurnal variation in MI incidence reported by several investigators.23,37 Participants in these prior studies, however, did not undergo overnight PSG to exclude OSA. There are limited data on whether patients with OSA have normal circadian rhythms. von Känel and colleagues38 measured prothrombotic markers (PAI-1 and d-dimer) in OSA patients. PAI-1 antigen was assessed and was found to be higher in patients with OSA even after adjusting for age. The day-night variability of PAI-1 antigen, with a peak in early morning and a nadir in the afternoon, was retained as seen in the PAI-1 rhythms of normal controls.
Our study is the first to collectively assess the complete circadian variability of t-PA antigen and activity, and PAI-1 antigen and activity in the population with OSA. We also show that PAI-1 maintains its direct inhibitory action against t-PA in the OSA population (as in normal controls) with lowest levels of t-PA activity seen when PAI-1 activity is at its peak. Our study therefore adds to the evidence that PAI-1 retains its circadian rhythm in OSA patients, albeit with a higher mesor. The amplitude and acrophase are not disrupted.
Similar to the findings in the study by von Känel and colleagues, our study shows a higher mean (mesor) PAI-1 antigen and activity levels in patients with OSA compared to controls and no difference in the time of the peak (acrophase) for PAI-1 between patients with OSA and controls.
We observed that in our patients with OSA, there is no decline of PAI-1 levels that is seen in normal controls during the day, resulting in a greater area under the curve. This occurrence is a likely consequence of OSA and the repeated hypoxia and/or arousals associated with respiratory events. t-PA circulates in plasma as a complex with PAI-1 in a 1:1 ratio. PAI-1 binds to fibrin within a clot and inhibits t-PA, thereby increasing the clot's resistance to lysis.39 In our study, the t-PA antigen levels did not vary over a 24-h period; however, there was circadian variability in the t-PA activity. These changes in t-PA activity are most likely a result of variation in PAI-1 antigen and activity, and subsequent inhibition of t-PA activity.
Correlation Analysis
PAI-1 activity levels correlated strongly with severity of OSA (as measured by AHI) and minimum oxygen levels during sleep (Table 5), suggesting a relationship between PAI-1 and hypoxia. The putative underlying mechanism responsible for altered PAI-1 levels in patients with OSA could be due to intermittent hypoxia which, via hypoxia-inducible factor 1-alpha transcription factors, drives transcription of PAI-1.40–42 Future studies can explore this hypothesis by studing changes in PAI-1 levels in patients with OSA after treatment with CPAP.
Studies have implicated the molecular components of the body's endogenous circadian clock, such as CLOCK: BMAL1 and CLOCK: BMAL2, as key determinants of PAI-1 rhythmicity.40,43 Repeated arousals and disrupted sleep in patients with OSA may trigger changes in these circadian clock genes that control fibrinolytic markers, in a manner that augments transcription rather than disrupts rhythmicity.
The effect of age on fibrinolysis is variable depending on the study. In most studies, t-PA and PAI-1 antigen levels appear to increase with age. However, the effect of age on PAI-1 activity (which is the active form) varies in different studies and was either not correlated, or weakly correlated.15,44,45 Our study did not find any significant correlations between age and mean PAI-1 or t-PA antigen or activity levels. There was a positive correlation between age and the amplitude (peak levels) of PAI-1 activity; however, amplitude of other fibrinolytic markers were not correlated with age. There was a trend for lower amplitude of PAI-1 activity in the OSA group despite higher amplitude of PAI-1 antigen and older mean age. The control group was younger than the OSA group. If amplitude of PAI-1 activity was associated with age, we would have expected a higher amplitude of PAI-1 activity in the OSA population.
The higher amplitude of PAI-1 activity in the non-OSA group as compared with the patients with OSA indicates the relative importance of circadian drivers of PAI-1 activity levels in normal controls, and importance of more constitutive drivers of gene regulation such as nocturnal intermittent hypoxia in the patients with OSA. This relative blunting of circadian variability of PAI-1 activity (as shown by a lower amplitude), coupled with higher rhythm adjusted mean PAI-1 activity levels (as shown by a greater under the curve) may support the observation that there is an increased risk for CVD events throughout the circadian period in patients with OSA.
Endothelial Function
In contrast with prior studies that have shown altered endothelial function in patients with OSA,30 our results did not show any significant difference in the markers of endothelial function as measured by brachial artery flow-mediated dilatation testing and radial artery tonometry studies. One reason for this observation may be due to the inclusion of patients with milder OSA (average AHI, 18 events/h) in our study, and use of a less stringent definition for diagnosis of OSA (including obstructive events with either 3% desaturations or arousals; as compared with other studies that included patients with 4% desaturation). This definition of OSA with the 3% desaturations or arousals criteria for obstructive hypopneas has now been adopted by the American Academy of Sleep Medicine as the organization's standard definition for hypopneas.46 A possible mechanism for the changes in endothelial function in patients with OSA shown in prior studies may be that more severe degrees of intermittent hypoxia, or sustained hypoxia (as seen in severe OSA), is required to alter endothelial function by promoting the formation of reactive oxygen species.47,48 The discrepancy between the endothelial function findings and effect on fibrinolytic process may be because the hypoxia sustained in mild to moderate OSA, although sufficient to cause activation of PAI-1 gene, is not sufficient to cause alterations in endothelial dependent vasodilation.
Importance of Study Findings
Acute cardiovascular events including strokes and acute coronary syndromes (ACS) cluster between 06:00 and 12:00.49 Ishibashi et al.50 reported that the peak time of onset of ACS in patients with moderate OSA (AHI > 15 events/h) was shifted compared with controls. ACS was observed more frequently between midnight and noon compared with controls, and was more evident in patients with greater severity of OSA. Changes in the peak timing and/or rhythm- adjusted mean levels of PAI-1 antigen, PAI-1 activity, and t-PA activity may underlie these observations. In our study we found higher mean PAI-1 activity and antigen throughout the circadian period, and substantially lower mean t-PA activity levels in patients with OSA compared with controls. This in turn may create an impaired fibrinolytic state that increases the risk for cardiovascular events.
We did not find a difference in the timing of peak PAI-1 activity and antigen levels in patients with OSA compared with controls. A possible explanation for this observation is that the drivers of increased fibrinolytic impairment do not disrupt the rhythmicity of expression, but rather the amount of expression of PAI-1. This does not exclude the possibility that in more severe cases of OSA, a shift in the timing of peak levels could occur; however, this observation was not made in our study population.
CONCLUSION
Apart from traditional risk factors, OSA is now being increasingly recognized as an important modifiable risk factor for CVD. It has been shown previously that successful treatment of OSA with CPAP can lower incidence of CVD in patients with OSA.51–53 The underlying mechanisms for this favorable effect are still unclear. Based on the data presented in this study, there appears to be a definite link between OSA and the fibrinolytic system. This link could underlie the association between OSA and cardiovascular events. Treatment of OSA with CPAP may result in a favorable change in the circadian rhythm of fibrinolytic markers. Further studies to evaluate this effect and correlation with these changes with reduction in cardiovascular events, in a larger sample size, are therefore warranted. Measurement of oxidative stress, catecholamines, and inflammatory markers will also help in determining the role of other mechanisms, such as inflammation, hypoxia, and sympathetic activation. Significant changes in levels and circadian rhythm of fibrinolytic markers were noted in our study, which included subjects with mild OSA. Mild OSA is a very prevalent condition, but is often unrecognized and untreated. Screening for this condition can have a tremendous effect on public health as a modifiable risk factor for the prevention of stroke and other CVDs.
DISCLOSURE STATEMENT
This was not an industry supported study. Grant support was provided by CTSA award No. UL1TR000445 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors thank Patience Bridges for assistance in the preparation of this manuscript.
SUPPLEMENTAL MATERIAL
Spearman correlation analysis between amplitude of fibrinolytic markers and age in OSA and non-OSA patients
Spearman correlation analysis between mean levels of fibrinolytic markers
REFERENCES
- 1.Lloyd-Jones D, Adams RJ, Brown TM, et al. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 121:e46–215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 2.Rosamond W, Flegal K, Furie K, et al. Heart disease and stroke statistics--2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- 3.Dyken ME, Somers VK, Yamada T, Ren ZY, Zimmerman MB. Investigating the relationship between stroke and obstructive sleep apnea. Stroke. 1996;27:401–7. doi: 10.1161/01.str.27.3.401. [DOI] [PubMed] [Google Scholar]
- 4.Bassetti C, Aldrich MS. Sleep apnea in acute cerebrovascular diseases: Final report on 128 patients. Sleep. 1999;22:217–23. doi: 10.1093/sleep/22.2.217. [DOI] [PubMed] [Google Scholar]
- 5.Bassetti C, Aldrich MS, Chervin RD, Quint D. Sleep apnea in pat ischemic attack and stroke: A prospective study of 59 patients. Neurology. 1996;47:1167–73. doi: 10.1212/wnl.47.5.1167. [DOI] [PubMed] [Google Scholar]
- 6.Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke. 2006;37:967–72. doi: 10.1161/01.STR.0000208215.49243.c3. [DOI] [PubMed] [Google Scholar]
- 7.Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;353:2034–41. doi: 10.1056/NEJMoa043104. [DOI] [PubMed] [Google Scholar]
- 8.Shahar E, Whitney CW, Redline S, et al. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med. 2001;163:19–25. doi: 10.1164/ajrccm.163.1.2001008. [DOI] [PubMed] [Google Scholar]
- 9.Mohsenin V, Valor R. Sleep apnea in patients with hemispheric stroke. Arch Phys Med Rehabil. 1995;76:71–6. doi: 10.1016/s0003-9993(95)80046-8. [DOI] [PubMed] [Google Scholar]
- 10.Peker Y, Carlson J, Hedner J. Increased incidence of coronary artery disease in sleep apnoea: a long-term follow-up. Eur Respir J. 2006;28:596–602. doi: 10.1183/09031936.06.00107805. [DOI] [PubMed] [Google Scholar]
- 11.Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet. 2005;365:1046–53. doi: 10.1016/S0140-6736(05)71141-7. [DOI] [PubMed] [Google Scholar]
- 12.Collet JP, Montalescot G, Vicaut E, et al. Acute release of plasminogen activator inhibitor-1 in ST-segment elevation myocardial infarction predicts mortality. Circulation. 2003;108:391–4. doi: 10.1161/01.CIR.0000083471.33820.3C. [DOI] [PubMed] [Google Scholar]
- 13.Scarabin PY, Aillaud MF, Amouyel P, et al. Associations of fibrinogen, factor VII and PAI-1 with baseline findings among 10,500 male participants in a prospective study of myocardial infarction--the PRIME Study. Prospective Epidemiological Study of Myocardial Infarction. Thromb Haemost. 1998;80:749–56. [PubMed] [Google Scholar]
- 14.De Taeye B, Smith LH, Vaughan DE. Plasminogen activator inhibitor-1: a common denominator in obesity, diabetes and cardiovascular disease. Curr Opin Pharmacol. 2005;5:149–54. doi: 10.1016/j.coph.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Angleton P, Chandler WL, Schmer G. Diurnal variation of tissue-type plasminogen activator and its rapid inhibitor (PAI-1) Circulation. 1989;79:101–6. doi: 10.1161/01.cir.79.1.101. [DOI] [PubMed] [Google Scholar]
- 16.Urano T, Sumiyoshi K, Nakamura M, Mori T, Takada Y, Takada A. Fluctuation of tPA and PAI-1 antigen levels in plasma: difference of their fluctuation patterns between male and female. Thromb Res. 1990;60:133–9. doi: 10.1016/0049-3848(90)90292-k. [DOI] [PubMed] [Google Scholar]
- 17.Juhan-Vague I, Alessi MC, Raccah D, et al. Daytime fluctuations of plasminogen activator inhibitor 1 (PAI-1) in populations with high PAI-1 levels. Thromb Haemost. 1992;67:76–82. [PubMed] [Google Scholar]
- 18.Andreotti F, Kluft C. Circadian variation of fibrinolytic activity in blood. Chronobiol Int. 1991;8:336–51. doi: 10.3109/07420529109059170. [DOI] [PubMed] [Google Scholar]
- 19.Brown NJ, Agirbasli MA, Williams GH, Litchfield WR, Vaughan DE. Effect of activation and inhibition of the renin-angiotensin system on plasma PAI-1. Hypertension. 1998;32:965–71. doi: 10.1161/01.hyp.32.6.965. [DOI] [PubMed] [Google Scholar]
- 20.Johansson L, Jansson JH, Boman K, Nilsson TK, Stegmayr B, Hallmans G. Tissue plasminogen activator, plasminogen activator inhibitor-1, and tissue plasminogen activator/plasminogen activator inhibitor-1 complex as risk factors for the development of a first stroke. Stroke. 2000;31:26–32. doi: 10.1161/01.str.31.1.26. [DOI] [PubMed] [Google Scholar]
- 21.Wiklund PG, Nilsson L, Ardnor SN, et al. Plasminogen activator inhibitor-1 4G/5G polymorphism and risk of stroke: replicated findings in two nested case-control studies based on independent cohorts. Stroke. 2005;36:1661–5. doi: 10.1161/01.STR.0000174485.10277.24. [DOI] [PubMed] [Google Scholar]
- 22.Juhan-Vague I, Pyke SD, Alessi MC, Jespersen J, Haverkate F, Thompson SG. Fibrinolytic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. ECAT Study Group. European Concerted Action on Thrombosis and Disabilities. Circulation. 1996;94:2057–63. doi: 10.1161/01.cir.94.9.2057. [DOI] [PubMed] [Google Scholar]
- 23.Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation. 1989;79:733–43. doi: 10.1161/01.cir.79.4.733. [DOI] [PubMed] [Google Scholar]
- 24.von Kanel R, Loredo JS, Ancoli-Israel S, Mills PJ, Dimsdale JE. Elevated plasminogen activator inhibitor 1 in sleep apnea and its relation to the metabolic syndrome: an investigation in 2 different study samples. Metabolism. 2007;56:969–76. doi: 10.1016/j.metabol.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 25.Zamarron C, Ricoy J, Riveiro A, Gude F. Plasminogen activator inhibitor-1 in obstructive sleep apnea patients with and without hypertension. Lung. 2008;186:151–6. doi: 10.1007/s00408-008-9076-8. [DOI] [PubMed] [Google Scholar]
- 26.Marfaing-Koka A, Roisman G, Nedelcoux H, Barbet C, Escourrou P, Bourgin P. Vasculopathy in sleep disordered breathing: PAI-1 and E-selectine correlate with SAS severity. Sleep. 2006;29:A205. Abstract Supplement. [Google Scholar]
- 27.Vogel RA. Heads and hearts - The endothelial connection. Circulation. 2003;107:2766–8. doi: 10.1161/01.CIR.0000080660.63404.68. [DOI] [PubMed] [Google Scholar]
- 28.Shimokawa H. Primary endothelial dysfunction: Atherosclerosis. J Mol Cell Cardiol. 1999;31:23–37. doi: 10.1006/jmcc.1998.0841. [DOI] [PubMed] [Google Scholar]
- 29.Duchna HW, Stoohs R, Guilleminault C, Christine Anspach M, Schultze-Werninghaus G, Orth M. Vascular endothelial dysfunction in patients with mild obstructive sleep apnea syndrome. Wien Med Wochenschr. 2006;156:596–604. doi: 10.1007/s10354-006-0341-2. [DOI] [PubMed] [Google Scholar]
- 30.Bayram NA, Ciftci B, Keles T, et al. Endothelial function in normotensive men with obstructive sleep apnea before and 6 months after CPAP treatment. Sleep. 2009;32:1257–63. doi: 10.1093/sleep/32.10.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oflaz H, Cuhadaroglu C, Pamukcu B, et al. Endothelial function in patients with obstructive sleep apnea syndrome but without hypertension. Respiration. 2006;73:751–6. doi: 10.1159/000094183. [DOI] [PubMed] [Google Scholar]
- 32.Ip MS, Lam B, Ng MM, Lam WK, Tsang KW, Lam KS. Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med. 2002;165:670–6. doi: 10.1164/ajrccm.165.5.2103001. [DOI] [PubMed] [Google Scholar]
- 33.Gami AS, Caples SM, Somers VK. Obesity and obstructive sleep apnea. Endocrinol Metab Clin North Am. 2003;32:869–94. doi: 10.1016/s0889-8529(03)00069-0. [DOI] [PubMed] [Google Scholar]
- 34.Iber C, Ancoli-Israel S, Chesson AL, Jr., Quan SF. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical specifications. 1st ed. Westchester, IL: American Academy of Sleep Medicine; 2007. [Google Scholar]
- 35.Corretti MC, Anderson TJ, Benjamin EJ, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257–65. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- 36.Cohn JN, Finkelstein S, McVeigh G, et al. Noninvasive pulse wave analysis for the early detection of vascular disease. Hypertension. 1995;26:503–8. doi: 10.1161/01.hyp.26.3.503. [DOI] [PubMed] [Google Scholar]
- 37.Kurnik PB. Circadian variation in the efficacy of tissue-type plasminogen activator. Circulation. 1995;91:1341–6. doi: 10.1161/01.cir.91.5.1341. [DOI] [PubMed] [Google Scholar]
- 38.von Kanel R, Natarajan L, Ancoli-Israel S, Mills PJ, Loredo JS, Dimsdale JE. Day/Night rhythm of hemostatic factors in obstructive sleep apnea. Sleep. 2010;33:371–7. doi: 10.1093/sleep/33.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;342:1792–801. doi: 10.1056/NEJM200006153422406. [DOI] [PubMed] [Google Scholar]
- 40.Schoenhard JA, Smith LH, Painter CA, Eren M, Johnson CH, Vaughan DE. Regulation of the PAI-1 promoter by circadian clock components: differential activation by BMAL1 and BMAL2. J Mol Cell Cardiol. 2003;35:473–81. doi: 10.1016/s0022-2828(03)00051-8. [DOI] [PubMed] [Google Scholar]
- 41.Uchiyama T, Kurabayashi M, Ohyama Y, et al. Hypoxia induces transcription of the plasminogen activator inhibitor-1 gene through genistein-sensitive tyrosine kinase pathways in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:1155–61. doi: 10.1161/01.atv.20.4.1155. [DOI] [PubMed] [Google Scholar]
- 42.Pinsky DJ, Liao H, Lawson CA, et al. Coordinated induction of plasminogen activator inhibitor-1 (PAI-1) and inhibition of plasminogen activator gene expression by hypoxia promotes pulmonary vascular fibrin deposition. J Clin Invest. 1998;102:919–28. doi: 10.1172/JCI307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naito Y, Tsujino T, Kawasaki D, et al. Circadian gene expression of clock genes and plasminogen activator inhibitor-1 in heart and aorta of spontaneously hypertensive and Wistar-Kyoto rats. J Hypertens. 2003;21:1107–15. doi: 10.1097/00004872-200306000-00010. [DOI] [PubMed] [Google Scholar]
- 44.Ranby M, Bergsdorf N, Nilsson T, Mellbring G, Winblad B, Bucht G. Age dependence of tissue plasminogen activator concentrations in plasma, as studied by an improved enzyme-linked immunosorbent assay. Clin Chem. 1986;32:2160–5. [PubMed] [Google Scholar]
- 45.Mehta J, Mehta P, Lawson D, Saldeen T. Plasma tissue plasminogen activator inhibitor levels in coronary artery disease: correlation with age and serum triglyceride concentrations. J Am Coll Cardiol. 1987;9:263–8. doi: 10.1016/s0735-1097(87)80373-x. [DOI] [PubMed] [Google Scholar]
- 46.Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8:597–619. doi: 10.5664/jcsm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foster GE, Poulin MJ, Hanly PJ. Intermittent hypoxia and vascular function: implications for obstructive sleep apnoea. Exp Physiol. 2007;92:51–65. doi: 10.1113/expphysiol.2006.035204. [DOI] [PubMed] [Google Scholar]
- 48.Lavie L. Obstructive sleep apnoea syndrome--an oxidative stress disorder. Sleep Med Rev. 2003;7:35–51. doi: 10.1053/smrv.2002.0261. [DOI] [PubMed] [Google Scholar]
- 49.Cohen MC, Rohtla KM, Lavery CE, Muller JE, Mittleman MA. Meta-analysis of the morning excess of acute myocardial infarction and sudden cardiac death. Am J Cardiol. 1997;79:1512–6. doi: 10.1016/s0002-9149(97)00181-1. [DOI] [PubMed] [Google Scholar]
- 50.Ishibashi Y, Osada N, Sekiduka H, et al. Peak time of acute coronary syndrome in patients with sleep disordered breathing. J Cardiol. 2009;53:164–70. doi: 10.1016/j.jjcc.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 51.Buchner NJ, Sanner BM, Borgel J, Rump LC. Continuous positive airway pressure treatment of mild to moderate obstructive sleep apnea reduces cardiovascular risk. Am J Respir Crit Care Med. 2007;176:1274–80. doi: 10.1164/rccm.200611-1588OC. [DOI] [PubMed] [Google Scholar]
- 52.Doherty LS, Kiely JL, Swan V, McNicholas WT. Long-term effects of nasal continuous positive airway pressure therapy on cardiovascular outcomes in sleep apnea syndrome. Chest. 2005;127:2076–84. doi: 10.1378/chest.127.6.2076. [DOI] [PubMed] [Google Scholar]
- 53.Milleron O, Pilliere R, Foucher A, et al. Benefits of obstructive sleep apnoea treatment in coronary artery disease: a long-term follow-up study. Eur Heart J. 2004;25:728–34. doi: 10.1016/j.ehj.2004.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spearman correlation analysis between amplitude of fibrinolytic markers and age in OSA and non-OSA patients
Spearman correlation analysis between mean levels of fibrinolytic markers